
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of geometric isomerism on the anticancer activity of the monofunctional platinum complex trans-[Pt(NH3)2(phenanthridine)Cl]NO3†
Wen
Zhou
 ab,
Mohammad
Almeqdadi
b,
Michael E.
Xifaras
b,
Imogen A.
Riddell‡
a,
Ömer H.
Yilmaz
*b and
Stephen J.
Lippard
*a
ab,
Mohammad
Almeqdadi
b,
Michael E.
Xifaras
b,
Imogen A.
Riddell‡
a,
Ömer H.
Yilmaz
*b and
Stephen J.
Lippard
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: lippard@mit.edu
bThe Koch Institute for Integrative Cancer Research, Massachusetts Institute of Tehcnology, Cambridge, MA 02139, USA. E-mail: ohyilmaz@mit.edu
First published on 27th February 2018
Abstract
A trans-DDP based monofunctional phenanthridine Pt(II) complex was synthesized and characterized. Its anticancer activity was studied in vitro on a panel of human cancer cell lines and mouse intestinal cancer organoids. This complex displays significant antitumor properties, with a different spectrum of activity than that of classic bifunctional cross-linking agents like cisplatin.
Initially the development of platinum-based anticancer agents was focused on the synthesis and evaluation of complexes that obeyed structure–activity relationships (SARs)1 set forth in the 1970's. From these studies, carboplatin and oxaliplatin were identified and both have gone on to receive worldwide approval as chemotherapeutics. Today they continue to play an important role in cancer treatment and management. However, the prevalence of inherent and acquired resistance to platinum treatment2–4 requires the development of new complexes that operate through different mechanisms. Monofunctional DNA binding platinum(II) complexes represent an alternative class of very potent anticancer agents.5 Unlike cisplatin and its derivatives, these complexes contain only one labile ligand and thus form a single covalent bond to DNA.
Pyriplatin, cis-diammine(pyridine)chloroplatinum(II), a monofunctional, cationic platinum(II) compound, displayed a spectrum of activity that differed from that of any of the clinically approved platinum drugs.6 However, the overall potency of pyriplatin was much less than that of cisplatin in all cancer cell lines tested. We subsequently pursued a rational, structure-based approach for improving the potency of monofunctional platinum anticancer drugs.7 By determining the X-ray structure of RNA polymerase II stalled at a pyriplatin adduct on a short DNA duplex containing a bound RNA, they deduced that the pyriplatin–DNA adduct may prevent translocation from the active site and block RNA chain elongation because of steric interactions and hydrogen bonding with the bridge helix, operating by a mechanism of transcription inhibition that differs from that of cis-DDP. Based on this rationale, phenanthriplatin was designed, synthesized, and discovered to have significantly greater efficacy for killing cancer cells than either cis-DDP or oxaliplatin.6 Moreover, according to the National Cancer Institute (NCI) cytotoxicity screening assay, phenanthriplatin exhibited a unique cancer cell-killing profile compared to all other platinum agents held in the NCI archives. The success of phenanthriplatin strongly encouraged further study of monofunctional Pt(II) compounds as anticancer drug candidates. In the present work, we report the synthesis and anticancer activity of a trans-DDP based monofunctional phenanthridine Pt(II) complex, in further pursuit of novel platinum cancer drug candidates.
trans-[Pt(NH3)2(phenanthridine)Cl]NO3, 1, was prepared following the procedure developed for phenanthriplatin6 and characterized by a variety of analytical methods, including 1H and 13C NMR spectroscopy, ESI-MS, and elemental analysis. Complex 1 was also structurally investigated by single crystal X-ray diffraction.
The solid-state study reveals that complex 1 adopts the expected square-planar coordination geometry with a trans arrangement of ammine ligands. Similar to phenanthriplatin, the plane of the phenanthridine ligand in complex 1 is approximately perpendicular to that of the platinum coordination plane (Fig. 1). The distance between the platinum atom and the overhanging carbon atom on the phenanthridine ligand (black arrow in Fig. 1) of complex 1 (3.224 Å) is nearly identical to that previously reported for phenanthriplatin (3.220 Å). This observation suggests that, like phenanthriplatin, complex 1 may show selectivity for nucleophilic binding sites on DNA while inhibiting unfavourable reactions with cytoplasmic sulphur nucleophiles.
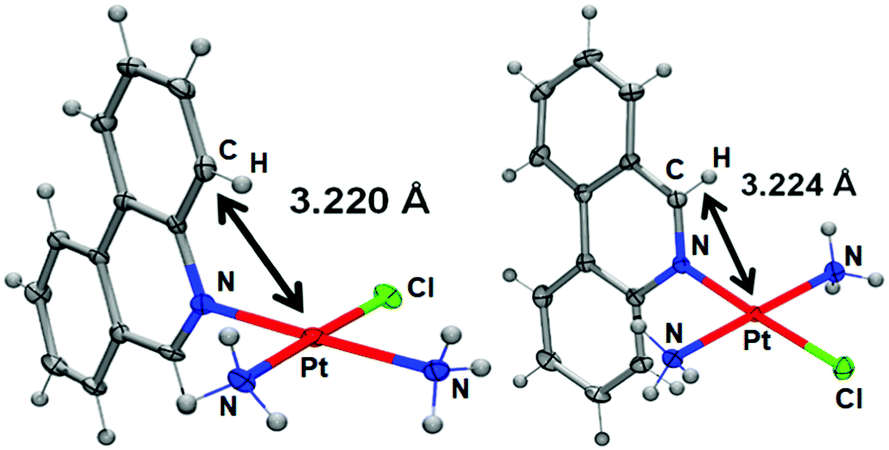 | ||
| Fig. 1 Comparison of the X-ray crystal structures of phenanthriplatin (left) and 1 (right). The platinum–carbon distances shown reveal that these two complexes exhibit similar degrees of steric protection from incoming nucleophiles at the axial positions. | ||
We anticipated that, as with phenanthriplatin, the steric bulk of the phenanthridine ligand in complex 1 would impede procession of both DNA8 and RNA9 polymerases and may interfere with other essential enzymatic functions,10 leading to potent anticancer activity. The in vitro anticancer activity of complex 1 was initially probed using fluorescence microscopy and the LIVE/DEAD cell assay, which is a combination of the ethidium homodimer-1 assay and staining with calcein AM (Fig. 2). Live cells stained with calcein AM yield a green fluorescence signal, whereas dead cells exhibit no fluorescence or a red signal due to the ethidium homodimer-1. As observed in Fig. 2, A2780 ovarian cancer cells treated with complex 1 (10 μM) for 48 h were mostly dead, whereas control cells without any drug treatment under the same conditions were alive. The IC50 for complex 1 against A2780 at 48 h is 4.94 ± 1.52 μM.
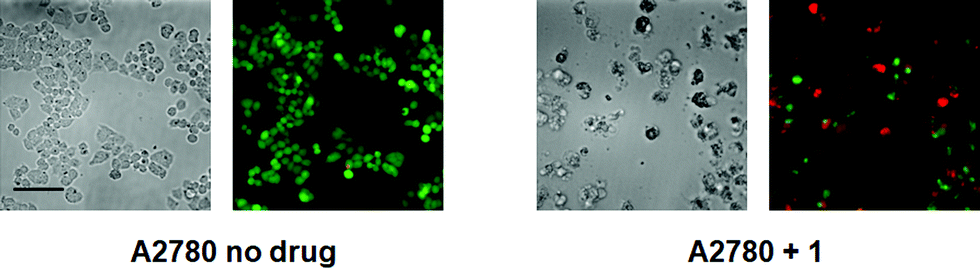 | ||
| Fig. 2 Calcein AM/ethidium homodimer-1 cell viability assay, details of which may be found in the main text and ESI† (scale bar, 44 μm). | ||
The cytotoxicity of the newly synthesized monofunctional complex 1 was next evaluated against a panel of four human cancer cell lines of different origin (A2780, ovarian cancer; A2780CP70, cisplatin–resistant ovarian cancer; MCF-7, breast cancer; HCT116, colorectal cancer) using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (Table 1). Cell viability was recorded after 72 h and compared to results obtained with cisplatin, oxaliplatin, and phenanthriplatin under identical conditions. Complex 1 displayed cytotoxicity similar to that of oxaliplatin but failed to perform as well as phenanthriplatin.
| IC50 (μM) | A2780 | A2780CP70 | MCF-7 | HCT116 |
|---|---|---|---|---|
| a Data reflect the mean and standard deviation of results from three separate experiments, each performed in triplicate. | ||||
| Cisplatin | 1.7 ± 1.0 | 9.9 ± 2.1 | 9.0 ± 3.2 | 16.8 ± 5.1 |
| Oxaliplatin | 0.21 ± 0.09 | 5.1 ± 2.2 | 12.2 ± 2.4 | 7.4 ± 1.7 |
| Phenanthriplatin | 0.19 ± 0.03 | 0.29 ± 0.06 | 0.80 ± 0.05 | 1.2 ± 0.5 |
| 1 | 1.9 ± 0.2 | 6.5 ± 1.4 | 14.9 ± 0.8 | 8.9 ± 2.3 |
The selectivity of complex 1 for tumour cells over healthy cells was probed next. Normal lung fibroblasts (MRC5) and cancerous lung (A549) cells were used to evaluate the selectivity of complex 1 for cancer vs. healthy cells. Based on the MTT assay, the ratio of IC50 values in healthy MRC5 cells to those in cancerous A549 cells was 0.9 for cisplatin compared with 2.9 for complex 1. The higher ratio obtained for complex 1 reveals its selectivity for cancer cells, at least in the cellular monolayer assays used in this study.
Next, we sought to quantitate the cellular uptake and the subcellular distribution of complex 1. The nuclei, cytoplasm, membrane, and whole cell concentrations of platinum were measured by graphite furnace atomic absorption spectroscopy (GFAAS) after treatment of the cells with the compounds for 5 h (Fig. 3). Phenanthriplatin was used as a comparison, and oxaliplatin and cisplatin were used as controls. After 5 h incubation with the Pt compounds, phenanthriplatin and complex 1 were taken up by cells more effectively than cisplatin or oxaliplatin. These results reflect the ability of the larger, hydrophobic heterocyclic phenanthridine ligand to facilitate uptake of the phenanthriplatin and complex 1 cations through the cytoplasmic membrane. However, the uptake of phenanthriplatin is, surprisingly, much higher than that of complex 1, indicating that geometric isomerism may play an important role in cellular uptake. Most of the platinum is found in the cytoplasm. The higher cellular uptake of phenanthriplatin most likely contributes to its enhanced cytotoxicity compared with the other platinum compounds.
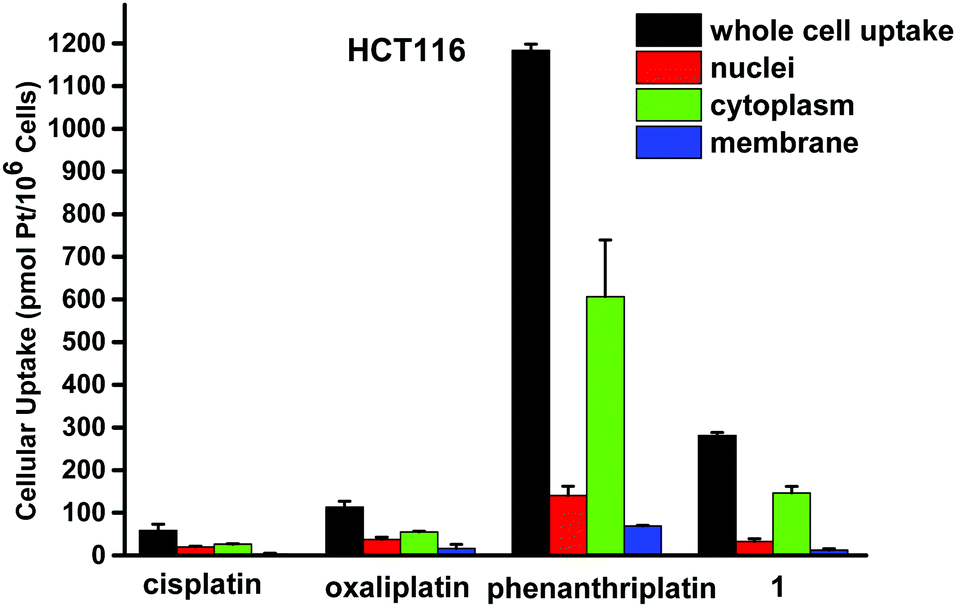 | ||
| Fig. 3 Cellular distribution of Pt in HCT116 treated with cisplatin, oxaliplatin, phenanthriplatin and 1. Cells were treated with 20 μM platinum complex for 5 h. Platinum uptake is presented in units of pmol of Pt per 106 cells. | ||
DNA is the cellular target of platinum-based anticancer agents, thus the ability of complex 1 to platinate nuclear DNA was assessed. Two million HCT116 cells were treated with growth medium containing 20 μM cisplatin, oxaliplatin, phenanthriplatin, or complex 1 for 5 h, followed by incubation in fresh media for an additional 16 h. The intracellular DNA was isolated and the quantity of bound platinum was measured by GFAAS. The extent of DNA platination was determined to be 23.2 ± 3.6 Pt adducts per 106 nucleotides for cisplatin, 6.7 ± 1.0 Pt adducts per 106 nucleotides for oxaliplatin, 87.8 ± 6.6 Pt adducts per 106 nucleotides for phenanthriplatin, and 6.3 ± 1.6 Pt adducts per 106 nucleotides for complex 1.
The effect of cisplatin, oxaliplatin, phenanthriplatin, and complex 1 on supercoiled pUC19 DNA in Tris–HCl buffer (0.1 M, pH 7.4) at 37 °C after incubating in the dark for 6 h was studied by agarose gel electrophoresis (Fig. 4). Comparing the migration of pUC19 in the presence of the different platinum agents we see that DNA modified by complex 1 behaves similarly to that modified by cisplatin and oxaliplatin. In contrast, migration of phenanthriplatin11 treated DNA is consistent with relaxation of the negatively supercoiled plasmid in response to intercalation of, and/or photocleavage by, the phenanthridine ring.12,13 Our results therefore indicate that both the choice of coordinating ligands as well as the stereochemistry at the platinum center dictate the ability of platinum compounds to unwind duplex DNA.14
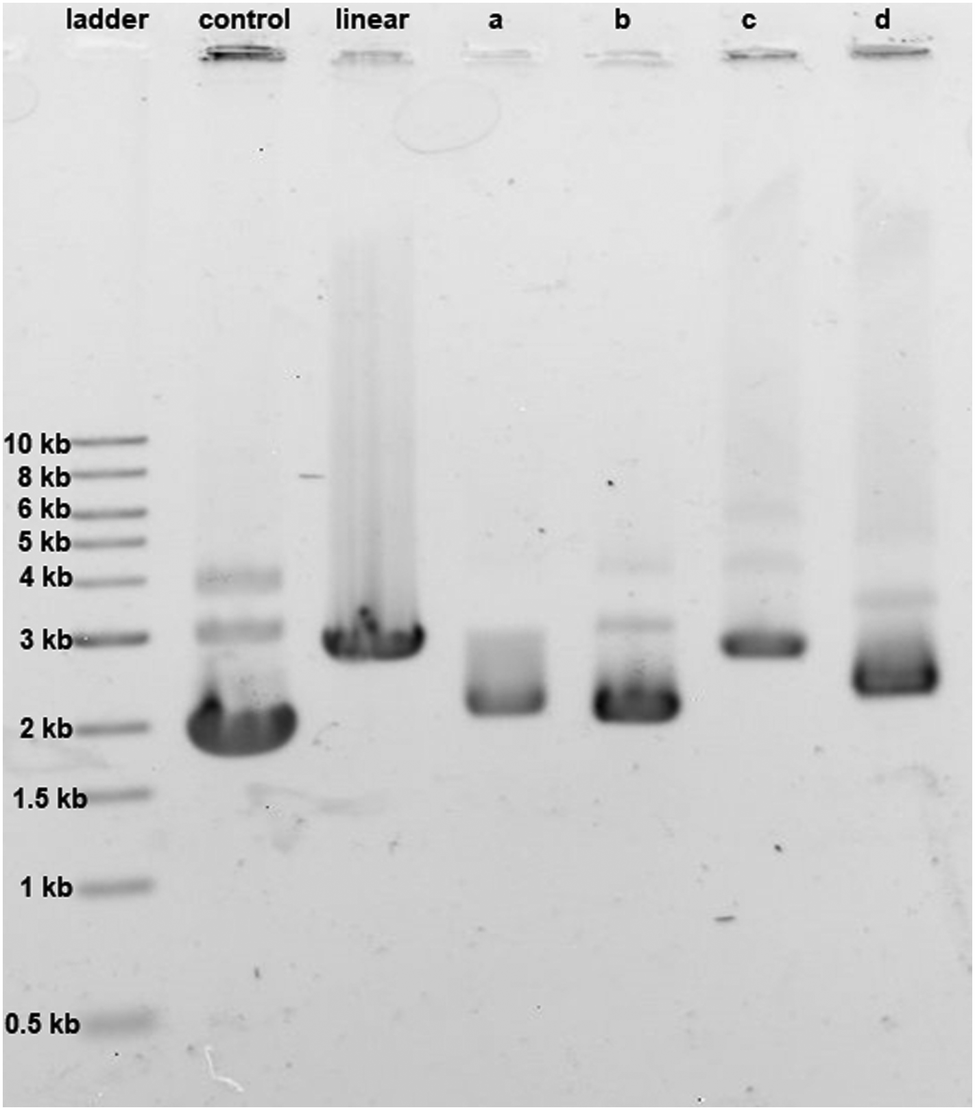 | ||
| Fig. 4 Unwinding effect of 30 μg mL−1 concentration of (a) cisplatin, (b) oxaliplatin, (c) phenanthriplatin, and (d) complex 1 on pUC19 DNA (15 μg mL−1). For control, no platinum compound was added; for linear, the pUC19 plasmid was restriction digested by HindIII. | ||
γH2AX, the phosphorylated form of histone protein H2AX, is a known biomarker of DNA damage induced by platinum drugs.15,16 γH2AX can be detected by immunostaining. As shown in Fig. 5, fluorescence microscopy was used to visualize the localization of γH2AX in the nucleus of HCT116 cells treated with complex 1, indicating genomic DNA damage. Apoptotic cells normally exhibit changes in cell morphology, such as blebbing, chromatin condensation, and nuclear fragmentation, which can be observed by fluorescence microscopy. As shown in Fig. 5, morphological changes occur following treatment of HCT116 cells with 20 μM complex 1 for 24 h. Chromatin condensation and nuclear fragmentation were also identified by Hoechst staining. All these cell-based experiments clearly indicate that complex 1 can effectively induce DNA damage and thus lead to cell cycle arrest and apoptosis in cancer cells.
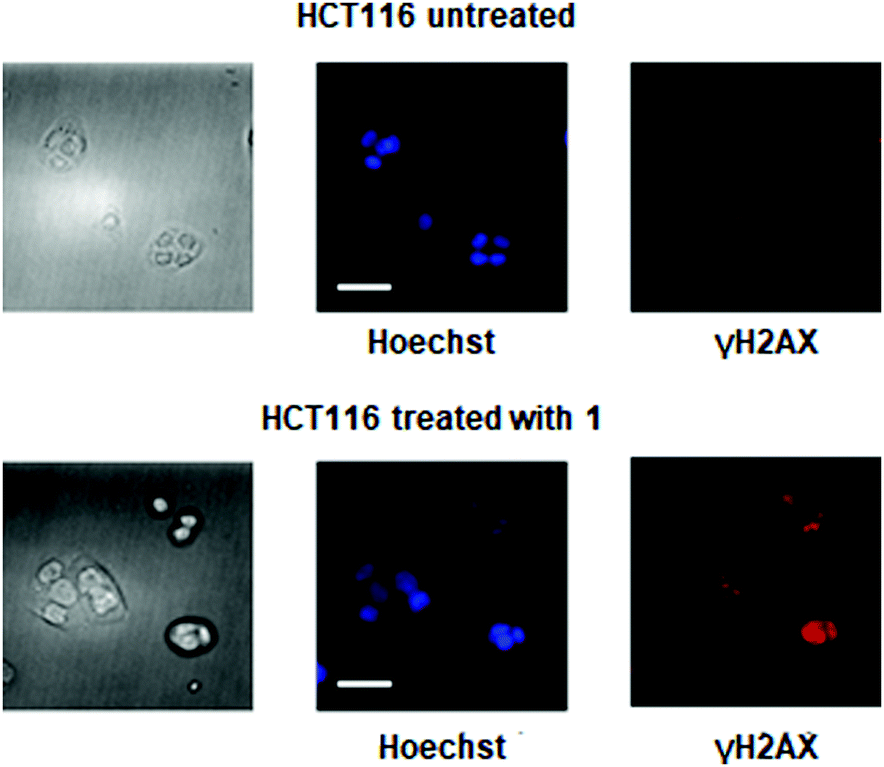 | ||
| Fig. 5 DNA damage and cellular responses of HCT116 cells treated with complex 1: cellular images, left; nuclear staining, blue; immunostaining of the biomarker of DNA damage, phosphorylatedH2AX (γH2AX), red (scale bar, 30 μm). | ||
Finally, intestinal organoids are three-dimensional primary cultures derived from intestinal stem cells that closely recapitulate the in vivo tissue structure.17 As previously described, organoid cellular composition closely mimics in vivo biology in terms of secretory function, absorptive function, and pathophysiologic states.18 Organoid assays have therefore gained in popularity for assessing drug toxicity and selectivity in vivo. Furthermore, organoid cultures are amenable for high-throughput drug screening assays that can accurately predict in vivo outcomes.19 The adenomatous polyposis coli (APC) gene product is essential for intestinal stem cell homeostasis as it regulates intracellular beta-catenin concentration and subsequent stem cell proliferation. Loss of function mutations in the APC gene occur as early events in a majority of colorectal cancers.20 Additional oncogenic mutations such as those involving KRAS and TP53 subsequently occur and are associated with more aggressive tumor phenotypes, permitting local invasion and metastasis to lymph nodes and the liver.
To validate the efficacy of phenanthriplatin and complex 1, we utilized the Apc-knockout KrasG12D p53-knockout (AKP) engineered mouse intestinal cancer organoid model. By quantifying organoid formation, morphology, and cell viability, we determined the cytotoxicity of each drug (Fig. 6). Phenanthriplatin and complex 1 both exhibited significantly lower IC50 values than that of both oxaliplatin and cisplatin. Taken together, these findings indicate that complex 1 has enhanced potency in treating intestinal cancers compared to oxaliplatin. In vivo studies are needed in order to understand how these results reflect the potential use of complex 1 clinically.
 | ||
| Fig. 6 Oxaliplatin, phenanthriplatin, and complex 1 inhibit AKP intestinal cancer organoid growth in vitro at different concentrations: (1) control AKP organoids exhibit a large cystic morphology, consistent with their highly prolific and poorly differentiated state; (2) oxaliplatin, phenanthriplatin, and complex 1 induce organoid death, as shown by cell necrosis within the organoid lumen. | ||
In conclusion, by replacing one of the chloride ligands of trans-DDP with the phenanthridine ligand, the monofunctional Pt(II) complex 1 was successfully synthesized, and it functioned as an effective anticancer agent. The results of cellular uptake, DNA platination, and DNA unwinding studies show very different activities between phenanthriplatin and its geometric isomer complex 1. This result indicates that the stereochemistry at the Pt(II) center may play a great role in affecting the properties of monofunctional platinum based anticancer agents.
This work was supported by grant CA034992 to S. J. L. and NIH grants AG045144, CA034992, CA211184 to O. H. Y. In addition, O. H. Y. is also a Pew-Stewart Scholar for Cancer Research and a Sidney Kimmel Scholar.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. W. Hambley, Coord. Chem. Rev., 1997, 166, 181 CrossRef CAS.
- G. Chu, J. Biol. Chem., 1994, 269, 787 CAS.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467 CrossRef CAS PubMed.
- D. W. Shen, L. M. Pouliot, M. D. Hall and M. M. Gottesman, Pharmacol. Rev., 2012, 64, 706 CrossRef CAS PubMed.
- T. C. Johnstone, G. Y. Park and S. J. Lippard, Anticancer Res., 2014, 34, 471 CAS.
- G. Y. Park, J. J. Wilson, Y. Song and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11987 CrossRef CAS PubMed.
- D. Wang, G. Y. Zhu, X. H. Huang and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9584 CrossRef CAS PubMed.
- M. T. Gregory, G. Y. Park, T. C. Johnstone, Y.-S. Lee, W. Yang and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9584 CrossRef PubMed.
- M. W. Kellinger, G. Y. Park, J. Chong, S. J. Lippard and D. Wang, J. Am. Chem. Soc., 2013, 135, 13054 CrossRef CAS PubMed.
- I. A. Riddell, K. Agama, G. Y. Park, Y. Pommier and S. J. Lippard, ACS Chem. Biol., 2016, 11, 2996 CrossRef CAS PubMed.
- K. S. Lovejoy, R. C. Todd, S. Zhang, M. S. McCormick, J. A. D'Aquino, J. T. Reardon, A. Sancar, K. M. Giacomini and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8902 CrossRef CAS PubMed.
- L. Gude, M. A.-J. Fernández, K. B. Grant and A. Lorente, Bioorg. Med. Chem. Lett., 2002, 12, 3135 CrossRef CAS PubMed.
- J. R. Choudhury and U. Bierbach, Nucleic Acids Res., 2005, 33, 5622 CrossRef CAS PubMed.
- M. V. Keck and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 3386 CrossRef CAS.
- P. L. Olive and J. P. Banath, Cytometry, Part B, 2009, 76B, 79 CrossRef CAS PubMed.
- Y. R. Zheng, K. Suntharalingam, T. C. Johnstone, H. Yoo, W. Lin, J. G. Brooks and S. J. Lippard, J. Am. Chem. Soc., 2014, 136, 8790 CrossRef CAS PubMed.
- J. Roper, T. Tammela, N. M. Cetinbas, A. Akkad, A. Roghanian, S. Rickelt, M. Almeqdadi, K. Wu, M. A. Oberli, F. Sánchez-Rivera, Y. K. Park, X. Liang, G. Eng, M. S. Taylor, R. Azimi, D. Kedrin, R. Neupane, S. Beyaz, E. T. Sicinska, Y. Suarez, J. Yoo, L. L. Chen, L. Zukerberg, P. Katajisto, V. Deshpande, A. J. Bass, P. N. Tsichlis, J. Lees, R. Langer, R. O. Hynes, J. Z. Chen, A. Bhutkar, T. Jacks and Ö. H. Yilmaz, Nat. Biotechnol., 2017, 35, 569 CrossRef CAS PubMed.
- A. Fatehullah, S. H. Tan and N. Barker, Nat. Cell Biol., 2016, 18, 246 CrossRef PubMed.
- H. Gao, J. M. Korn, S. Ferretti, J. E. Monahan, Y. Wang, M. Singh, C. Zhang, C. Schnell, G. Yang, Y. Zhang, O. A. Balbin, S. Barbe, H. Cai, F. Casey, S. Chatterjee, D. Y. Chiang, S. Chuai, S. M. Cogan, S. D. Collins, E. Dammassa, N. Ebel, M. Embry, J. Green, A. Kauffmann, C. Kowal, R. J. Leary, J. Lehar, Y. Liang, A. Loo, E. Lorenzana, E. Robert McDonald Iii, M. E. McLaughlin, J. Merkin, R. Meyer, T. L. Naylor, M. Patawaran, A. Reddy, C. Roelli, D. A. Ruddy, F. Salangsang, F. Santacroce, A. P. Singh, Y. Tang, W. Tinetto, S. Tobler, R. Velazquez, K. Venkatesan, F. Von Arx, H. Q. Wang, Z. Wang, M. Wiesmann, D. Wyss, F. Xu, H. Bitter, P. Atadja, E. Lees, F. Hofmann, E. Li, N. Keen, R. Cozens, M. R. Jensen, N. K. Pryer, J. A. Williams and W. R. Sellers, Nat. Med., 2015, 21, 1318 CrossRef CAS PubMed.
- L. N. Kwong and W. F. Dove, Adv. Exp. Med. Biol., 2009, 656, 85 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1582154. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc00393a |
| ‡ Current address: School of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. |
| This journal is © The Royal Society of Chemistry 2018 |