
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quinoline-para-quinones and metals: coordination-assisted formation of quinoline-ortho-quinones†
Mario
Kubanik
a,
Nelson Y. S.
Lam
 a,
Hannah U.
Holtkamp
a,
Tilo
Söhnel
a,
Robert F.
Anderson
ab,
Stephen M. F.
Jamieson
b and
Christian G.
Hartinger
*a
a,
Hannah U.
Holtkamp
a,
Tilo
Söhnel
a,
Robert F.
Anderson
ab,
Stephen M. F.
Jamieson
b and
Christian G.
Hartinger
*a
aSchool of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: c.hartinger@auckland.ac.nz; Web: http://hartinger.auckland.ac.nz
bAuckland Cancer Society Research Centre, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
First published on 11th January 2018
Abstract
The reaction of the para-quinone 6,7-dichloroquinoline-5,8-dione with various transition metal dimers led to the unexpected formation of quinoline-ortho-quinone metal complexes. Systematic variation of the reaction conditions helped identify the solvent as the source of the carbonyl oxygen.
Organometallic anticancer agents based on bioactive ligand systems have attracted a lot of attention due to their tuneable bioactivity against primary tumours or metastases and their general versatility in drug development.1,2 Recently, we reported a series of naphthoquinone complexes featuring an O,O-bidentate coordination motif where metal coordination induced the formation of reactive oxygen species in cancer cells in dependence of the nature of the metal centre.3,4 However, we found that these compounds undergo ligand exchange reactions in the presence of biological ligands which results to some extent in the cleavage of the ligand from the metal centre.3 In order to overcome this issue, we turned our research efforts to N,O-chelating quinoline ligands, which were found to form extremely stable and anticancer-active complexes.4,5 To combine the benefits of quinoline ligands with the interesting biological properties offered by quinone structures, we aimed to explore the coordination chemistry and biological activity of 5,8-quinolinediones. Moreover, a literature search showed that only a few metal complexes of such ligands have been reported.6,7
5,8-Quinolinedione derivatives are biologically active and exhibit antitumor, antibacterial and antimalarial effects.8 This structural motif is part of the pharmacophore found in several antitumor antibiotics, such as streptonigrin and lavendamycin.9 The cytotoxicity of quinolinediones results from its inhibition of the electron transport chain in mitochondria, effectively halting aerobic respiration and producing a semiquinone radical in a redox reaction.8
The 5,8-quinolinedione derivative 6,7-dichloro-5,8-quinolinedione (DQQ) was selected as the ligand and used to synthesize RuII-, OsII-, IrIII- and RhIII-based metallopharmaceuticals. In order to prepare DQQ, 8-hydroxyquinoline was oxidized using sodium chlorate in HCl (Fig. 1).10 The yield of this reaction after recrystallization was low (15–18%) which can be explained by the occurrence of several side reactions competing with the formation of the desired product, also noted by Shen et al.11,12 The by-product 5,6,7-trichloro-2-alkoxy-8-hydroxyquinoline (HHQCl) was isolated and characterized by NMR spectroscopy, as was its methoxy derivative MeHQCl by single crystal X-ray diffraction analysis (ESI†). The latter species surprisingly formed during recrystallization with methanol. Due to the reduced electron density of the 2-position of the pyridine ring proximal to the nitrogen, this position is amenable to oxidative nucleophilic substitution resulting in the formation of compounds HQCl under the oxidative reaction conditions used.13
 | ||
| Fig. 1 Preparation of DQQ, HQCl, and the ortho-quinoid metal complexes 1–5. R = H (HHQCl), methyl (MeHQCl), ethyl (EtHQCl), n-propyl (nPrHQCl), i-propyl (iPrHQCl), n-butyl (BuHQCl). | ||
To improve the yield of DQQ and to minimize the side reactions occurring, methanol was replaced with ethanol, n-propanol, i-propanol and n-butanol in the recrystallization step. While the yields of DQQ did not improve, the ethyl, n-propyl, i-propyl and n-butyl derivatives of 5,6,7-trichloro-2-alkoxy-8-hydroxyquinoline were isolated in up to 8% yield (ESI† for 1H NMR spectra). Furthermore, HPLC analysis of DQQ dissolved in a mixture of MeOH/H2O revealed the conversion of DQQ to 6- and 7-methoxy-5,8-quinolinedione derivatives by replacing one of the chloro substituents with a methoxy group (Fig. 2). This is indicated by the presence of two peaks with close retention times and the same accurate mass as determined by HPLC-ESI-QToF-MS, indicating isomer formation. The chloro/methoxy substitution reaction was found to be water content-dependent. While the substitution was suppressed in pure water, acetonitrile and dichloromethane, it was completed within 32 h in 25% H2O/methanol and even accelerated in a 1/1 mixture to be complete in 10 h. In pure methanol, the reaction was to some extent suppressed and only 50% conversion could be observed after 95 h.
 | ||
| Fig. 2 (top left) Time course of the conversion of DQQ to the methoxy derivative in 25% H2O/methanol. (bottom) Time-dependent chromatograms showing the growth of a second peak at lower retention time which was identified by high resolution ESI-MS as the methoxy derivative (top right). | ||
As mentioned above, there are only a few 5,8-quinolinedione complexes known.6,7 In order to prepare complexes of the general structure A (Fig. 1) from the 5,8-quinolinedione ligand DQQ, it was reacted with dimeric [M(L)Cl2]2 precursors (M = Ru, Os, Rh, Ir; L = η6-p-cymene [cym], η6-biphenyl, η5-pentamethylcyclopentadienyl [Cp*]) in alcoholic solution under reflux for 1 h (Scheme 1), which resulted in the formation of purple precipitates. While NMR spectroscopy suggested the formation of A, X-ray diffraction analysis of single crystals of 1, 2, 4 and 5 surprisingly revealed the formation of the ortho-quinone complexes (Fig. 3; ESI†). All complexes featured the pseudo-tetrahedral piano stool structure with the metal centre coordinated to the pyridine nitrogen and the adjacent oxygen in a bidentate fashion. The first coordination sphere is completed by chlorido and p-cymene (1 and 2) or Cp* (4 and 5) ligands. The conversion of DQQ to an ortho-quinone was confirmed by high resolution ESI-MS indicating the absence of a chloro substituent after complexation and substitution with an oxygen atom. The 1H NMR spectral shifts observed for the pyridine proton H2 were found downfield for the Ru and Os complexes 1–3 as compared to DQQ, whereas the Ir and Rh congeners showed a slight upfield shift for the proton adjacent to the nitrogen atom.
 | ||
| Fig. 3 Molecular structure of one enantiomer of 1 drawn at 50% probability level. Solvent molecules were deleted for clarity. Selected bond lengths are given in the ESI.† | ||
A similar reaction was observed by Corey and König who showed that anhydrides form from metal carboxylates, such as Cu(acetate)2, in the presence of DQQ,14 while in the presence of alcohols esters were obtained. In both cases a metal-coordinated ortho-quinone formed. It was suggested that the addition of water to the intermediate yields a polymeric structure but elemental analysis data indicated the formation of a Cu(ortho-quinone)2 complex,14 which we confirmed by ESI-MS ([M + Na]+m/zexp 501.882, m/ztheor 501.879). These Cu(ortho-quinone)2 species may feature the same coordination mode as observed in 1–5 for the formed ortho-quinone and the metal centres used. Using pseudo-octahedral organometallic moieties in our experiments where the ligands cannot take part in the reaction, avoided the formation of polymers or higher metal-to-ligand ratio complexes.
To distinguish between the solvent or dissolved oxygen as the source of the oxygen atom introduced in position 6 of the ortho-quinone, we performed the reaction between DQQ and [M(L)Cl2]2 in dry protic and aprotic, deoxygenated solvents and under different conditions, i.e., reflux, microwave-assisted and at room temperature (Table 1). 1H NMR spectroscopy experiments with DQQ and [Ru(cym)Cl2]2 in CDCl3 showed neither conversion of DQQ into the ortho-quinone nor complex formation, even when refluxing the reaction mixture. Addition of MeOD to the mixture on the other hand led almost instantaneously to the formation of Ru compound 1. These studies demonstrate that the ortho-quinone–metal complex only forms in the presence of protic solvents. As the use of dry degassed solvents had no influence on the reaction, the solvent should be the source of the oxygen atom in position 6 of the ortho-quinone. To further verify this assumption, 18O-labelled water was used as a solvent in the reaction. Under these conditions, the formation of 1 was confirmed by ESI-MS. The mass spectrum featured a signal at m/z 507.9641 as the base peak. This ion was identified to contain three 18O atoms ([M + Na]+m/ztheor 507.9648), which is evidence that the protic solvents used were the source of the oxygen atom. Performing the same reactions but with OsII, RhIII and IrIII precursors supported this conclusion. This is also in line with previous reports that other Lewis acids can be used in the derivatization of DQQ.15,16
| Solvent | Conditions | 1 |
|---|---|---|
| a 1 was detected by 1H NMR spectroscopy but not isolated. M.W., microwave. | ||
| Methanol (abs), under N2 | Reflux 1 h | Yes |
| M.W. 30 min, 80 °C | Yes | |
| Ethanol (abs), under N2 | Reflux 1 h | Yes |
| M.W. 30 min, 80 °C | Yes | |
| Isopropanol (abs), under N2 | M.W. 30 min, 80 °C | Yes |
| n-Butanol (abs), under N2 | M.W. 30 min, 80 °C | Yes |
| Water | Reflux 1 h | Yes |
| M.W. 30 min, 80 °C | Yes | |
| THF (abs), under N2 | Reflux 1 h | No |
| Reflux 36 h | No | |
| R.T. 5 d | No | |
| M.W. 30 min, 80 °C | No | |
| Acetonitrile (abs), under N2 | Reflux 21 h | No |
| R.T. 24 h | No | |
| Chloroform (abs), under N2 | Reflux 4 h | No |
| M.W. 30 min, 80 °C | No | |
| Chloroform/methanol | Reflux 5 min | Yes |
| DCM (abs), under N2 | Reflux 4 h | No |
Based on these observations, the following mechanism for the formation of ortho-quinones is suggested (Fig. 4): similar to Corey and König,14 the initial step involves coordination of the metal centre to DQQ with DQQ acting as a bidentate ligand, where the metal acts as a Lewis acid. The coordination of the metal centre activates C6 to a nucleophilic attack by ROH into the conjugated π-electron system. Subsequent decomposition of the formed tetrahedral intermediate affords an oxonium ion, to which the R–O bond is cleaved to yield the ortho-quinone–metal complex in a metal coordination-assisted reaction and RCl, as determined for the reaction with n-butanol by ESI-MS.
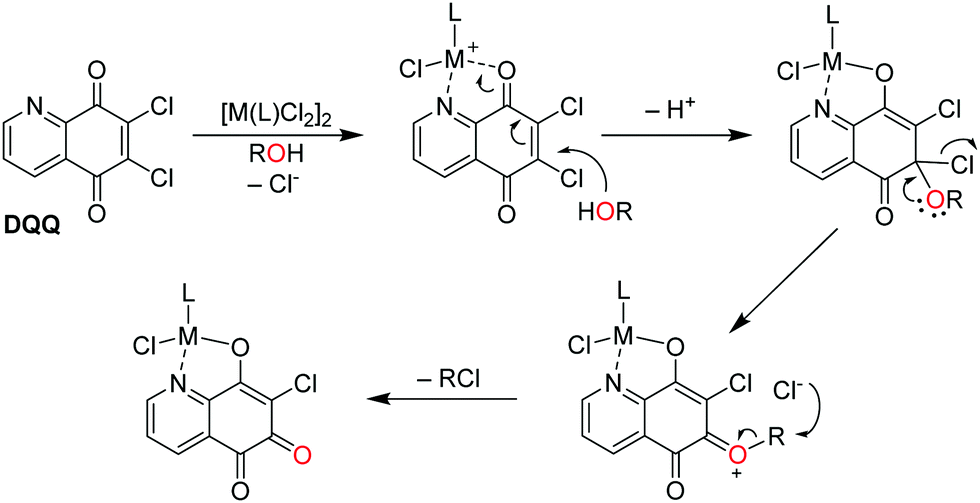 | ||
| Fig. 4 Proposed reaction mechanism for the conversion of the quinoline-5,8-dione DQQ to an ortho-quinone derivative upon complexation to metal centres in protic solvents. | ||
Having identified the solvent choice as essential for the formation of the ortho-quinone from DQQ, the preparation of the originally anticipated quinolinedione complexes was attempted by using a silver salt to activate [Ru(cym)Cl2]2 in acetonitrile to form [Ru(cym)(CH3CN)2Cl]PF6. NMR spectroscopy and MS experiments indicated the presence of the desired products in the reaction mixture but we were not successful in isolating them. As the compounds were designed as anticancer agents with redox active ligand systems and this requires stability in aqueous solution, this route was not further pursued, since in biological medium the DQQ complexes would be converted into ortho-quinone compounds in situ. Instead, the ortho-quinone complexes 1–5 were evaluated on their cytotoxic properties. Surprisingly only the osmium complex showed promising activity in HCT116 human colorectal, NCI-H460 non-small cell lung and SiHa cervical carcinoma cells (Table 2) with IC50 values in the low μM range.
The cytotoxicity of quinone-based compounds has been suggested to be related to them undergoing redox reactions.8 In order to study the impact of metal complexation on the redox properties of the ortho-quinone system, pulse radiolysis data was collected (Fig. 5 for 1). As the free ortho-quinone ligand of 1–5 is not accessible, the redox properties of the complexes were compared with that of DQQ. While the one-electron reduction potential E(1) of DQQ is found in the positive range at 96 mV, the metal complexes feature around −100 mV (ESI†), which is similar to that of the anticancer drug candidate KP1019 whose mode-of-action is based on the activation by reduction mechanism.17 Compared to ortho- and para-naphthoquinones the E(1) of DQQ is significantly higher, while the E(1) of para-naphthoquinone is lower than that of the ortho derivative.18 When comparing the para-quinone DQQ with the complexes, where the ligand is present in an ortho-quinoid structure, the difference in E(1) is significantly larger. This indirectly demonstrates an impact of the metal centre on the E(1) of the ligands, although no direct coordination occurs to either of the oxygen atoms of the ortho-quinone. This can be explained by the conjugated electron system being present in the ligands and involving the metal centre.
 | ||
| Fig. 5 Changes in the absorption spectrum recorded for 1 following a one-electron reduction, ○. Spectrum corrected for ground-state absorption (dashed line), ●. | ||
In summary, when aiming to prepare organometallic compounds from DQQ we observed the surprising activation of the C–Cl bond in meta-position to the quinone oxygen aimed for coordination to the metal centre. Variation of the reaction conditions helped to identify the solvent as the source of the oxygen atom introduced to form a metal–quinoline-ortho-quinone complex. The reaction mechanism to form this complex involves bidentate coordination of DQQ to the metal centre, and thereby conversion of the para-quinone DQQ into a quinolinolato ligand featuring an ortho-quinone. Pulse radiolysis studies revealed an impact of metal coordination on the redox potentials of the ligands and the Os complex 2 was identified as the most cytotoxic derivative with IC50 values in the range of 1.8–4.4 μM in human cancer cells.
We thank the University of Auckland (Doctoral Scholarships to M. K. and H. H.). The authors are grateful to Tanya Groutso for collecting the single crystal X-ray diffraction data, and Tony Chen for ESI-MS analyses. We thank Auckland Science Analytical Services of the University of Auckland for access to their facilities and Dr Dan Furkert (University of Auckland) for useful discussions.
Conflicts of interest
There are no conflicts to declare.References
- C. S. Allardyce and P. J. Dyson, Dalton Trans., 2016, 45, 3201 RSC.
- A. Kurzwernhart, W. Kandioller, C. Bartel, S. Bachler, R. Trondl, G. Muhlgassner, M. A. Jakupec, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 4839 RSC.
- W. Kandioller, E. Balsano, S. M. Meier, U. Jungwirth, S. Goschl, A. Roller, M. A. Jakupec, W. Berger, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2013, 49, 3348 RSC.
- M. Kubanik, W. Kandioller, K. Kim, R. F. Anderson, E. Klapproth, M. A. Jakupec, A. Roller, T. Söhnel, B. K. Keppler and C. G. Hartinger, Dalton Trans., 2016, 45, 13091 RSC.
- M. Kubanik, H. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658 CrossRef CAS.
- A. Paretzki, H. S. Das, F. Weisser, T. Scherer, D. Bubrin, J. Fiedler, J. E. Nycz and B. Sarkar, Eur. J. Inorg. Chem., 2011, 2413 CrossRef CAS.
- Y. Prieto, M. Munoz, V. Arancibia, M. Valderrama, F. J. Lahoz and M. L. Martin, Polyhedron, 2007, 26, 5527 CrossRef CAS.
- C.-K. Ryu and H.-J. Kim, Arch. Pharmacal Res., 1994, 17, 139 CrossRef CAS.
- A. N. Pearce, E. W. Chia, M. V. Berridge, G. R. Clark, J. L. Harper, L. Larsen, E. W. Maas, M. J. Page, N. B. Perry, V. L. Webb and B. R. Copp, J. Nat. Prod., 2007, 70, 936 CrossRef CAS PubMed.
- I. A. Shaikh, F. Johnson and A. P. Grollman, J. Med. Chem., 1986, 29, 1329 CrossRef CAS PubMed.
- D. Q. Shen, Y. Cheng, L. K. An, X. Z. Bu, Z. S. Huang and L. Q. Gu, Chin. Chem. Lett., 2008, 19, 533 CrossRef CAS.
- Q.-M. Chen, G.-B. Yi, L.-K. An and X.-L. Feng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o1108 CAS.
- M. M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) kosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631 CrossRef PubMed.
kosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631 CrossRef PubMed. - E. J. Corey and H. Koenig, J. Am. Chem. Soc., 1962, 84, 4904 CrossRef CAS.
- A. Defant, G. Guella and I. Mancini, Eur. J. Org. Chem., 2006, 4201 CrossRef CAS.
- A. Defant, B. Rossi, G. Viliani, G. Guella and I. Mancini, J. Raman Spectrosc., 2010, 41, 1688 CrossRef.
- P. Schluga, C. G. Hartinger, A. Egger, E. Reisner, M. Galanski, M. A. Jakupec and B. K. Keppler, Dalton Trans., 2006, 1796 RSC.
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 18, 1637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1571650–1571654. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc09478g |
| This journal is © The Royal Society of Chemistry 2018 |