
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective protein unfolding: a universal mechanism of action for the development of irreversible inhibitors†
Samuel
Askin
a,
Thomas E. H.
Bond
a,
Alanna E.
Sorenson
 a,
Morgane J. J.
Moreau
a,
Helma
Antony
a,
Rohan A.
Davis
b and
Patrick M.
Schaeffer
*a
a,
Morgane J. J.
Moreau
a,
Helma
Antony
a,
Rohan A.
Davis
b and
Patrick M.
Schaeffer
*a
aCentre for Biodiscovery & Molecular Development of Therapeutics, James Cook University, 142, James Cook Drive, Townsville, QLD 4811, Australia. E-mail: patrick.schaeffer@jcu.edu.au
bGriffith Institute for Drug Discovery, Griffith University, Brisbane, QLD 4111, Australia
First published on 22nd January 2018
Abstract
High-throughput differential scanning fluorimetry of GFP-tagged proteins (HT-DSF-GTP) was applied for the identification of novel enzyme inhibitors acting by a mechanism termed: selective protein unfolding (SPU). Four different protein targets were interrogated with the same library to identify target-selective hits. Several hits selectively destabilized bacterial biotin protein ligase. Structure–activity relationship data confirmed a structure-dependent mechanism of protein unfolding. Simvastatin and altenusin were confirmed to irreversibly inactivate biotin protein ligase. The principle of SPU combined with HT-DSF-GTP affords an invaluable and innovative workflow for the identification of new inhibitors with potential applications as antimicrobials and other biocides.
Enzyme inhibitors are invaluable for the development of drugs, herbicides and pesticides. The potency and specificity of inhibitors are key to their successful applications in modern medicine and agriculture. Inhibitors can be either reversible or irreversible. Reversible inhibitors bind non-covalently to the target through a variety of mechanisms e.g. competitive, uncompetitive, non-competitive and mixed inhibition. Irreversible inhibitors often react with the enzyme via covalent bond formation. In the last decades very few fundamentally new mechanisms of inhibition leading to inactivation of protein targets have been discovered. A new mechanism of action was recently unveiled for nucleozin, a potent inhibitor of the influenza nucleoprotein.1 Nucleozin aggregates nucleoprotein rendering its packaging in the virus impossible.2 Destabilizing compounds are commonly dismissed as they are assumed to be non-specific (e.g. chaotropic agents) but a few studies have shown that organometallic complexes3,4 and nanoparticles5 can selectively destabilize proteins. Other systems such as PROTAC can flag target proteins to the proteasome.6,7 DSF and DSF-GTP are perfectly suited to screen compounds in high-throughput (HT) by measuring their effect on the thermal stability of a target protein.8–10
In this work we screened an identical library of 940 compounds with four different proteins in order to identify target selective hits (Fig. 1 and Fig. S1, ESI†). We used HT-DSF-GTP to compare the effect of each compound on the thermal stability of E. coli (Ec) biotin protein ligase11 (BirA), dengue virus RNA-dependent RNA polymerase9 (RdRp), the putative replicative DNA helicase (DnaB) and primase (DnaG)12 of Burkholderia pseudomallei (Bp).
 | ||
| Fig. 1 HT DSF-GTP screening of 940 pure compounds with Ec BirA-GFP. Compounds (1 μL at 5 mM in DMSO, Compounds Australia) were dispensed in 96-well clear PCR plates (Biorad, HSP9601). Ec BirA-GFP (49 μL at 1 μM) was added to each compound for 15 min at RT prior subjecting the reactions to a melt-curve protocol (30–85 °C at 0.5 °C/30 s) in a IQ5 iCycler (Bio-Rad). Tm were analyzed as previously described.10 Structures of selected compounds and respective ΔTm are shown. See ESI,† for detailed methods and Fig. S1 and S2A for extended data. | ||
All targets were produced as fusion proteins including both N-terminal hexahistidine and C-terminal GFP tags as previously described for RdRp-GFP, Bp DnaG-GFP,9 and BirA-GFP.11 Proteins (1 μM) were reacted with compounds (100 μM) in 96-well PCR plates for 15 min at room temperature before subjecting the reactions to a melt-curve protocol in a real-time PCR cycler (Fig. 1 and Fig. S1, ESI†). Transition midpoint (Tm) values were recorded and processed as described previously using an automated Tm peak recognition software.10 For each compound, the net change in Tm (ΔTm) was determined by subtracting the Tm of ‘Target-GFP’ from the Tm of ‘Target-GFP + compound’ in identical buffer conditions. After the initial screen (801 compounds from Open Collection (Scaffolds) and 139 from Open Access Natural Product-Based Library, Compounds Australia) it became evident that of the compounds that affected a protein, the majority decreased the proteins’ Tm. This is particularly obvious for Ec BirA-GFP (Fig. 1). From the initial screen, 94 hits were identified (i.e. the most stabilizing or destabilizing hits) and rescreened with all four targets to evaluate the reproducibility of the assay. Only four compounds that produced no distinguishable peak in the first screen were subsequently identified in the second screen.
The study was focussed on the hits identified in the Ec BirA-GFP screen as they had the largest effect on protein stability. A positive ΔTm cut-off value was set to +3 °C which is equal to the increase in Tm of Ec BirA-GFP in the presence of 2 μM of biotin.11
We also set a negative ΔTm cut-off value at −7 °C due to the high number of destabilizing compounds identified for Ec BirA-GFP. Eleven selective hits were identified, with only one hit that significantly increased Ec BirA-GFP's Tm (Fig. 1 and 2A). The remaining 10 compounds thermally destabilized Ec BirA-GFP between −7 and −11 °C. These hits do not affect GFP (internal control) suggesting a target selective destabilizing mechanism. This is further supported by the fact that the remaining targets were mostly unaffected although in a different buffer. After rescreening and identification of false positives no compound destabilized GFP or any of the remaining protein targets ≥5 °C (see ESI,† Fig. S1).
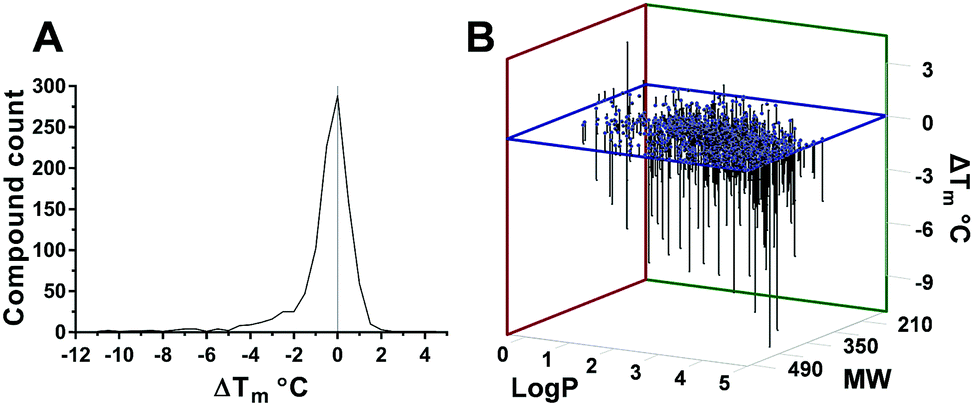 | ||
Fig. 2 (A) Statistical distribution of Ec BirA-GFP stabilizing and destabilizing compounds. (B) Correlation between log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and MW of compounds, and their effect on Ec BirA-GFP Tm. P and MW of compounds, and their effect on Ec BirA-GFP Tm. | ||
The distribution of ΔTm values obtained for Ec BirA-GFP (Fig. 2A) shows that the majority of compounds does not significantly affect the Tm of Ec BirA-GFP (i.e. ±2 °C) with maximal effects recorded between −11 and +4.5 °C (Fig. 2A). Only one stabilizing hit 1 (ΔTm = +4.5 °C) was identified with a MW of 341 and logP value of 0.9 (Fig. 1 and 2B). No Ec BirA-GFP Tm peak was observed with structural analogues 2 and 3 (Fig. 1). For destabilizing hits, a clear correlation can be seen between increasing MW and logP values, and decreasing Tm (Fig. 2B). Destabilizing compounds are commonly written off as being non-specific. Strikingly, several of the Ec BirA selective destabilizing compounds were either serrulatanes134a–e (Fig. 1 and Fig. S2B, ESI†) or tetrahydroanthraquinones14,155a–l (Fig. 1 and Fig. S2C, ESI†) suggesting a structure-specific mechanism of protein destabilization. The strong structure–activity relationship (SAR) and striking correlation between the logP and MW of Ec BirA selective destabilizing compounds lead us to hypothesize that they may present an untapped source of inhibitors.
First, we examined the concentration dependence of Ec BirA-GFP hits (see Fig. 3A and Fig. S3A, ESI†), including stabilizing hit 1; destabilizing hits 2 and 3 which eliminated the Tm peak of Ec BirA, RdRp and Bp DnaG; serrulatanes 4a and 4b; 6; mitchellene B167, casticin 8, altenusin 9, lovastatin 10a, 11; and inactive compound Ia (see ESI,† Fig. S2A–D). All hits reduced the Tm of Ec BirA-GFP in a concentration-dependent fashion including 2 and 3 highlighting that for these the loss of Tm peak was due to protein unfolding at 100 μM.
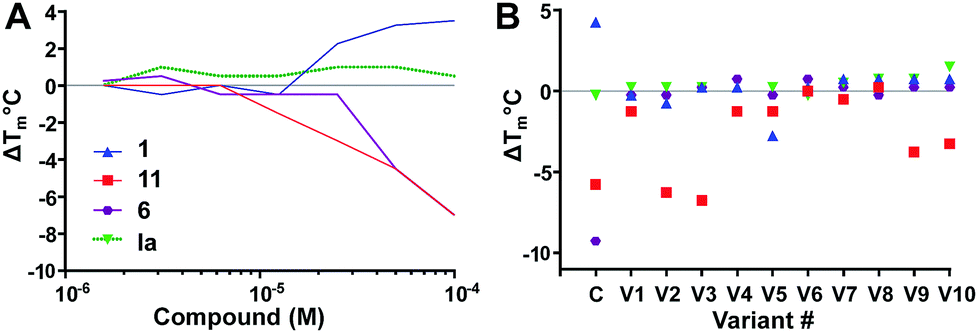 | ||
| Fig. 3 Concentration dependence (A) and SAR studies (B). (A) Effect of two-fold serially diluted compounds on Ec BirA-GFP Tm. One inactive compound Ia (see ESI,† Fig. S2D) and eleven hits were selected from the initial screen. Only four compounds are shown for clarity (see ESI,† Fig. S3A for full data). (B) Effect of scaffold analogues on Ec BirA-GFP Tm. For each selected inactive compound and hits (Open Collection Scaffolds Library), ten analogues (V1–V10) were tested. The same four compounds are shown in panel A and B for clarity (see ESI,† Fig. S3B for full data). | ||
Next, we examined SAR of four Ec BirA-GFP selective hits and four inactive compounds (see Fig. 3B and ESI,† Fig. S3B for extended data). For each selected compound, we tested ten structural analogues on Ec BirA-GFP's Tm (V1–V10). No analogue of 1 and 6 affected Ec BirA-GFP but several analogues of both 11 and 12 significantly decreased its Tm (ΔTm range: −3.25 to −6.75 °C; see ESI,† Fig. S2A and S3B). Finally, with the exception of two analogues of inactive compound Ib (see Fig. S2D and S3B, ESI†) being slightly destabilizing (≤2 °C), none of the others affected Ec BirA-GFP. The data supported that target selectivity of destabilizing hits is structure dependent.
Lovastatin 10a, a HMG CoA reductase inhibitor was identified as a Ec BirA-GFP destabilizing hit (ΔTm = −7 °C) in the initial screen. 10a was only little to moderately destabilizing towards DnaG-, DnaB- and RdRp-GFP (ESI,† Fig. S1B). Simvastatin 10b (Fig. 4) has broad antimicrobial activity against various Gram-positive pathogens and is also active against several Gram-negative pathogens e.g. Ec, Acinetobacter baumannii, Salmonella typhimurium, Klebsiella pneumoniae, and Pseudomonas aeruginosa with MIC ranging from 8–32 μg mL−1 when their outer membrane permeability is compromised using sub-inhibitory concentrations of colistin.17 This prompted us to perform a small scale statin-focussed SAR study. For this, 10a, 10b, pravastatin 10c and fluvastatin 10d were tested on Ec BirA-GFP (Fig. 4 and Fig. S2A, ESI†). 10a, 10b and 10d destabilized Ec BirA-GFP, however 10b was more potent at lower concentrations and 10d was the least potent. 10c, an open lactone form, did not destabilize Ec BirA-GFP. This observation suggests an important role for the lactone ring of statins in BirA destabilization. It is important to note that a methyl to hydroxyl group substitution in 10c could also be the cause of its inactivity. With 10d, it is more difficult to draw any parallel conclusions as its structure is very different to the three other statins (see ESI,† Fig. S2A). Nevertheless we can hypothesize that the polyaromatic ring structure of 10d is most likely responsible for Ec BirA-GFP destabilization rather than its open lactone structure, and that its protein binding site is thus different to the site of binding of 10a and 10b.
 | ||
| Fig. 4 SAR and concentration dependence of statins 10a–d on Tm of Ec BirA-GFP. Error bars represent SD (N = 2). See ESI,† Fig. S2A for structure of 10d. | ||
Statins were further investigated with GFP-Basta18 in isothermal reactions to assess if they induce BirA aggregation (Fig. 5A). Ec BirA-GFP (1 μM) was reacted with statins (100 μM) in the presence of biotin, ATP and MgCl2 for 30 min at 37 °C before centrifugation and quantification of the soluble fluorescent protein fraction (Ffold) by fluorimetry of the GFP domain (see ESI,† for detailed methods). 10b was most destabilizing, leading to ∼70% protein aggregation after 10 min incubation at 37 °C followed by 10a. Neither 10c nor 10d caused significant aggregation of Ec BirA-GFP over 30 min in these conditions. 10a and 10b were further tested on two orthologous BirA, i.e. Bp and Mycobacterium tuberculosis (Mt) BirA-GFP.19,20 These proteins share ∼30% sequence identity with each other and the Ec BirA. Both statins showed very little effect on Mt BirA-GFP at 100 μM (ΔTm = −2 °C) and moderate destabilizing effects on Bp BirA-GFP (cf. ESI,† Fig. S4A) revealing a preferential destabilizing effect of 10a and 10b on Ec BirA. Using a GFP-based biotinylation activity assay,11,21 we confirmed that 10b leads to Ec BirA-GFP inactivation (Fig. 5B). For this, Ec BirA-GFP was pre-incubated at 37 °C with 10b or 10c for 1 h prior addition of the GFP-AviTag substrate and biotinylation reaction. Under these conditions, 10b inhibited Ec BirA-GFP activity by 83% (Fig. 5B, see ESI,† for detailed methods). As expected, 10c did not affect Ec BirA-GFP. Taken together, the data show that inhibition of biotinylation activity is associated with 10b-induced unfolding and aggregation of Ec BirA-GFP.
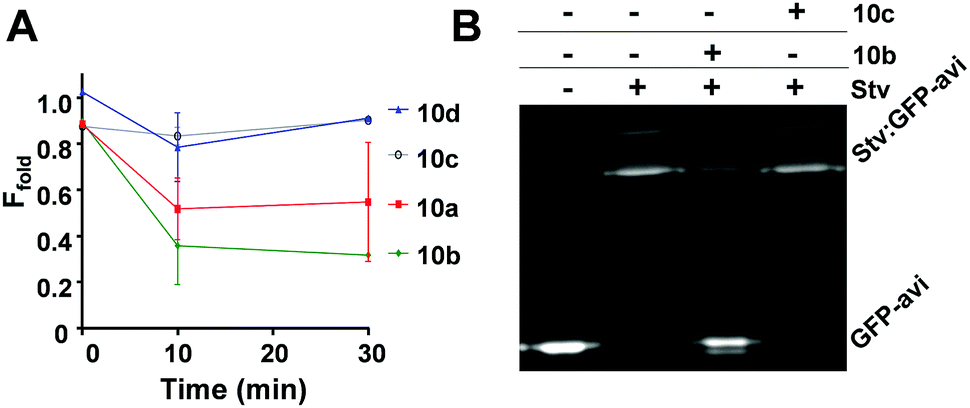 | ||
| Fig. 5 Effect of statins on aggregation and biotinylation activity of Ec BirA-GFP. (A) Isothermal aggregation reactions18 were performed at 37 °C with statins 10a–d at 100 μM. Soluble fluorescent protein fraction (Ffold) was determined by measuring residual fluorescence of Ec BirA-GFP in the reaction supernatant after centrifugation. Control reactions were performed without a statin. Error bars represent SD (N = 2). (B) Inhibition of Ec BirA-GFP biotinylation activity by 10b and 10c at 37 °C. Inhibition of GFP-AviTag substrate biotinylation was determined by streptavidin (Stv)-induced band-shift and SDS-PAGE analysis.11 See Fig. 4 for structures of statins and ESI,† for detailed methods. | ||
Altenusin 9 was identified as a potent and highly selective Ec BirA-GFP destabilizing hit (ΔTm = −8.5 °C). Indeed, 9 showed no effect on any other proteins (Fig. S1B, ESI†). It has previously been shown to exert broad antimicrobial activity against several multidrug-resistant bacterial and fungal strains albeit not against Ec.22,23 Concentration dependence curves for Ec and the two orthologous Bp and Mt BirA-GFP showed that 9 was Ec selective. Only a small destabilizing effect was observed with Bp and Mt BirA-GFP in the presence of 100 μM of 9 (Fig. 6A and Fig. S4B, ESI†). The data clearly demonstrate that 9 inactivates Ec BirA-GFP at 25 °C (Fig. 6B, see ESI,† for detailed methods) through a highly selective protein unfolding (SPU) mechanism.
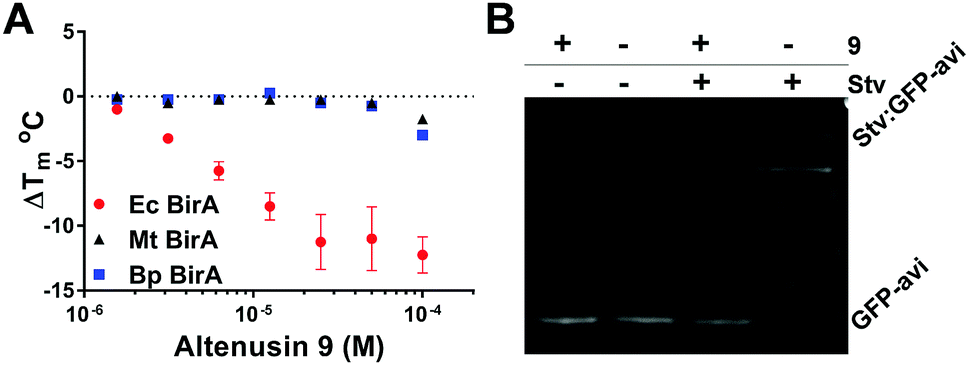 | ||
| Fig. 6 (A) Concentration dependence of 9 on Tm of Ec, Bp and Mt BirA-GFP. Melt-curve protocol: 25–85 °C at 0.5 °C/30 s. Error bars represent SD (N = 2). (B) Inhibition of Ec BirA-GFP biotinylation activity by 9 at 25 °C (see Fig. 5B and ESI,† for detailed methods).11 | ||
Target engagement in the cell24 as well as non-specific binding to serum proteins25 are major hurdles in drug development. We examined if 9, 10a and 10b were able to exert their destabilizing effect in a clarified bacterial cell lysate and in human serum (see ESI,† for detailed methods). DSF-GTP is the only DSF method that can selectively measure the Tm of a protein in the presence of other protein contaminants. DSF-GTP is thus perfectly suited for target engagement and serum interaction studies. All hits lost their ability to destabilize Ec BirA-GFP in the bacterial lysate even after heat treatment (95 °C for 5 min) and clarification (see ESI,† Fig. S5). Since heat treatment denatures all but the most thermostable proteins, the loss of destabilizing activity could be due to a non-proteinaceous soluble factor in the bacterial lysate. In serum, significant differences in Ec BirA-GFP Tm were observed with the three different control reactions, with the heat-treated serum being the most destabilizing condition (see ESI,† Fig. S5). This suggest that one or more factors generated in the heat-treated serum are significantly affecting the Tm of Ec BirA-GFP. The heat-inducible destabilizing factor(s) can be partially eliminated after clarification of the heat-treated serum suggesting an aggregated proteinaceous factor. Most importantly and in stark contrast with the statins, 9 significantly destabilised Ec BirA-GFP in human serum. This effect is most pronounced in the heat treated and clarified serum (ESI,† Fig. S5).
10b has been shown to display a multitude of biological activities and clearly promiscuously binds several protein targets. In Staphylococcus aureus (SA), 10b significantly inhibits DNA, protein and lipid synthesis and to a lesser extent RNA synthesis and cell wall synthesis.17 BirA is the sole enzyme responsible for covalently attaching biotin at the active site of important metabolic enzymes, the biotin carboxylase and decarboxylases.26 The biotin carboxylases are essential enzymes in gluconeogenesis, lipogenesis, amino acid metabolism and energy transduction. Although the two orthologous Mt20 and Bp19 BirA tested in this study were not significantly destabilized by 10b, it is important to note they are both type I enzymes whereas Ec BirA is a type II enzyme like Sa BirA.27 Thus, it is still tempting to propose BirA as a major target of 10b in susceptible bacteria whose inactivation would lead to a systematic inhibition of metabolic processes such as in Sa.17 The fact that the bacterial lysate abolished the destabilizing activity of 10b on Ec BirA-GFP could indicate that it binds to further targets present in the bacteria or released after lysis. The high MIC value of 10b limits its use as a topical agent17 which is supported by our data in serum. Whether the antibacterial activity of 9 could also be improved against Gram-negative bacteria with the help of sub-inhibitory concentrations of colistin remains to be tested. Nevertheless, if entry of 9 into the bacteria can be achieved, and based on its activity in serum as well as its moderate cytotoxicity, it could become a promising lead.
Overall, our data clearly demonstrate that protein destabilizing compounds can be target- and species-selective (ESI,† Fig. S1B and Fig. 6A), and thus may offer quality lead compounds with applications in various fields of life sciences. Our data also highlight the unique application of HT DSF-GTP for screening protein–ligand interactions in complex matrices and identifying non-promiscuous hits. SPU as a mechanism of action combined with the power of HT DSF-GTP offers a fast and efficient platform for screening and identifying an exciting class of new inhibitors.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Antony and P. M. Schaeffer, Analyst, 2013, 138, 6073–6080 RSC.
- R. Y. Kao, D. Yang, L. S. Lau, W. H. Tsui, L. Hu, J. Dai, M. P. Chan, C. M. Chan, P. Wang and B. J. Zheng, et al. , Nat. Biotechnol., 2010, 28, 600–605 CrossRef CAS PubMed.
- A. J. Wilson, K. Groves, R. K. Jain, H. S. Park and A. D. Hamilton, J. Am. Chem. Soc., 2003, 125, 4420–4421 CrossRef CAS PubMed.
- K. Groves, A. J. Wilson and A. D. Hamilton, J. Am. Chem. Soc., 2004, 126, 12833–12842 CrossRef CAS PubMed.
- N. O. Fischer, C. M. McIntosh, J. M. Simard and V. M. Rotello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5018–5023 CrossRef CAS PubMed.
- T. K. Neklesa, J. D. Winkler and C. M. Crews, Pharmacol. Ther., 2017, 174, 138–144 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- M. W. Pantoliano, E. C. Petrella, J. D. Kwasnoski, V. S. Lobanov, J. Myslik, E. Graf, T. Carver, E. Asel, B. A. Springer and P. Lane, et al. , J. Biomol. Screening, 2001, 6, 429–440 CrossRef CAS PubMed.
- M. J. J. Moreau, I. Morin, S. P. Askin, A. Cooper, N. J. Moreland, S. G. Vasudevan and P. M. Schaeffer, RSC Adv., 2012, 2, 11892–11900 RSC.
- M. J. Moreau and P. M. Schaeffer, Mol. BioSyst., 2013, 9, 3146–3154 RSC.
- S. P. Askin, T. E. H. Bond and P. M. Schaeffer, Anal. Methods, 2016, 8, 418–424 RSC.
- P. M. Schaeffer, M. J. Headlam and N. E. Dixon, IUBMB Life, 2005, 57, 5–12 CrossRef CAS PubMed.
- E. C. Barnes, A. M. Kavanagh, S. Ramu, M. A. Blaskovich, M. A. Cooper and R. A. Davis, Phytochemistry, 2013, 93, 162–169 CrossRef CAS PubMed.
- V. Choomuenwai, K. T. Andrews and R. A. Davis, Bioorg. Med. Chem., 2012, 20, 7167–7174 CrossRef CAS PubMed.
- K. D. Beattie, N. Ellwood, R. Kumar, X. Z. Yang, P. C. Healy, V. Choomuenwai, R. J. Quinn, A. G. Elliott, J. X. Huang and J. L. Chitty, et al. , Phytochemistry, 2016, 124, 79–85 CrossRef CAS PubMed.
- E. C. Barnes, A. R. Carroll and R. A. Davis, J. Nat. Prod., 2011, 74, 1888–1893 CrossRef CAS PubMed.
- S. Thangamani, H. Mohammad, M. F. Abushahba, M. I. Hamed, T. J. Sobreira, V. E. Hedrick, L. N. Paul and M. N. Seleem, Sci. Rep., 2015, 5, 16407 CrossRef CAS PubMed.
- M. J. Moreau, I. Morin and P. M. Schaeffer, Mol. BioSyst., 2010, 6, 1285–1292 RSC.
- T. E. H. Bond, A. E. Sorenson and P. M. Schaeffer, Microbiol. Res., 2017, 199, 40–48 CrossRef CAS PubMed.
- T. E. H. Bond, A. E. Sorenson and P. M. Schaeffer, Microbiol. Res., 2017, 205, 35–39 CrossRef CAS PubMed.
- A. E. Sorenson, S. P. Askin and P. M. Schaeffer, Anal. Methods, 2015, 7, 2087–2092 RSC.
- J. Xiao, Q. Zhang, Y. Q. Gao, J. J. Tang, A. L. Zhang and J. M. Gao, J. Agric. Food Chem., 2014, 62, 3584–3590 CrossRef CAS PubMed.
- J. Kjer, V. Wray, R. Edrada-Ebel, R. Ebel, A. Pretsch, W. Lin and P. Proksch, J. Nat. Prod., 2009, 72, 2053–2057 CrossRef CAS PubMed.
- D. Martinez Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef PubMed.
- M. Fasano, S. Curry, E. Terreno, M. Galliano, G. Fanali, P. Narciso, S. Notari and P. Ascenzi, IUBMB Life, 2005, 57, 787–796 CrossRef CAS PubMed.
- A. Chapman-Smith and J. E. Cronan, Jr., J. Nutr., 1999, 129, 477S–484S CAS.
- N. R. Pendini, M. Y. Yap, S. W. Polyak, N. P. Cowieson, A. Abell, G. W. Booker, J. C. Wallace, J. A. Wilce and M. C. Wilce, Protein Sci., 2013, 22, 762–773 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary figures. See DOI: 10.1039/c8cc00090e |
| This journal is © The Royal Society of Chemistry 2018 |