
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatically-stable oxetane-based dipeptide hydrogels†
Laura
McDougall
a,
Emily R.
Draper
 a,
Jonathan D.
Beadle
b,
Michael
Shipman
b,
Piotr
Raubo
c,
Andrew G.
Jamieson
*a and
Dave J.
Adams
*a
a,
Jonathan D.
Beadle
b,
Michael
Shipman
b,
Piotr
Raubo
c,
Andrew G.
Jamieson
*a and
Dave J.
Adams
*a
aSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: andrew.jamieson.2@glasgow.ac.uk; dave.adams@glasgow.ac.uk
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
cMedicinal Chemistry, Oncology, IMED Biotech Unit, AstraZeneca, Cambridge, UK
First published on 24th January 2018
Abstract
Low molecular weight gelators that are not easily degraded by enzymes have a range of potential applications. Here, we report new Fmoc-protected dipeptides in which the amide carbonyl group has been replaced by an oxetane ring. Remarkably one of these peptidomimetics, but not the corresponding dipeptide, is an effective gelator, forming hydrogels at a concentration of 3 mg mL−1. On assembly, there is a lack of beta-sheet structure, implying that there is no requirement for this motif in such a gel. Furthermore, the modified dipeptide is also stable to proteolysis compared to the parent dipeptide.
Low molecular weight gels are being widely investigated currently for a range of applications.1,2 These gels arise from the self-assembly of small molecules into fibres. This type of gel has interesting properties compared to polymer gels. For example, they are usually reversible and tend to form at low gelator concentrations.
There are a significant number of reports demonstrating biocompatibility of low molecular weight gels.3 A range of these gels have been used for applications such as cell culturing, drug delivery and controlled release. For some of these applications, in vivo proteolytic degradation would be beneficial but for a number of others, it is desirable that the self-assembled scaffold is not easily degraded by enzymes.
Recently, amino acid or dipeptide-based gelators have become common.3,4 These can be highly effective gelators of water, and there is now a large library of these molecules that can form gels using a range of gelation methods. For these gelators, a number of approaches have been developed to slow down the enzymatic degradation. These include using D-amino acids as the constituent parts of the gelator;5 generally synthesising the gelator with either the L- and D-amino acids results in equally effective gelation, but the D-amino acids are significantly more resistant to enzymatic action. Another approach is to use β-amino acids such as carnosine that are less susceptible to enzymatic cleavage.6 Finally, non-natural amino acids can be used. For example, Panda et al. have used an α-carbon to β-carbon unsaturated analogue of the naturally occurring phenylalanine, which imparted proteolytic stability.7 Similarly, other dehydrodipeptides have also been shown to be both effective gelators and resist proteolytic degradation.8,9 Whilst these approaches can be used to impart stability to enzymes, it is beneficial to find other potential methods and gelators. When used, the gels will also need specific mechanical properties, diffusion rates, and stability to shear. For low molecular weight gelators, it is very difficult to predict in advance the properties, so increasing the range of materials available is of benefit.
Some of us have recently reported the synthesis of a new class of peptidomimetic where one of the backbone amide carbonyl bonds is replaced by a four-membered oxetane ring.10–12 A number of Fmoc-protected dipeptides were prepared containing this oxetane ring.10 Since Fmoc-dipeptides are a well-known, effective class of gelator,3,4,13,14 we reasoned that we could prepare gels from these dipeptides (Fig. 1a). Importantly, the oxetane ring was expected to render the gelators resistant to enzymatic cleavage through deletion of the central amide bond.
 | ||
| Fig. 1 (a) Concept of oxetane modification of Fmoc dipeptide gelators. (b) Chemical structures of modified dipeptides used in this study. | ||
For low molecular weight gelators, it is very difficult to predict in advance which will form gels and which will not.1 We therefore used a computational descriptor approach15 to predict the gelation ability of three modified dipeptides (Fig. 1b). 1 was predicted to be an effective gelator (84% likely), with 2 and 3 expected not to form gels. All of the predictions were within the domain space of the model, and hence expected to be correct.15 The synthesis of 1–3 was readily achieved in three steps from oxetan-3-one and the appropriately protected L-amino acids (for full details, see ESI†).
To test the effectiveness of 1, 2 and 3 as gelators, we used a pH-triggered approach, which is commonly successful for Fmoc-dipeptides.16,17 The different dipeptides were suspended in water and a molar equivalent of base added to deprotonate the terminal carboxylic acid. As the base, we used sodium carbonate to avoid deprotection of the Fmoc group. 1 and 2 formed transparent solutions at 3 mg mL−1, but 3 formed a turbid suspension (Fig. 2a). To form gels, the pH of the solutions was decreased. To do this, we exploited the hydrolysis of glucono-δ-lactone (GdL) as we have done for a number of Fmoc-dipeptides and related gelators.18 Here, the dissolution of GdL is fast, and the subsequent hydrolysis of GdL to gluconic acid results in a homogeneous pH decrease.
 | ||
| Fig. 2 (a) Photographs of the solutions of 1, 2 and 3 at a concentration of 3 mg mL−1 at high pH. (b) Photographs of the samples shown in (a) after the pH has been lowered via the hydrolysis of GdL. The white bands in (b) are labels on the sample tubes. | ||
Using this approach, 1 formed a self-supporting material after standing overnight (Fig. 2b). Decreasing the pH of a solution of 2 resulted in some of the sample forming a gel over 18 hours (Fig. 2b), but this was transient with precipitation occurring over two days. The solution of 3 did not form a gel, but rather precipitation occurred as the pH decreased (Fig. 2b). Hence, the descriptor-based approach15 correctly predicted the gelation propensity of these three molecules (see above). We note that it is often difficult to explain why some molecules form gels and some do not,1 which is why prediction of the effectiveness of a specific molecule as a gelator is often difficult a priori. Remarkably, the impact of replacing a carbonyl with an oxetane ring is immediately apparent as the dipeptide analogue of 1; Fmoc-GlyPhe is known to not be able to form gels.16,17
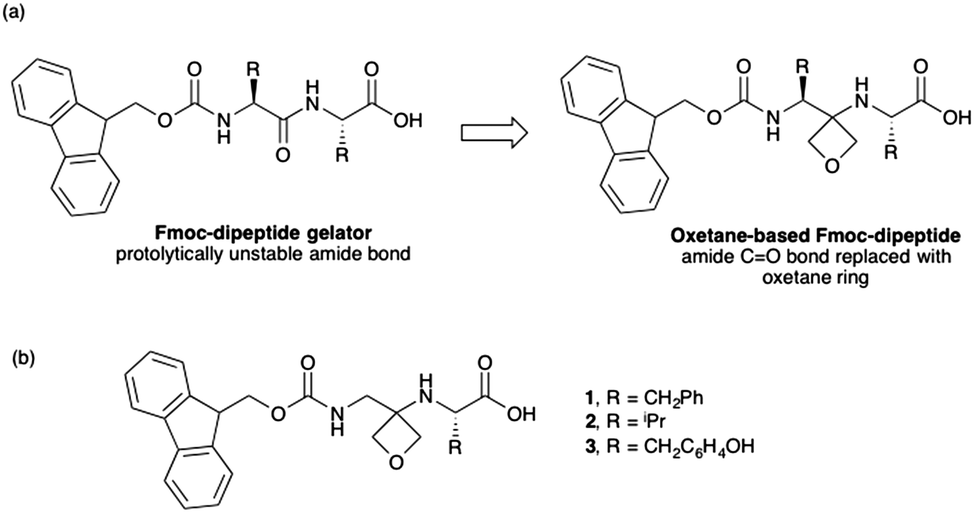
The hydrolysis of GdL is slow, meaning that the process of gelation can be followed with time. The assembly can be linked to the pH of the sample; as the pH decreases, the apparent pKa of the gelator is reached and it is typically at this point that gelation begins.19 Here, for 1, the pH drops slowly (Fig. 3a). From these data, the apparent pKa of 1 is around 6.8; such a high apparent pKa is consistent with this class of gelator.19,20 The mechanical properties of the sample can be followed using rheology. As can be seen from Fig. 3a, gelation begins after around 20 minutes by the increase of the storage modulus (G′) and the loss modulus (G′′). At this point, the pH is below the pKa of 1. The gel develops over time, with both G′ and G′′ stabilising after 120 minutes. At this point, tan δ (G′′/G′) is 0.13, which is typical of such a gel.21 Frequency sweeps (Fig. 3b) and strain sweeps (Fig. 3c) are also as expected for such a gel. From the strain sweeps, it can be seen that the gel is insensitive to strain until 10%, at which point the gel begins to break. The gels are relatively insensitive to frequency.
 | ||
| Fig. 3 (a) Change in pH (red data), storage modulus (G′, full circles, top data), and loss modulus (G′′, open circles, bottom data) with time for a solution of 1 after the addition of GdL. (b) Frequency sweep of gels of 1 after 18 hours (G′, full circles; G′′, open circles). (c) Strain sweep of gels of 1 after 18 hours (G′, full circles; G′′, open circles). For (b) and (c), the data are shown as an average from three gels, with the error bars representing the standard deviation of the data. (d) SEM image of a xerogel of 1. The scale bar represents 500 nm. The contrast has been improved on this image for clarity. The original is provided in the ESI.† | ||
Scanning electron microscopy (SEM) imaging of the dried gel showed the expected network of fibres (Fig. 3d, additional images are shown in Fig. S1, ESI†). The fibre widths were measured by image analysis, with the average diameter being 21.3 ± 4.4 nm (Fig. S2, ESI†). Importantly, 1 showed no sign of chemical reaction after the gelation process as shown by the NMR of the gelator obtained by freeze-drying a gel (Fig. S3–S5 and Table S1, ESI†). HPLC of the freeze-dried 1 implies that there is around 6% oxetane ring opening (Fig. S6, ESI†), although this ring-opening may occur during the analysis.
As noted above, there has been significant interest in Fmoc-dipeptides as hydrogelators. In one case for Fmoc-diphenylalanine, a detailed model of the assembly has been proposed,22 although more recently some of the assignment of the supporting infra-red data has been shown to have been mis-assigned.23 Data for Fmoc-dialanine is also not consistent with this model, and the authors also highlight how extrapolating from spectroscopic assignments from proteins to dipeptides should be carried out with caution.24 Since in this example we have a dipeptide-based gelator where one of the carbonyls has been replaced by an oxetane ring, we investigated the assembly process to allow comparison with the known family of Fmoc-dipeptides. The suggestion is that Fmoc-dipeptides assemble by a so-called π-beta structure, which has been shown by fluorescence, circular dichroism (CD), and infra-red (IR) spectroscopy.22
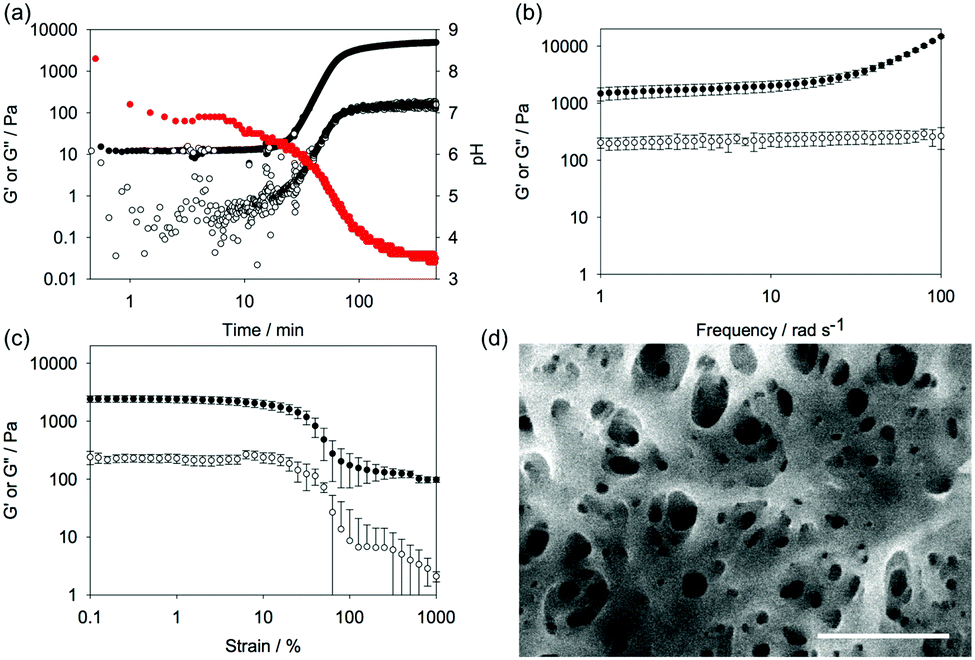
Here, the fluorescence emission for 1 in water at high pH shows the expected fluorescence from the fluorenyl ring (Fig. 4a). After gelation, significant quenching of the fluorescence has occurred (Fig. 4a), consistent with aggregation and self-quenching of the fluorenyl group. Unlike for reported Fmoc-dipeptides,25 no apparent red shift in the emission was observed.
 | ||
| Fig. 4 (a) Fluorescence for a solution of 1 at high pH (black data) overlaid with the data after gelling overnight (blue data). The excitation wavelength was 265 nm. (b) Circular dichroism spectrum at high pH of a solution of 1 (black data) overlaid with the data after gelling overnight (blue data). (c) IR spectrum of freeze-dried xerogel of 1. (d) Kinetics plot showing change in pH (red data), fluorescence intensity at 320 nm (blue data) and circular dichroism intensity at 270 nm (black data) with time. | ||
Before gelation, a solution of 1 shows little apparent CD or linear dichroism signal (CD data shown in Fig. 4b). This is typical for such gelators.18 After adding the GdL, strong signals appear in the CD. The minimum at around 255 nm and the positive signals above 260 nm are consistent with the fluorenyl group being assembled into a helical stack and similar to data elsewhere.18,24 The data below 240 nm is harder to assign, as there will be a contribution from the hydrolysis products of GdL.26 No linear dichroism was observed, showing that the signals are true CD signals and not arising from alignment (see Fig. S7, ESI†). The IR shows a broad peak centred at 1600 cm−1, which is not present in the IR of the non-assembled precursor (Fig. 4c and Fig. S7, ESI†); however, this may be due to the carbonate species added as a base (Fig. S8, ESI†). There is a peak at 1701 cm−1, which can be assigned to the carbonyl of the Fmoc carbamate and is also present in the precursor. A peak at 1722 cm−1 can be assigned to the protonated carboxylic acid.
Again, as the use of GdL allows the kinetics of the assembly to be followed, we can compare the rates of change in the pH, fluorescence, CD, and rheology (Fig. 4d). From these data, it is clear that the fluorescence and circular dichroism essentially do not change after around 60 to 90 minutes. This is just as the rheological moduli are starting to increase. Hence, the data show that the molecular-level assembly is complete as the pKa of the carboxylic acid is reached, presumably into fibrous structures. The gel properties therefore arise as these fibres entangle. The entanglement does not lead to further changes in the CD or fluorescence data. This is consistent with our work on naphthalene–dipeptides.19 Drawing all of this together, assembly begins as the carboxylic acid is protonated. The fluorescence and CD data are consistent with the assembly placing the Fmoc groups sufficiently close to one another to quench the fluorescence and in to a helical arrangement. This is consistent with work on Fmoc-dipeptides.18,24 However, despite this similarity and the fact that gels are formed here despite a lack of apparent beta-sheet structure, our data imply that the assignment as requiring a beta-sheet for assembly and gelation is not correct.
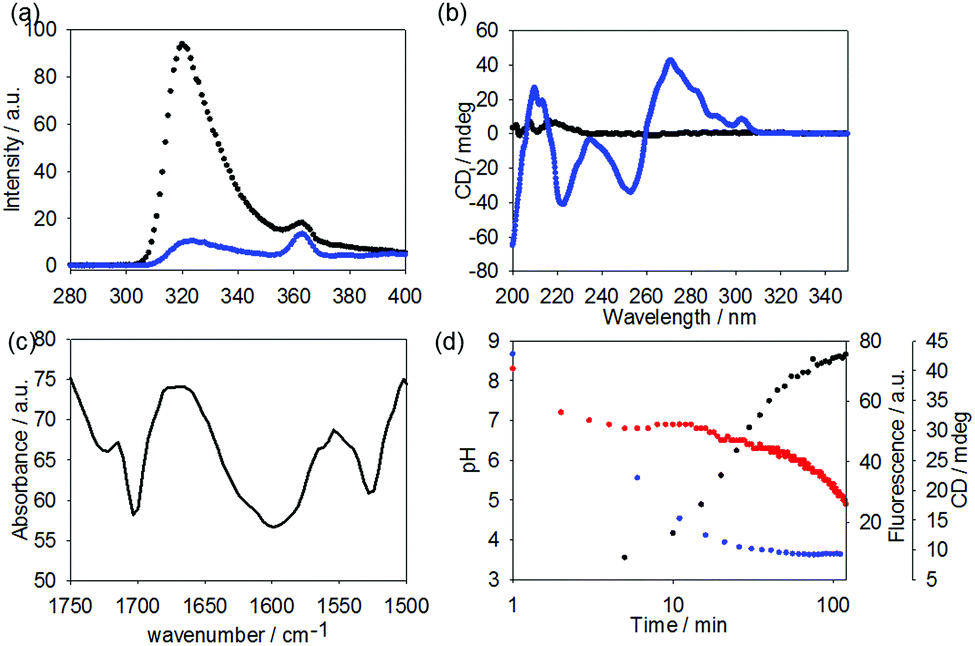
Finally, we tested how prone to proteolysis these materials are. As a comparison, we chose Fmoc-GlyPhe. Whilst this dipeptide does not form gels, it is the closest structural analogue to 1. Stability in blood serum was initially assessed, however no degradation was observed. Carboxypeptidase A is an exopeptidase that hydrolyses peptide bonds of C-terminal residues with aromatic side-chains such as phenylalanine and thus was chosen to challenge the stability of Fmoc-GlyPhe and 1. As such, 1 or Fmoc-GlyPhe were suspended in Tris buffer at pH 7.9. This is the optimal pH for the enzyme and additionally the molecules are unassembled at this pH. Hence, this is the most demanding test. To these solutions, Carboxypeptidase A was added. After set periods of time, an aliquot was removed, acetonitrile added to quench the enzymatic activity and the products probed by HPLC after centrifugation to remove the enzyme. Fmoc-GlyPhe degraded extremely quickly, with 98% conversion to Fmoc-Gly after 1 minute and complete conversion after 5 minutes (Fig. 5a).
 | ||
| Fig. 5 (a) Degradation of FmocGlyPhe over 5 min to Fmoc-G-OH. (b) 1 was sampled at t = 0, 1 min, 5 min, 4 h and 24 h. No degradation was observed over this timescale. Negative control containing no enzyme was conducted in parallel. | ||
1 showed significant stability, with no enzymatic conversion after 24 hours (Fig. 5b). A small amount of hydrolysis of the oxetane ring had occurred, but this is due to suspension in water as opposed to enzymatic activity as shown by control experiments in buffer alone.
In conclusion, we have shown that gels can be formed from a new class of peptidomimetic. The modified dipeptide is significantly more resistant to proteolysis as compared to the native dipeptide and so this class of material may have applications in vivo.
LM thanks the University of Glasgow and EPSRC for a studentship (EP/N509668/1). ERD thanks the Leverhulme Trust for an Early Career fellowship (ECR-2017-223) and the University of Glasgow for a LKAS leadership fellowship. DJA thanks the EPSRC for a Fellowship (EP/L021978/1). JDB thanks AstraZeneca and the University of Warwick for financial support. We thank Sharon Kelly (University of Glasgow) for help in collecting the CD spectra.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem., 2017, 3, 390–410 CAS.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- J. Li, Y. Kuang, Y. Gao, X. Du, J. Shi and B. Xu, J. Am. Chem. Soc., 2013, 135, 542–545 CrossRef CAS PubMed.
- J. Nanda and A. Banerjee, Soft Matter, 2012, 8, 3380–3386 RSC.
- J. J. Panda, A. Mishra, A. Basu and V. S. Chauhan, Biomacromolecules, 2008, 9, 2244–2250 CrossRef CAS PubMed.
- H. Vilaca, G. Pereira, T. G. Castro, B. F. Hermenegildo, J. Shi, T. Q. Faria, N. Micaelo, R. M. M. Brito, B. Xu, E. M. S. Castanheira, J. A. Martins and P. M. T. Ferreira, J. Mater. Chem. B, 2015, 3, 6355–6367 RSC.
- C. K. Thota, N. Yadav and V. S. Chauhan, Sci. Rep., 2016, 6, 31167 CrossRef CAS PubMed.
- J. D. Beadle, A. Knuhtsen, A. Hoose, P. Raubo, A. G. Jamieson and M. Shipman, Org. Lett., 2017, 19, 3303–3306 CrossRef CAS PubMed.
- N. H. Powell, G. J. Clarkson, R. Notman, P. Raubo, N. G. Martin and M. Shipman, Chem. Commun., 2014, 50, 8797–8800 RSC.
- M. McLaughlin, R. Yazaki, T. C. Fessard and E. M. Carreira, Org. Lett., 2014, 16, 4070–4073 CrossRef CAS PubMed.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- W. T. Truong, Y. Su, D. Gloria, F. Braet and P. Thordarson, Biomater. Sci., 2015, 3, 298–307 RSC.
- J. K. Gupta, D. J. Adams and N. G. Berry, Chem. Sci., 2016, 7, 4713–4719 RSC.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- C. Tang, R. V. Ulijn and A. Saiani, Langmuir, 2011, 27, 14438–14449 CrossRef CAS PubMed.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- S. Fleming, P. W. J. M. Frederix, I. Ramos Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510–9515 CrossRef CAS PubMed.
- X. Mu, K. M. Eckes, M. M. Nguyen, L. J. Suggs and P. Ren, Biomacromolecules, 2012, 13, 3562–3571 CrossRef CAS PubMed.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, T. Tuttle and R. V. Ulijn, Chem. Commun., 2013, 49, 10587–10589 RSC.
- K. L. Morris, L. Chen, A. Rodger, D. J. Adams and L. C. Serpell, Soft Matter, 2015, 11, 1174–1181 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc09701h |
| This journal is © The Royal Society of Chemistry 2018 |