
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Artificial molecular and nanostructures for advanced nanomachinery
Elizabeth
Ellis
ab,
Suresh
Moorthy
ac,
Weng-I Katherine
Chio
 ad and
Tung-Chun
Lee
*ac
ad and
Tung-Chun
Lee
*ac
aDepartment of Chemistry, University College London (UCL), UK
bInstitute for Materials Research and Engineering (IMRE), Agency for Science Technology and Research (A*STAR), Singapore
cInstitute for Materials Discovery, University College London (UCL), UK. E-mail: tungchun.lee@ucl.ac.uk
dSingapore Bioimaging Consortium (SBIC), Agency for Science Technology and Research (A*STAR), Singapore
First published on 19th February 2018
Abstract
Artificial nanomachines can be broadly defined as manmade molecular and nanosystems that are capable of performing useful tasks, very often, by means of doing mechanical work at the nanoscale. Recent advances in nanoscience allow these tiny machines to be designed and made with unprecedented sophistication and complexity, showing promise in novel applications, including molecular assemblers, self-propelling nanocarriers and in vivo molecular computation. This Feature Article overviews and compares major types of nanoscale machines, including molecular machines, self-assembled nanomachines and hybrid inorganic nanomachines, to reveal common structural features and operating principles across different length scales and material systems. We will focus on systems with feature size between 1 and 100 nm, where classical laws of physics meet those of quantum mechanics, giving rise to a spectrum of exotic physiochemical properties. Concepts of nanomachines will be illustrated by selected seminal work along with state-of-the-art progress, including our own contribution, across the fields. The Article will conclude with a brief outlook of this exciting research area.
1. Introduction
Sophisticated nanomachinery has been a goal for the scientific community since the idea of nanorobots swimming through our bloodstreams to cure illness was first proposed by Richard Feynman in his ‘There's plenty of room at the bottom’ speech in 1959.1 The idea is that these tiny machines could seek out, identify and fix problems, like miniature surgeons. Other uses of nanomachines have been envisaged such as scavenging environmental toxins,2,3 molecular computation4 and beyond. These futuristic ideas are closer and closer to becoming a reality. Recent years have seen the development and characterisation of structures able to perform controlled functions at smaller and smaller length scales, culminating in the work of the 2016 Nobel laureates, who pioneered the development of molecular machines that could function as rotors, shuttles and pumps.5,6 Recently, larger and more complex nanomachines have also been designed and made, including DNA nanorobots that are capable of serving as switches, rotors, joints and logic gated carriers.7–9 The success in creating molecular and nanomachines indeed opens up tremendous opportunities of performing unprecedented tasks at the molecular length scale. Nevertheless, we are still a few steps behind the Feynman-type nanorobots, which probably require the combination of nanomachinery parts across various length scales for performing intelligent and sophisticated functions.This Feature Article aims to offer insights into common features and operating principles shared by a broad range of nanoscale machines across different length scales and material systems, from molecular shuttles to soft nanomotors and from molecular assemblers to hybrid nanopropellers. We will focus on artificial nanosystems that are capable of performing useful functions, very often, by means of doing mechanical work at the nanoscale, and will consider the above as a general definition of “nanomachines”. While the term “nano” has been widely used to describe systems of 1–1000 nanometers (i.e. 10−9–10−6 meters), our scope will be on nanosystems between 1 and 100 nm – the length scale where classical laws of physics meet the laws of quantum mechanics, giving rise to exotic phenomena, such as quantum-size effects and localised surface plasmon resonance. This is also a length scale at which enzymes, the natural nanomachines, operate under strong stochastic forces. The Article will start by overviewing three major groups of nanomachines, namely molecular machines, self-assembled nanomachines and hybrid inorganic nanomachines (Fig. 1), where the boundary between these groups can sometimes be vague and subject to a specific context. The aim is not to provide an exhaustive list of precedent work, but to illustrate the operation of nanomachines by seminal work and state-of-the-art research across the fields. It will be followed by discussions on features and design rules that are common across different length scales and material systems, and will conclude by an outlook of this exciting research area.
 | ||
| Fig. 1 Molecular and nanomachines classified based on their length scales and material systems. Subclasses based on function types are labelled on the left of each subclass. Length scale of the feature size of the nanomachines is shown at the bottom for reference. For hierarchical structures, e.g. rotor driven liquid crystal, it is difficult to specify the feature size. In those cases, approximated length scale of the basic functional unit will be considered. Panels (from left to right, top to bottom) – molecular machines: stereodivergent assembler,20 four-wheeled car,25 rotor,14 shuttle,13 rotor-driven liquid crystal film,35 azobenzene-functionalised liquid crystal elastomer;32 self-assembled nanomachines: single polymer chain NP,91 DNA origami logic gated nanobot,78 asymmetric polymersome,61 Pt NP loaded stomatocyte;62 inorganic & hybrid nanomachines: dynamic nanoflasks,118 CBs capped by Au NPs,116 host–guest complex of Au NP inside a SiO2 nanocup,119 Au NP with a Janus shell,101 Pt–Au Janus NP,94 Pt–mesoporous SiO2 Janus NP,99 Au–Fe nanohelix111 and SiO2–Ni nanopropeller.106 Adapted by permission from the corresponding references. Copyright NPG, AAAS, ACS and Wiley-VCH. | ||
2. Molecular machines
The advance of human civilisation has led to mechanisation across multiple length scales. This includes the development of sophisticated devices, from smartphones to jet engines, which have had a considerable impact on our lives. We are now seeking to push the limits of mechanisation at small scales by constructing increasingly minute machines. Nevertheless, in fields such as information processing, top-down approaches are reaching their fundamental limits, due to the associated increase in power consumption. Feynman's idea of using a bottom-up approach involves “manoeuvring things atom by atom”1 to build nanoscale machines. This was not initially considered feasible by chemists due to the fact that atoms are highly reactive and do not normally stay isolated, i.e. they cannot be transferred from one place to another individually.10 As an alternative to the atomic bottom-up approach, supramolecular chemistry offered the idea that self-assembly of molecules is much more convenient than that of atoms for constructing molecular devices. This approach opened up promising possibilities in designing molecules capable of performing non-trivial mechanical movements, giving rise to artificial molecular machines and motors.Why do we need molecular machines? Like every macroscopic machine which reduces human effort, molecular machines might be useful in the following context: (1) in synthetic organic chemistry these machines can perform multiple-step synthesis, starting from simple chiral molecules to complex natural products, in a programmable and scalable manner. Ultimately, molecular machines will be capable of building complex molecules more efficiently than via conventional synthetic routes. (2) From an information processing point of view, switching behaviours of molecular machines can be exploited to perform computation in a parallel and even analogue manner. (3) Directed molecular motion, such as that seen in molecular rotors and shuttles, can be used to physically extract mechanical work, through synchronised or collective motion, to move macroscopic objects. The movements of components in these systems can be powered by light, chemical reactions, pH and temperature gradients.
Enabled by advances in synthetic organic techniques and analytical instrumentation, the progress achieved in molecular machines by Jean-Pierre Sauvage, J. Fraser Stoddart and Ben L. Feringa was recognised by the Nobel Prize in Chemistry in 2016. In these systems, supramolecular interactions within multicomponent molecular structures were used to perform non-trivial mechanical movement and put into widespread use in a variety of nanoscale molecular devices.11
This section highlights recent examples of nanoscale molecular devices and discusses molecular systems that can perform useful work in solution, on interfaces, and in the solid state. Two major technological advances have proven particularly useful in addressing the complex challenge of constructing machines at the molecular scale. The first one involves topological entanglement and so-called mechanical bonds, while the second is based on isomerisable (unsaturated) chemical bonds. Both advances have resulted in a wide range of complex structures with machine-like functions.
2.1. Molecular machines in solution
In 1983, Jean-Pierre Sauvage successfully connected two ring-shaped molecules together to form catenane. This was widely considered the first step towards making a molecular machine.12 Later, J. Fraser Stoddart threaded a ring-shaped molecule on to a molecular axle through mechanical bonds and formed a supramolecular architecture named rotaxane.13 In parallel to the advances based on mechanically interlocked structures for molecular machines, Ben L. Feringa developed a molecular motor, that rotates around an overcrowded alkene, to spin continually in one direction.14 Since then, mostly driven by academic curiosity, chemists have developed various types of molecular machines that operate in solution, which can be categorised based on (1) the types of motion they perform, including rotors, switches, propellers, shuttles, walkers, ratchets and tweezers, and (2) the types of energy input used to operate the machine including chemical, light and electrochemical inputs.15Beyond chemical curiosities, molecular machines containing moving parts can do various useful tasks in solution. One potential advantage is that such machines can act as molecular factories that synthesise or build things from individual molecules in a programmable manner. In the laboratory environment, such stepwise syntheses in solution might produce unconventional products inaccessible by current methods.
In 2003, Nolte et al. reported16 a molecular machine based on an interlocked molecular structure capable of synthesising epoxides from double bonds. In this supramolecular structure, a wheel is formed from a manganese porphyrin based cage which is threaded onto a polybutadiene chain. The catalytically active manganese(III) site present in the porphyrin wheel progressively oxidises the double bonds present in the polybutadiene chain, along which the wheel moves. The chemical structure of porphyrin wheel consists of a manganese(III) site at the top and a glycoluril clip at the bottom is shown in Fig. 2. Incorporation of a Mn(III) catalytic site into the macrocyclic wheel ensures the conversion of alkenes to epoxides. When a polybutadiene polymer (Mn = 300 kDa, 98% cis) substrate is threaded through the macrocyclic wheel, the catalyst converts polybutadiene into polybutadiene epoxide. To ensure the catalysis occur inside the cavity of the macrocyclic wheel in the rotaxane-like architecture, a bulky axial ligand such as tert-butylpyridine is used. The bulky ligand axially coordinates from the top of the catalyst and thus allows the reaction to occur inside the sterically demanding cavity of the rotaxane-like catalyst. The cavity induces stereoselective synthesis and produces predominantly trans product (80% trans- and 20% cis-epoxide polymer) from polybutadiene, preventing the polymer substrate from forming the other stereoisomeric cis product (22% trans, 78% cis in the control experiment which used an Mn–porphyrin without a macrocyclic cavity). The synthesis is carried out at dilute mM concentrations to avoid intermolecular reactions. Products were analysed using 1H NMR. Although it remains unclear whether the catalytic process is sequential or random, it was deduced, from the rate of the reaction and polymer length, that the macrocycle wheel moves around the polymer thread at an average speed of 1–700 pm s−1.
 | ||
| Fig. 2 Schematic representation of the catalytically active rotaxane for epoxidation of polybutadiene to polybutadiene-epoxide.16 Adapted by permission from ref. 16. Copyright 2003 Nature Publishing Group. | ||
Metal-free macrocyclic molecules cucurbit[n]urils (CBn) are also known to catalyse coupling reactions, serving as a convenient route towards rotaxane-based molecular machines in aqueous media. Mock et al. reported in the 1980s that CB6 can catalyse 1,3-dipolar cycloaddition by encapsulating azide and alkyne reactants and form a 1,2,3-triazole with a preference to 1,4-regioisomer.17 This idea shed light on the preparation of polyrotaxanes through 1,3-dipolar cycloaddition from properly designed diazide and dialkyne monomers. In particular, polyrotaxanes have been reported by combing CB6 and diazide/dialkyne monomers with a long dodecamethylene diammonium spacer.18 Upon formation of polymer, the CB6 is threaded by a triazole ring with two ammonium groups binding at the carbonyl portals, resulting in the Stoddart-type molecular shuttles. Deprotonation of the ammonium group at appropriate pH shuttles the CB6 to dodecamethylene spacers, whereas protonation of the amino moieties, shifts the CB6 back to the triazole ring. In 2013, Lee and Nau et al. further explored, using mass spectrometry and molecular modelling, the capability of the CB cavity to mediate chemical reactions. Interestingly, they observed the first inner-phase chemical reaction in the gas phase, where the encapsulated guest molecules (bicyclic azoalkanes) undergo a cascade of retro-Diels Alder reactions inside the CB cavity, meanwhile the competing supramolecular dissociation pathway becomes insignificant.19 Their work opens up new possibilities in studying chemical reactivity and supramolecular interactions of small molecules under nanoscale confinement, aiding the design of novel molecular machines.
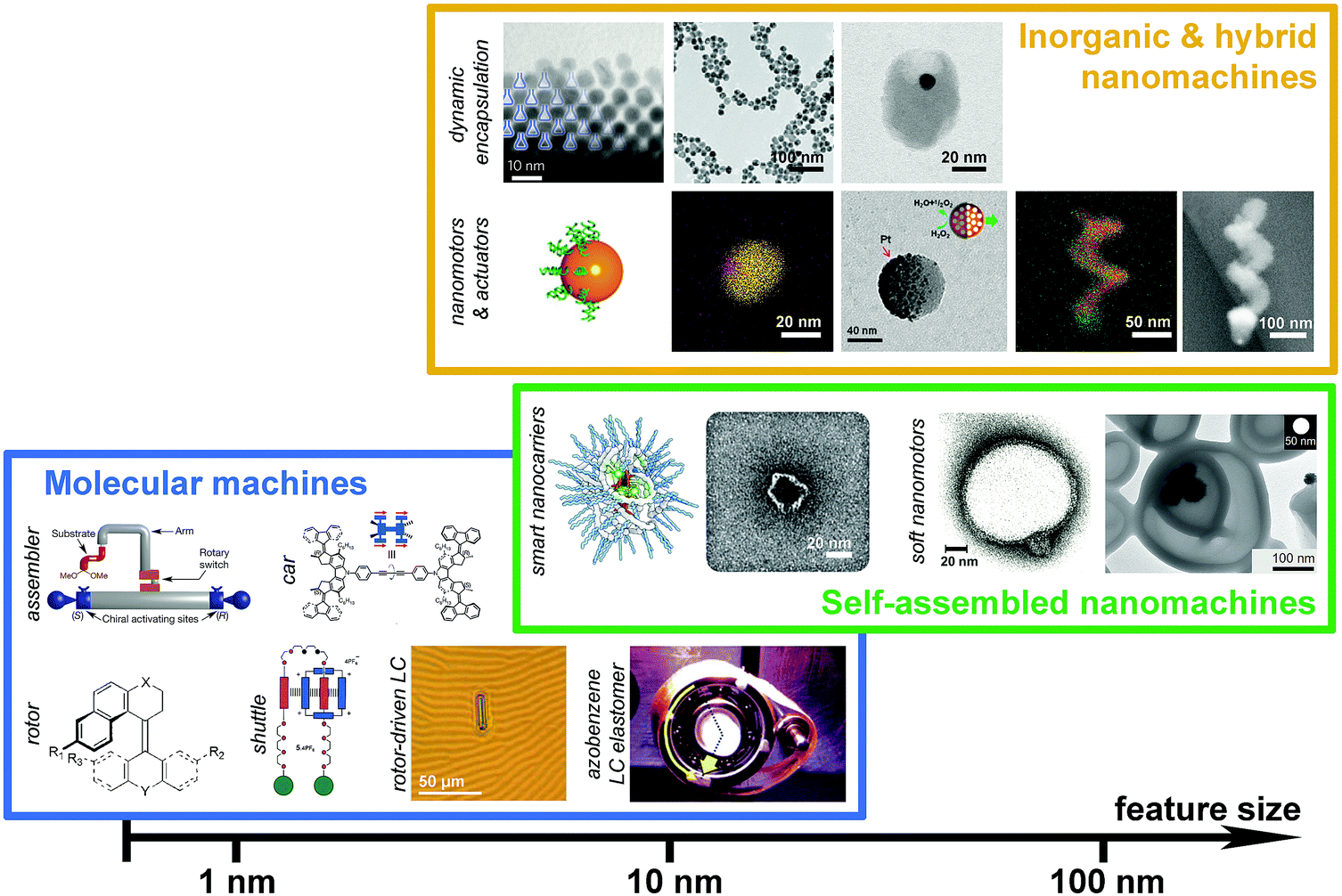
Recently, Leigh et al. reported20 a molecular robot that can perform stereoselective synthesis and produces uncommon stereoisomers, for example anti-diastereomers, which are not selectively produced by conventional synthesis; the conventional methods only allow access to the syn-diastereoisomers. In particular, the reaction consists of a tandem addition of a thiol and an alkene to an α,β-unsaturated aldehyde in presence of iminium–enamine catalyst such as chiral prolinol silyl ethers to synthesise an excess of any one of four possible diastereoisomers in one-pot. In principle four different isomers are possible in this reaction but syn-diastereoisomers are commonly produced.
The molecular robotic design consists of three parts; (1) a rotary switch on stator, (2) two terminals and (3) a rotatable arm (Fig. 3). The two terminals are made up of chiral activating sites (chiral prolinol silyl ethers group); the left terminal being S isomers and the right one being R isomers. A rotary switch (hydrazone), which can go from E to Z under the condition (trifluoroacetic acid or trimethylamine) of machine operation, supported on a quinoline stator is placed in between the terminals via triazole linkages. An arm (para-substituted phenyl rings) is covalently attached to the rotary switch. The function of the arm is to load substrates (α,β-unsaturated aldehyde) through an ester linkage. Depending on the input, this arm can be rotated through E–Z configurational changes of the hydrazone switch. Hence, the substrates loaded on the arm can be brought either to the left or the right terminal on demand, to trigger the catalytic reaction. Therefore, the stereochemical outcome of this tandem reaction is dictated by the relative position of the arm with respect to the two terminals.
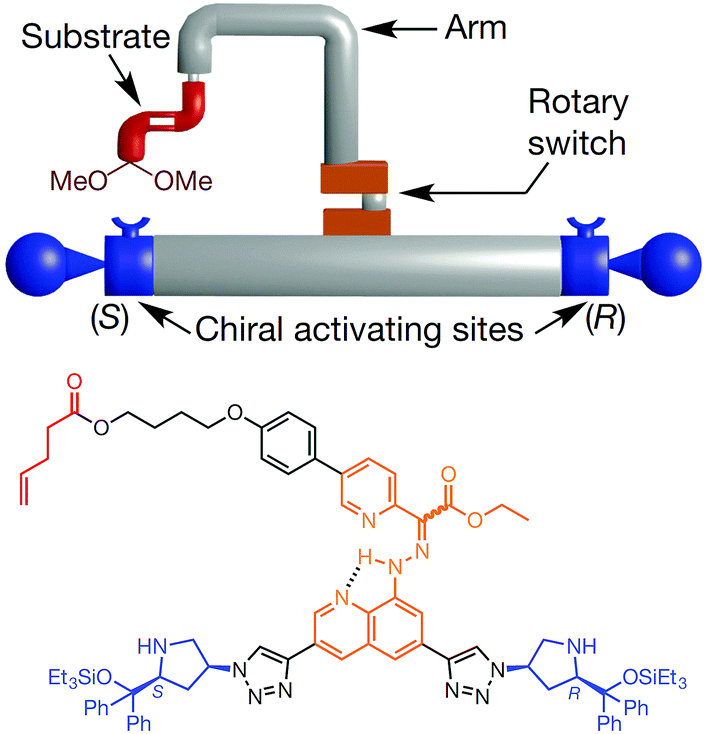 | ||
Fig. 3 Representation of a molecular machine that synthesises two different products by moving a substrate attached to rotor to two different sides. The rotary hydrazone switch in the middle of the machine stays predominantly in E configuration (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ∼ 80:20). The switch can be switched from E to Z to bring the substrate to either the (S) or the (R) to form chiral iminium centre for the first bond formation of a tandem reaction. Once iminium is released, the machine forms enamine intermediates that initiate the second bond formation of the tandem reaction.20 Adapted by permission from ref. 20. Copyright 2017 Nature Publishing Group. :Z ∼ 80:20). The switch can be switched from E to Z to bring the substrate to either the (S) or the (R) to form chiral iminium centre for the first bond formation of a tandem reaction. Once iminium is released, the machine forms enamine intermediates that initiate the second bond formation of the tandem reaction.20 Adapted by permission from ref. 20. Copyright 2017 Nature Publishing Group. | ||
Leigh et al. also synthesised21 a molecular machine that can stitch small molecules together to form desired new molecules such as amino acid sequences. The machine has rotaxane type structure that mimics the function of ribosome (a massive biological molecular machine) to synthesise amino acid sequences in the desired order. In the rotaxane design, a synthetic thread type molecular rod, of a few nanometers long, is attached to three amino acids pre-assembled covalently. When a wheel type molecular ring bearing thiol group is threaded on a rod, the thiol group picks up the amino acids one by one as it moves along the molecular rod. After picking up all the three pre-assembled amino acids, the ring slips off the thread and completes the amino acid synthesis in a sequence that was pre-assembled in the rod. Unlike the biological ribosome which can stitch 20 building blocks a second, the new synthetic molecular machines connect up to four amino acid building blocks. Once the molecular machine is initiated, it completes the synthesis in 12 hours without any additional interventions.
2.2. Molecular machines on surfaces and interfaces
Molecular machines capable of performing useful work on solid surface are more difficult to realise than those in solution. The advantage of anchoring a molecular machine on solid surfaces is the resultant coherent functioning of molecules that allows more efficient extraction of work in a collective manner; this is not possible in solution, where discrete molecules move in an incoherent manner. Molecular switches on surfaces can also control bulk properties of the material, such as wettability and conductivity.22,23 This section discusses molecular machines that work on surfaces and interfaces such as air–water and solid–liquid. In particular, it highlights stimuli-responsive molecular rotors working on solid surfaces that undergo rotational motion to perform useful functions, for example, moving a molecular car or gating the release of cargoes. The stimuli include chemical, light and electricity.Feringa et al. reported the first example of surface bound molecular rotary motors in 2005.24 The rotary motion of this motor is similar to the one discussed in Fig. 3, but the rotary motion does not induce any subsequent translational motion. The rotary motor design has four parts: a rotor, a stator, an axle and legs (Fig. 4). A chiral helical alkene with an upper half that serves as a rotor and is connected through a carbon–carbon double bond (axle) to a lower half that serves as a stator. The stator at the bottom is derivatised with two thiol-functionalised legs. These thiol legs attach the whole system onto the gold surface and the two-point attachment avoids uncontrollable rotation of the motor. Input sequences such as shining light and subsequent heating rotate the upper half of the molecule in an anticlockwise fashion to 180° with respect to the bottom half. The net 180° rotation is due to photo-induced E–Z isomerisation of the central double bond, followed by a subsequent thermal helix inversion. Repeating the same input sequences completes a full rotary cycle of 360°. The motor is unidirectional as the rotor in the upper half always rotates anticlockwise and it does not rotate back clockwise due to the helical inversion step.
 | ||
| Fig. 4 A molecular rotary motor driven by light attached to gold surface.24 Adapted by permission from ref. 24. Copyright 2005 Nature Publishing Group. | ||
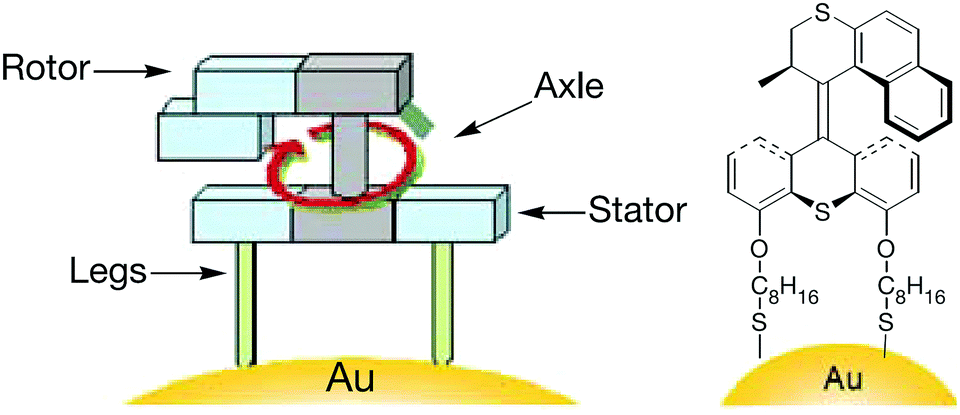
In 2011, Feringa et al. reported25 the smallest nano-car in the world which can be electrically driven. The car is a single molecular device that consists of four wheels and can travel almost in a straight line on copper surface when powered by electricity (Fig. 5). Designing molecular cars could be useful to carry out work at the nanoscale. The single molecular car consists of four chiral wheels based on unidirectional rotary motors. Upon excitation, the double bond present in the chiral units undergoes double bond isomerisation and helix inversions and thus rotates the chiral wheel unit in only one direction. In other words, the wheel cannot go back in the reverse direction. The concept of unidirectional rotary motors has been well established in previous studies in solution.
 | ||
| Fig. 5 Structure of the four-wheeled molecular car – the meso-(R,S–R,S) isomer.25 Adapted by permission from ref. 25. Copyright 2011 Nature Publishing Group. | ||
On a surface, the single molecule functions as follows: the molecule is deposited onto a Cu(111) surface by vacuum sublimation. It can move in one direction when electrical energy is fuelled through the tip of a scanning tunnelling microscope (STM); the STM tip with voltage larger than 500 mV activates conformational and configurational changes of the chiral wheel units, resulting in paddlewheel-like motion pushing the entire molecule forward in a stepwise manner. All four wheels should be activated at the same time, to avoid the car from either spinning or uncontrolled movement.
For proper functioning, the correct starting configuration of the meso-(R,S–R,S) isomer is shown in Fig. 5. The car should rotate all the four wheels in the same direction to move forward in a straight line. It is noted that, however, the central portion of the molecular car can undergo rotation around the single and triple bonds. Therefore there is only a 50% chance where the meso-(R,S–R,S) isomer is deposited on copper surface in a desirable configuration. In the case of undesirable landing, the rotary motion of the four wheels will cancel out each other, resulting in no displacement of the nano-car. Per STM tip activation, the car moved 0.7 nm forward by paddlewheel-like motion and, ten such activation events result in the molecular car travelling around 6 nm across the surface in one direction.
Ariga et al.26 used mechanical pressure stimuli to control the shape of the steroid cyclophane based molecular machine, that can form the Langmuir monolayer at the air–water interface. An air–water interface provides a two-dimensionally confined environment, where hydrophilic and hydrophobic molecules could be manipulated using external pressure. In the chemical structure of steroid cyclophane molecules, the cyclophane forms the central core and is connected to four steroid moieties, cholic acid. The cholic acid has both hydrophilic and hydrophobic faces, and stays flat in water through its hydrophilic face. When the steroid cyclophane molecules form a monolayer at the air–water interface, they respond differently to low and high pressure. In particular, in the compressed (high pressure) state the steroid cyclophane in the monolayer adopts “cavity shape”, whereas in the expanded state (low pressure) it adopts “flat shape”. Such a dynamically controlled cavity structure was used to accommodate aromatic guests such as the naphthalene derivatives in a 1:1 stoichiometry. The selective capture and release of aromatic guest molecules from the water phase in the air–water interface was possible through shape conversion of the molecular machine that forms a monolayer, using macroscopic pressure stimuli. Eight such compression and expansion cycles were repeated.
Similarly, axial rotation of molecules induced by changes in macroscopic pressure has been observed. Liu et al.27 reported the pressure-induced formation of spiral morphology from non-spiral barbituric acid derivatives, when their chloroform solution was spread on the air–water interface. In particular, when a Langmuir–Blodgett film of barbituric acid derivatives was compressed, directional hydrogen bonding between molecules will induce them to curve in a certain direction, either clockwise or anticlockwise, resulting in the formation of spiral nanostructures.
Finally, molecular machines can also be anchored at the pore entrance and the pore surface of mesoporous silica. For instance, integration of mechanically interlocked molecules such as pseudorotaxane and rotaxane at the entrance of mesoporous silica can produce nanovalves, i.e. a stimuli responsive device that can encapsulate and release guest molecules in the 2 nm sized pores of silica. These nanovalves are useful for transport of cargo molecules in drug delivery. In a seminal paper, Zink et al.28,29 prepared a nanovalve containing a bistable[2]rotaxane, in which the thread consists of two different electron-rich stations (naphthalene and tetrathia-fulvalene TTF); naphthalene located close to the pore entrance, whereas TTF stays away from the mesopores. The gate at the mesopore entrance is opened when an electron-deficient ring (cyclobisparaquat-p-phenylene, shown in blue in Fig. 6) encircles TTF, whereas the gate is closed when the ring encircles naphthalene station. Chemical oxidation and reduction of TTF station move the ring up and down and it can be harnessed to open and close the pore entrance, respectively. In addition to chemical gating at the mesopore entrance, photoactive azobenzene moieties can be anchored within the mesopores and serve as light-powered molecular impellers that aid expelling the cargo.30 Here, both light and pH responsiveness have been employed for controlled release of guests or drugs from the nanopores.31
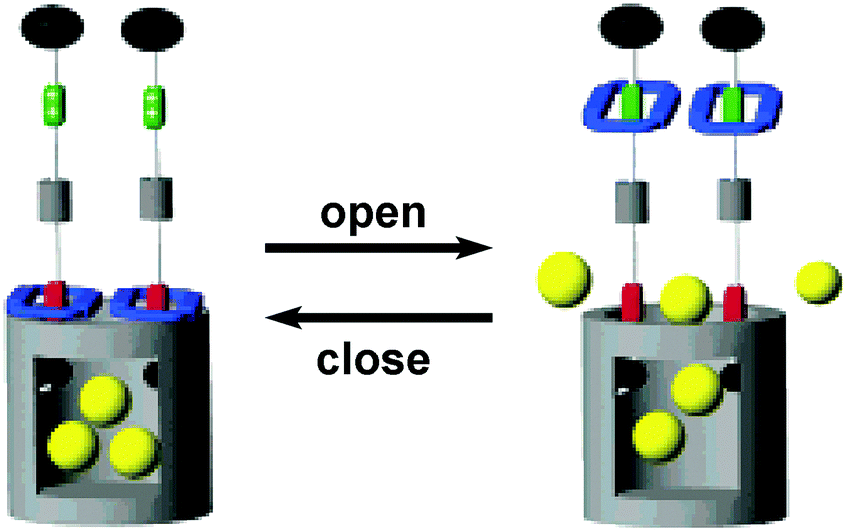 | ||
| Fig. 6 Graphical representations of the surface attachment of redox switchable bistable rotaxanes to mesoporous silica particles.29 Adapted by permission from ref. 29. Copyright 2007 American Chemical Society. | ||
2.3. Molecular machines in the solid state – from microscopic to macroscopic behaviour
Molecules that absorb light and change their shape can potentially be used to convert light energy directly into mechanical work. Cooperative conformational changes at a single molecular level can be harvested to generate significant macroscopic movements. This section discusses examples of such behaviour.Ikeda et al. reported flexible robotic arms made of crosslinked liquid-crystalline polymers (CLCP) containing azobenzene.32 The azobenzene in CLCP undergoes light-induced E–Z isomerisation and causes three-dimensional bending and translational movements. Initially, the free standing film made up the CLCP is bent in shape. When films are irradiated with UV light (366 nm), the bent film extends to a flat shape; when irradiated with visible light (>540 nm), the film reverts to the initial bent shape. It is demonstrated that under UV-vis irradiation cycles at room temperature, the film can walk unidirectionally on a planar surface. Unlike simple photo-induced bending movements, more sophisticated light-driven motion can also be achieved by designing the film structure and irradiation conditions. For instance, a plastic motor driven by light based on azobenzene-functionalised liquid-crystalline elastomer (LCE) has been developed which can convert light energy directly into a continuous rotation without the aid of batteries (Fig. 1 bottom row, right panel).33 In particular, the authors prepared a millimetre-sized continuous ring of the LCE laminated films that can undergo a 360° rolling movement upon simultaneous exposure to both UV and visible light on the opposite ends of the rollers. To test that the materials generate mechanical power, polyethylene films were coated with LCE films to create a belt size of 36 mm × 5.5 mm and the belt was looped around two wheels with 10 mm and 3 mm in diameter. Upon shining light, the belt began to turn around the wheels and rotated them like a conveyor belt.
Fischer et al. developed34 microswimmers that are able to propel through water by synchronised movement without employing complex driving elements or external forces such as magnetic fields. The swimming micro-robot was formed by combining the properties of light responsive liquid crystals and elastomers. It was capable of propelling upon exposure to structured light produced by a digital micromirror device (DMD). Two types of micro-robots were fabricated: an elongated cylinder of 1 cm long and a tiny disk of 50 μm thickness. These micro-robots undergo biomimetic shape change in water when they are exposed to light. Projecting a light pattern across the cylindrical robot causes the photo-responsive units (azobenzene) in the liquid crystal molecules to bend in U-shape and aggravates the deformation, forming protrusions that are responsible for travelling wave generation. These waves move the liquid along with them so that the microswimmer propels through liquid at a speed of 2.1 μm s−1. Upon varying the sequence of light patterns, the microswimmers followed a wide range of custom movements such as a rectangular trajectory or rotation within the same micro-robot.
In the above examples, bending or rotation of polymeric films or liquid crystal elastomer is induced by photochemical isomerisation around unhindered azo-based double bonds. Since there is no control over the kinetics of the photo-isomerisation, the direction of movement is determined by the shape of the film or the illumination patterns. Remarkably, however, it is possible to control the direction of macroscopic motion through directional photo-isomerisation across a sterically engineered double bond. In a seminal work, rotary motion of the molecular motor that rotates around an overcrowded alkene was demonstrated to induce the rotation of larger microscopic objects. Feringa et al. achieved35 torque generation on a microscopic glass rod (5 × 28 μm, i.e. 104 time bigger than the molecular motor) in a cholesteric liquid-crystal film containing only 1% (w/w) of the rotary nanomotors. Upon UV light irradiation, the molecular rotors rotate in a single direction, inducing the rod to rotate in a clockwise fashion which could be observed under an optical microscope (Fig. 1 bottom row, second panel from the right). Notably, exchanging the molecular motor with its enantiomer causes rotation in an anti-clockwise direction. This pioneering work illustrated that it is indeed possible to harvest mechanical work from an artificial molecular rotary machine.
3. Self-assembled nanomachines
Self-assembled nanomachines are a step up in size from molecular machinery and rely on the autonomous organisation of an ensemble of molecules. This can result in larger, standalone nanostructures that are capable of independently performing functions such as serving as a switch, hinge, logic gate or producing active propulsion in liquid media. Traditional self-assembly of small molecules, such as lipids or block copolymers, into larger structures produces highly symmetric configurations, such as micelles or vesicles.36,37 In order to act as nanomachinery, assemblies need to be more complex, compartmentalised, and often anisotropic to facilitate directional movement or carry out coordinated functions. Hierarchical self-assembly can be achieved through the use of multicomponent molecules, such as block copolymers, DNA or peptide chains, that can assemble into diverse structures including α-helices and β-sheets, which then assemble further into higher order structures.38 Nature exploits this strategy to make motor proteins such as kinesin and myosin, which perform work in the form of transport along fibres, powered by hydrolysis of ATP.39 It is in theory possible to mimic this level of complexity in self-assembly via DNA origami approaches, the design and folding of DNA chains, and through self-assembly of sophisticated block copolymers.Cargo delivery is regarded as an important capability of nanomachinery whereby molecular species are encapsulated and released in a controllable manner. This function can be accomplished using self-assembled structures such as micelles and vesicles, which can incorporate hydrophobic molecules within hydrophobic parts of their structures and hydrophilic molecules within aqueous cavities in the self-assembled structures.40 Compartmentalised self-assembled structures have been designed with the ability to encapsulate different molecules in different sections of their structure. For instance, polymer–lipid Janus particles can encapsulate drugs of different solubility within the two separate compartments and their effectiveness was tested against lung tumours in mice.41
Stimuli-responsiveness is considered another critical property of functional nanomachines. The ability to sense and respond to the surrounding environment can allow nanomachines to perform more dynamic and smart tasks. Supramolecular nanostructures are particularly well-suited for this purpose. Here, the self-assembled constructs are held together only by weak and dynamic intermolecular interactions (incl. hydrogen bonding, π–π interaction hydrophobic forces and dispersion forces), these nano-objects are capable of changing their shapes or disassembling–reassembling in a reversible manner.42–45 For instance, Lee and Scherman et al. have demonstrated the release of cancer drugs from hierarchical nanocarriers can be controlled by three orthogonal, biologically relevant triggers (pH, temperature and a specific molecular cue) that can result in disassembly of the micelles, releasing doxorubicin.46 This controlled release reduces toxicity to other areas of the body. A wide range of stimuli-responsive polymeric structures have been designed, governed by hydrophobic interactions, which can selectively release molecules in response to certain stimuli.47 For example, pH responsive polymers anchored on gold nanoparticles have been shown to respond to the low pH of cancer cells and selectively release doxorubicin.48
Active propulsion is arguably the most sought-after function of nanomachines in recent years, as it allows nanomachines to move themselves actively against randomising Brownian forces and chemical gradients to reach desirable regions of a given system. Propulsion at the nanoscale is particularly challenging due to the low Reynolds number environment, where inertia of motion becomes negligible and conventional methods of propulsion become ineffective. At the nanoscale, one effective mechanism of self-propulsion is self-diffusiophoresis,49,50 which involves propulsion forces produced by a concentration gradient of neutral molecular species across a nano-object. This concentration gradient is typically generated and sustained by catalytic chemical reactions occurring on only one face of an anisotropic nanostructure, for instance the catalytic decomposition of hydrogen peroxide on the Pt-face of a Pt–SiO2 Janus nanoparticle (see Section 4 for details). The concentration gradient will then induce slipping of solvent molecules over the surface of the particle, to neutralise the gradient. This phoretic fluid flow will result in active propulsion of the particle through liquid media. An essential feature for nanostructures to exhibit self-diffusiophoresis is structural anisotropy. If a reaction takes place uniformly around the entire particle, no concentration gradient is created and hence no propulsion takes place.51 While breaking of symmetry at the nanoscale can be difficult, however, self-assembly of block copolymers has been used to create anisotropic structures, which have been shown to display self-diffusiophoretic behaviour.
3.1. Block copolymer nanomachines
While cargo delivery and stimuli-responsiveness of self-assembled nanostructures have been well established, this subsection mainly explores recent advances of self-propelling nanomotors based on anisotropic block copolymer constructs. In general, the behaviour of block copolymers in solution depends on a number of factors, such as the length of each block, the block length ratio, the interactions between each block and the solvent and between each block and each of the other blocks. In aqueous media, if the hydrophilic block is longer than the hydrophobic block, this will drive the micelle towards a high curvature spherical morphology. If the hydrophilic block is short, however, this can lead to lower curvature morphologies such as cylindrical micelles and vesicles. Furthermore, in order for micelles to form, the concentration of block copolymers in solution must be above the critical micelle concentration of the polymers. When designing block copolymers for micelle formation it is considered that the packing parameter can be used to estimate block ratios for specific morphologies.52 The tendency for polymers to phase separate when exposed to other non-compatible polymers can be exploited when attempting to form asymmetric structures that are necessary for self-propulsion.A few different methods of making asymmetric micelles from block copolymers have been developed.53 They can largely be classified into methods using two diblock copolymers,54,55 or methods using one triblock copolymer with three different blocks.56,57 In order for an asymmetric particle to be formed the core or corona must consist of two different polymer blocks. The symmetry-breaking step therefore relies on the incompatibility between blocks and can be ‘locked-in’ via subsequent crosslinking. One method, developed by Mueller et al., involves the stepwise reduction of degrees of freedom in the micelles. In particular, ABC triblock copolymers are placed in a poor solvent of the middle B block, producing a micelle with a B core and a mixed AC corona. The micelles are then transferred to a solvent which preferably interacts with the C block, so the A blocks self-segregate to form a compact patch on the micelles which then stick together via these patches, in order to minimise unfavourable solvent interactions. The B blocks can then be crosslinked and the “supermicelles” dispersed in a good solvent, leaving Janus micelles with a crosslinked B core and a segregated A and C corona. The corona ratio can be tuned by altering the block ratios of the polymers.58,59 This method has been used to form micelles in organic solvents from polymers synthesised by living anionic polymerisation, and to form pH responsive micelles in aqueous solution from polymers synthesised by Atom Transfer Radical Polymerisation (ATRP). Other methods also exist, for example a method has been developed where a diblock copolymer was synthesised, self-assembled into a spherical micelle and the core crosslinked. Another monomer was then used to swell the crosslinked core, then polymerised to form a single area of the new monomer on the surface of the micelles.60
A self-assembled polymeric nanomotor was recently designed and self-assembled from a mixture of two vesicle-forming block copolymers, poly(ethylene oxide)–poly(butylene oxide) (PEO–PBO) and poly(oligoethyleneglycol methyl ether methacrylate)–poly(diisopropylamino ethyl methacrylate) POEGMA–PDPA or poly(methacryloxyphosphorylcholine)–poly(diisopropylamino ethyl methacrylate) (PMPC–PDPA). The polymers self-assembled into vesicles of PDPA–POEGMA/PMPC mixed with PEO–PBO. Over time the PEO–PBO phase-separated from the PDPA–POEGMA/PMPC within the membrane to form a small PEO–PBO bulge, due to the incompatibility of the different types of polymers (Fig. 7a). Enzymes were encapsulated within the vesicle and used to catalyse reactions. The reaction products selectively diffuse through the more permeable PEO–PBO patch, creating a concentration gradient of products over the surface of the vesicle, which leads to a slip velocity and induces movement. The vesicles were found to exhibit chemotactic behaviour, in which they move in the direction of an increasing concentration of substrate. In addition, it was shown that the chemotactic vesicles were able to penetrate more effectively through the blood brain barrier than standard vesicles.61
 | ||
| Fig. 7 Self-propelling soft nanomotors from block copolymer self-assembly. (a) Asymmetric POEGMA–PDPA or PMPC–PDPA vesicles with PED–PBO patch, containing glucose oxidase and catalyse.61 (b) PEG–PS stomatocytes encapsulating Pt NPs in cavity62 which is compatible to peptide functionalisation for cell targeting.66 Adapted by permission from ref. 61 and 62. Copyright 2017 AAAS and 2011 Nature Publishing Group. | ||
Self-assembly of poly(styrene)–poly(ethylene glycol) (PS–PEG) block copolymers was also used to form self-propelling vesicles (Fig. 7b).62 The polymers self-assembled into vesicles in THF, which then collapsed to form stomatocyte-like structure upon the removal of organic solvent via dialysis against water. The stomatocytes had a central compartment with a small opening to the outer solution. Platinum nanoparticles, which function as catalysts, were encapsulated within the central cavity of the stomatocyte. A concentration gradient over the surface of the nano-object can be created when fuel of hydrogen peroxide is catalytically decomposed by the platinum nanoparticles to form oxygen molecules which were then expelled through the opening of the cavity. This mechanism results in active self-propulsion of stomatocytes in liquid media. Owing to the relatively fast rotational Brownian motion, the motion is practically in the enhanced-diffusion regime. This group has also shown that the vesicles can be biodegradable by using PCL instead of polystyrene63 and can encapsulate drugs such as doxorubicin.64 Moreover, they can be powered via enzymes instead of platinum,65 be tagged with cell targeting receptors and penetrate cell membranes in an efficient manner.66
Block copolymer structures are also easily functionalised and can be designed to be responsive to certain stimuli, such as pH, temperature and oxidation state. They could therefore potentially be incorporated into other nanomachinery (see introductory paragraphs of Section 3 for details).
3.2. DNA nanomachines
Deoxyribonucleic acid (DNA) is a polynucleotide that is found in most living organisms for storing genetic information in the form of a well-defined sequence of nucleotides. Remarkably, owing to the capability of encoding and transferring information and the ease of synthesis, DNA can be engineered to perform artificial tasks, including molecular computation67 and nanoscale walkers.68 For instance, DNA walkers that mimic the motion of walking motor proteins have been designed that are able to move in a selected direction along a track69 and even transport cargo.70 Like motor proteins the nanomachines have two ‘feet’ that successively coordinate to the fibre. Nevertheless, instead of using ATP for fuel, the motors use a fuel made from nucleotide strands. Directional progressive motion is achieved by use of a Brownian ratchet mechanism to prevent the walkers from reverting to the wrong direction along the track.68Exquisite control over the self-assembly of DNA can be gained by using DNA origami68,71,72 and related techniques.73 DNA origami involves the controlled self-assembly of DNA strands using Watson–Crick base pairing to produce biocompatible nanostructures and is extremely versatile. The interactions are predictable and selective, where a long DNA strand (typically a circular genomic DNA from the virus M13mp18) can be selectively folded by pairing with many short staple strands that fold it into place.74,75 The necessary strands required for folding into particular designs can be calculated computationally and added in excess. Owing to the specificity of DNA hybridisation, virtually any position on the resultant DNA construct can be selectively targeted and functionalised.
A DNA origami approach has been used to design two-dimensional (2-D) tracks that are more rigid and well-defined in shape compared to other DNA tracks. DNA origami tracks can guide DNA walkers to move along a programmable 2-D trajectory and to follow instructions such as ‘start’, ‘turn’, ‘follow’ and ‘stop’.76 Moreover, their larger and more complex scaffolds allow DNA origami structures to be efficiently coupled to larger nano-objects and to perform more sophisticated tasks. For instance, a DNA origami approach has been used to design and make nanoscale assembly lines to transport and put together gold nanoparticles.77 The DNA walker is made of a triangular structure with three hands and four feet, with the feet allowing locomotion and the hands moving the cargo. They can move across a tile of DNA origami with switchable stations for controlled assembly. This and similar types of nano-assembly lines could allow synthesis of structures that were previously challenging to make.
In addition to “DNA-walkers”, other DNA nanomachines have been designed, including DNA boxes, switches and joints. A free-floating DNA origami nanobox was created with its hinge programmed to open in response to certain keys.78 Various biologically active substrates could be contained within this nanocarrier, and the opening mechanism entailed an aptamer encoded logic gate which could be activated by various combinations of stimuli, such as antigens produced by cancer cells. The opening of the box was entropically driven (Fig. 8a). DNA switches have been designed that open and close depending on pH.79 These switches operate based on Hoogsteen base pairing (an alternative type to Watson–Crick) and can be tuned to respond to a wide pH range of 6.5 to 10.2, depending on the amount of TAT/CGC triplets present in the sequence. The switches demonstrated an extremely fast response time (<100 ms) and could potentially be incorporated into larger nanomachines to trigger response to external stimuli. Cancer-related gene sensing DNA nanomachines have also been designed.80 These machines operate using a two wheel drive based system. They were shown to be able to detect genes on single strand DNA and fluoresce in their presence. Joints could also form an integral part of nanomachines and have been constructed from DNA origami. Examples of slider and hinge joints have been reported,81 which demonstrate programmable motion when exposed to DNA strand inputs. The combination of these sliding and hinge joints leads to a wide range of motion (Fig. 8b).
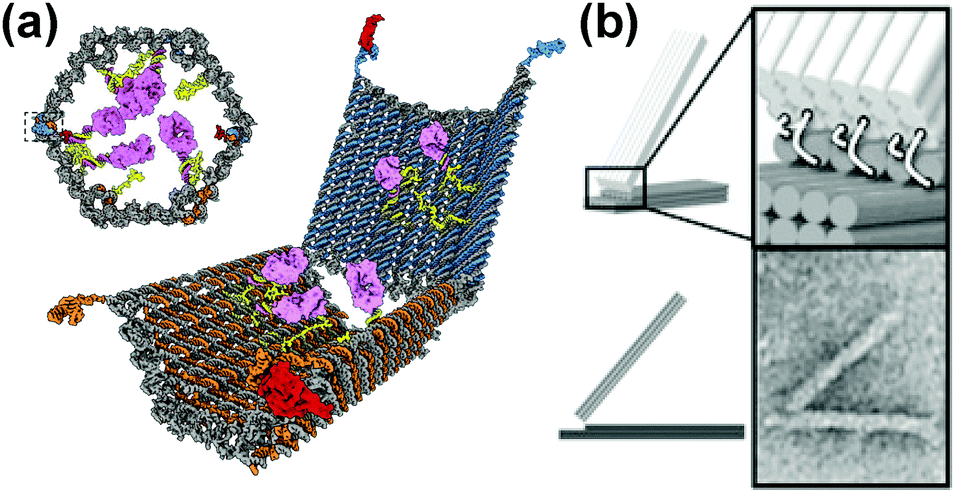 | ||
| Fig. 8 DNA nanomachines. (a) Logic gated nanorobot for targeted payload delivery.78 (b) Hinge joint schematics and TEM image (bottom right).81 Adapted by permission from ref. 78 and 81. Copyright 2012 AAAS and 2015 PNAS. | ||
In further interesting developments, DNA origami nanorobots have been engineered to perform molecular computation within living animals. The logic gated DNA nanoboxes mentioned previously78 were modified to interact with one another and tested inside living insects. Three different types of box were used, containing payloads which interacted with the other boxes. It was suggested that similar systems could be used for diagnostics or computational control of therapeutics.82 These nanorobots were modified further for use in the brain. Bioactive molecules were switched on and off in response to cognitive changes via electrically controlled heating of conjugated metal nanoparticles.83
3.3. Enzyme and enzyme-like nanomachines
Synthetic peptide chain self-assembly is leading to well controlled nanostructures which have potential applications as nanomachines.84–86 Natural peptide chains self-assemble to form enzymes, some of which have been recently shown to exhibit nanomotor behaviours.87 Enzymes are formed via the self-assembly of peptide chains. These chains are made up of a sequence of amino acids, the primary structure, which fold into structures such as alpha-helices and beta-sheets known as the secondary structures. These structures then self-assemble further into tertiary and quaternary structures and form binding sites for substrates, containing transition metals which help catalyse reactions. Due to the localised87 nature of the active sites on enzymes, the catalysis reaction is thought to result in a self-diffusiophoresis mechanism, similar to a Janus nanoparticle. In the presence of an appropriate substrate, enzymes in action have been shown to exhibit enhanced diffusion in their own right. Enzymes such as catalase have even been shown to exhibit chemotaxis. Therefore these free floating enzymes are now described as nanomotors. Understanding how enzymes accomplish active motion would be very valuable, yet the mechanism is still subject to debate. It is thought to involve heat dissipation from exothermic reactions, self-diffusiophoresis and conformational changes that enzymes undergo during catalysis.50,88,89 Other examples of use of enzymes as nanomachines include immobilised enzymes as microfluidic pumps.90The self-assembly of single chain polymer nanoparticles is similar to peptide chain folding in that a single chain of molecules is folded into a larger structure and could also lead to novel ways of making nanomachinery. The chains are folded in a controlled manner using techniques such as selective point folding, where complementary units are placed along the chain and come together to produce folds. These single polymer chains form protein-like structures which could potentially be used for similar applications.91
4. Hybrid inorganic nanomachines
Nanomachines based predominantly on inorganic materials have been reported. They are often made of metal, metal oxide or hybrid nanoparticles (NPs). Compared to nanomachines self-assembled from soft materials, inorganic nanomachines are denser, structurally rigid and less biocompatible. Nevertheless, owing to the presence of noble metal (e.g. Pt) and/or semiconductor (e.g. TiO2), they generally exhibit a stronger capability of extracting chemical energy from the surroundings through catalysis to perform mechanical work, mainly in the form of active propulsion in liquid media. These propelling nanomachines are typically known as nanomotors.49,51,92An additional advantage of inorganic NPs comes from their strong interactions with external stimuli, e.g. light, magnetic field and electric field, enabling precise external control and remote powering mechanisms. For instance, an external rotating magnetic field can be used to power and orient the motion of ferromagnetic chiral-shaped NPs in liquid media. The large scattering cross section of inorganic NPs also allows convenient observation of them using conventional optical techniques, e.g. dark-field optical microscopy for single particle tracking analysis and dynamic light scattering for ensemble measurement.
While the above properties make inorganic nanoparticles an outstanding candidate for nanomotors, challenges in designing, making, observing and controlling these active nanosystems remain, in particular for those smaller than 100 nm. Propulsion at the nanoscale requires exertion of a continuous drag force because inertia diminishes in the low-Reynolds-number regime as described by the Navier–Stokes equations. In this context, anisotropic nanostructures are generally required to generate and sustain directional drag force. However, the smaller the length scale, the more difficult it is to realise the anisotropic structure and the more dominant the randomising Brownian forces become (the translational diffusion constant of a Brownian particle scales with 1/r, the rotational diffusion constant with 1/r3, where r is the radius of the particle). For small nanomotors, e.g. d < 50 nm, the persistent length of motion is often very small as they suffer from significant rotational diffusion; hence, these small nanomotors operate practically in the enhanced diffusion regime rather than the ballistic regime.
The following subsections highlight some recent advances in inorganic nanomotors and hybrid nanomachines with feature size <100 nm. Two major types of nanomotors will be reviewed and discussed, namely Janus nanomotors and chiral nanomotors. Other hybrid structures exist, such as tubular nanojets,93 however these generally have larger feature sizes than 100 nm. The last subsection discusses non-propelling nanomachines that are either actively driven by an external field or thermally driven via biased stochastic processes.
4.1. Janus nanomotors
Janus nanomotors are based on the construct of Janus nanoparticles (JNPs), which are NPs with two chemically distinct faces, named after the two-face Roman God. They are arguably the simplest form of inorganic nanomotors and can be fabricated by various interface-based techniques. The anisotropic structure of JNPs allows the possibility of creating a chemical potential or thermal gradient across the JNP, which is typically achieved by chemical catalysis and laser light irradiation, respectively. The resultant potential gradient will then induce a phoretic flow of fluid along the JNP surface. The slipping of fluid will lead to the active motion of the Janus nanomotors in the opposite direction.Lee and Fischer et al. reported 30 nm nanomotors based on Pt–Au JNPs that exhibit enhanced diffusion behaviour under strong Brownian forces.94 The motility of the nanomotor is fuelled by the classical catalytic decomposition of H2O2 by Pt–Au JNP, which consists of a Pt NP embedded in one face of a Au NP. This represents the smallest artificial chemical nanomotor reported thus far with its size approaching that of enzymes. The nanomotor shows bursts of short linear ballistic motion, resulting in enhanced diffusion that saturates at ∼1.5% H2O2 concentration. Since the overall structure resembles that of the first self-electrophoretic micromotor,95 it was proposed that this Pt–Au JNP nanomotor also self-propels by the same mechanism. Nevertheless, molecular dynamics96 and theoretical studies97 suggest that other mechanisms, e.g. self-diffusiophoresis, can be simultaneously in action in this system.
He et al. reported Janus mesoporous silica nanomotors (JMSNMs) with size below 100 nm powered by self-diffusiophoresis and their use as nanocarriers for drug delivery.98 Mesoporous silica nanoparticles (MSNPs) of 65 nm are synthesised with pore size of 3 nm, a 7 nm Pt layer is deposited onto the MSNPs by sputter coating with a Cr adhesive layer to form JNPs (Fig. 9). JMSNMs are self-propelled by releasing oxygen gas from the catalytic decomposition of H2O2 triggered by Pt. Rhodamine–poly(allylamine hydrochloride) (Rhd-PAH) is used to label the JMSNMs for tracking movements under confocal laser scanning microscope (CLSM). The speed of the JMSNMs is significantly enhanced when the concentration of fuel is increased from zero (Brownian motion) to 30%. The mesoporous structure of nanomotors allows loading of cargo molecules and transport them to target areas. Doxorubicin hydrochloride (DOX), an anticancer drug, is loaded into the mesopores in vitro and functionalised with an egg phosphatidycholine (PC) bilayer. Cellular enzymes trigger the catalytic hydrolysis of lipid bilayers to release DOX encapsulated in the JMSNMs. Similarly, Sánchez et al. reported JMSNMs with tuneable size of 40, 65 and 90 nm and their application as active cargo delivery system. JMSNMs are propelled by self-diffusiophoresis in dilute H2O2 solution (<3 wt %).99 The diffusion coefficient is enhanced up to 100% when comparing to that of the Brownian motion. In order to examine the cargo loading capability of JMSNMs, MSNPs are labelled with fluorescein isothiocyanate (FITC) before forming JMSNMs. The cargo molecules, rhodamine B (RhB) and methylene blue (MB), are then loaded into the JMSNMs by diffusion into the mesopores. Cargo release of RhB and MB are observed under CLSM, illustrating the potential for JMSNMs to serve as active drug nanocarriers.
 | ||
| Fig. 9 Self-propelling Janus mesoporous silica nanomotors (JMSNMs) as active drug nanocarriers.98 (a) Sputter coating to form Janus structure, (b) nanomotors can be powered by H2O2 fuel, (c) drug loading and lipid bilayer functionalisation, (d) active drug delivery. Adapted by permission from ref. 98. Copyright 2014 Wiley-VCH. | ||
Subsequently, He et al. reported the fabrication of fuel-free JMSNMs with size of 50, 80 and 120 nm.100 The nanomotors are powered by self-thermophoresis using near-infrared light (NIR) in water without the use of toxic H2O2 fuel which makes them more compatible for biomedical applications. A 10 nm Au layer is deposited by vacuum sputtering on the MSNMs to form Janus nanomotors. A thermal gradient is generated across the JMSNMs when the Au layer is being illuminated by the NIR laser. JMSNMs are driven by self-thermophoresis at much higher speed than their previously reported work.98 By varying the NIR laser power, it is possible to modulate the speed of JMSNMs and switch them on/off on demand. Cargo transportation has also been demonstrated with FITC-loaded JMSNMs. Very recently, Li et al. reported the design of sub-20 nm Janus plasmonic nanomotors driven by catalytically induced self-thermophoresis.101 Au NPs are coated with bis(p-sulfonatophenyl)phenyl-phosphine (BSPP) to inhibit the catalytic activity, which are then absorbed onto APTES-coated silica beads via electrostatic interaction. BSPP ligands on the outer surface of Au NPs are replaced by thiolated oligonucleotide (T5) to form the catalytically active side. Janus nanomotors are formed by etching silica beads with HF. The redox reaction between Fe(CN)63− and S2O32− is catalysed by the T5 coated side which results in a thermal gradient across the Janus nanomotors. They showed diffusion coefficient of the nanomotors is linearly related to the rate of catalytic reaction.
4.2. Chiral nanomotors
Chiral nanoparticles, in particular ferromagnetic nanohelices,102,103 are promising candidates for nanomotors because they exhibit effective rotation–translation mechanical coupling which allows them to be remotely powered and oriented by an external rotating magnetic field under zero-field gradient conditions. Compared to chemical nanomotors, the advantages of fuel-free nanomotors are that they can be controlled remotely and precisely in terms of direction of motion and speed, i.e. they can stop/become Brownian on-demand. Nevertheless, controlling multiple nanomotors simultaneously remains a challenge.Helical nanostructures, especially those with a well-defined pitch and handedness, are difficult to fabricate or synthesise because crystallisation forces and minimisation of surface energy generally result in highly homogeneous and symmetric structures. In 2013, Lee and Fischer et al. reported a wafer-scale fabrication scheme for making nanohelices and other three-dimensional hybrid nanoparticles.104 This technique, later known as nano-GLAD, is based on conventional glancing angle deposition (GLAD) which is a physical vapour deposition technique with real time control over the angle of incidence and the type of material flux. Nano-GLAD can typically produce 1012 identical nanohelices of pre-programmed handedness and pitch in a single one-hour growth cycle. The nanostructures can then be easily detached from the wafer surface to produce a nanocolloidal solution with a concentration (1010–1011 NPs per mL) comparable to that of commercial Au NP samples. By coupling to atomic layer deposition, nano-GLAD can produce corrosion-protected nanohelices, allowing the use of a wider range of magnetic materials (e.g. cobalt) in the construct, and enhancing the stability of these nanomotors in more hostile environment.105
Fischer et al. reported the fabrication of magnetically actuated helical nanopropellers with a filament diameter of ∼70 nm, which are small enough for potential applications in both extra and intracellular environments (Fig. 10a).106 400 nm long silica helices containing a magnetic Ni-section are grown on a Au nano-dot pattern with spacing of ∼135 nm. The nanohelices show significantly enhanced propulsion velocities than larger micropropellers in viscoelastic hyaluronan (HA) gels, which is a polymeric gel present in biological tissues, because they are of the same size range as the gel's mesh size. Controllable navigation of nanopropellers with higher speeds than in Newtonian fluid has also been achieved.
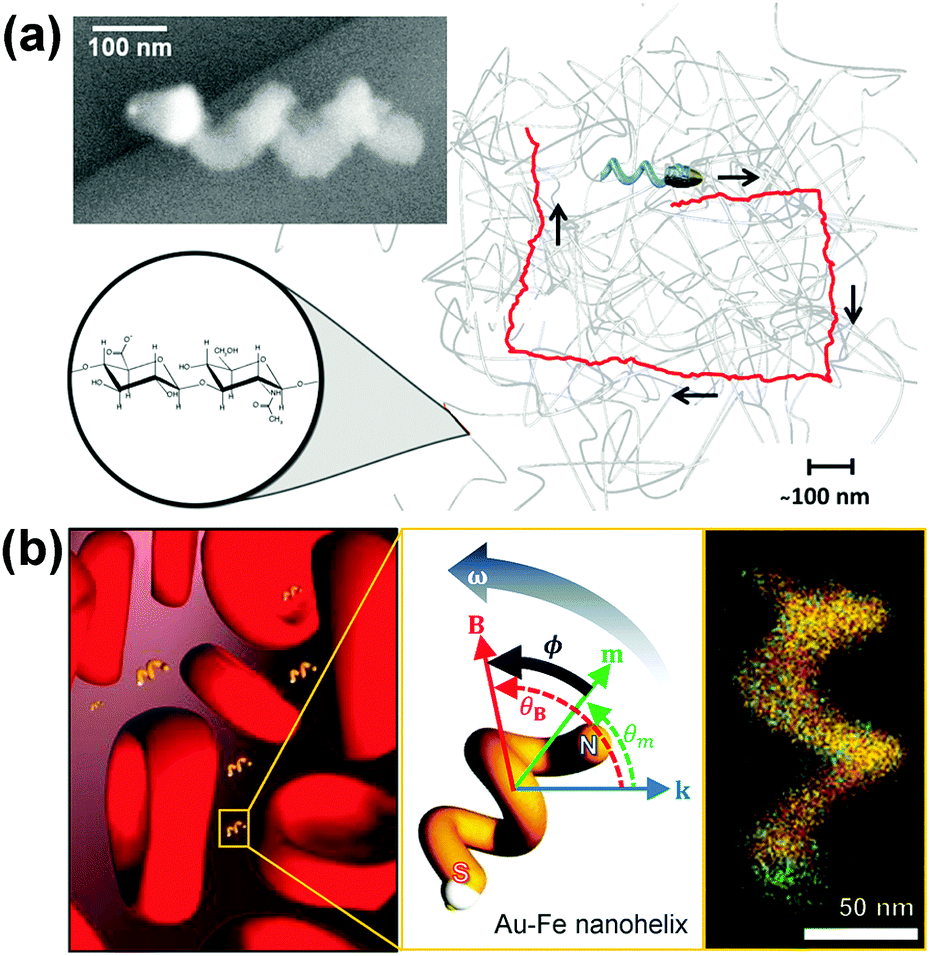 | ||
| Fig. 10 (a) Chiral magnetic nanopropeller that is sufficiently small to swim through the mesh of viscoelastic hyaluronan gels. Inset: SEM image of a nanopropeller.106 (b) Magneto-plasmonic Au–Fe nanohelices for direct active nanorheology in full blood samples.111 Adapted by permission from ref. 106 and 111. Copyright 2014, 2016 American Chemical Society. | ||
4.3. Hybrid nanomachines
Hybrid nanomachines consist of more than one type (molecular/self-assembled/inorganic) of functional moieties that work together to perform a specific task. Representative examples include the above-mentioned stomatocytes loaded with Pt NPs62 or enzymes64 (Section 3.1) and nano-assembler of Au NPs based on DNA origami (Section 3.2).77 Hybrid nanomachines are envisioned to emerge as the next-generation nanomachines with cross-scale and smart functionality. This subsection will highlight some additional examples of promising candidates.By using enzymes to replace inorganic catalysts, Sánchez et al. have reported Janus hollow mesoporous silica nanomotors powered by biocatalysis of three different enzymes, including catalase, urease and glucose oxidase in H2O2, urea and glucose, respectively.107 SiO2 NPs are modified to form hollow mesoporous silica NPs with amine groups. JNPs are formed by deposition of a 10 nm SiO2 layer on the hollow mesoporous silica NPs, with the enzymes being conjugated onto the other side via a glutaraldehyde linker molecule. The translational diffusion coefficient of the nanomotors can be enhanced up to 83% at 1.5 wt% H2O2. They also measured the effective driving force of the nanomotors using optical tweezers. As urease and glucose oxidase based nanomotors are driven in non-toxic fuels, they are fully biocompatible and promising for biomedical applications such as drug delivery. Other enzyme-powered micro/nanomachines have been discussed in another review article.108
The combination of plasmonic and magnetic materials within the same nanoparticles can enable magnetic modulation of plasmonic signals using a weak magnetic field. For instance, Wei et al. reported gyromagnetic imaging based on 100 nm Au nanostars with a 13 nm superparamagnetic (Fe3O4) core and NIR-active arms.109 The Au nanostars are adsorbed onto coated glass substrates and imaged under polarised dark-field microscope. The plasmonic properties of Au nanostars upon NIR excitation generate optical contrast from their rotational motion in the presence of a rotating magnetic field. A periodic scattering response is produced and converted into Fourier-domain images with lower background signals. Moreover, the gyration of these gold nanostars can be gated neurochemically upon functionalisation with polymer with pendant viologen groups and cucurbiturils.110
Using a similar principle, Lee and Fischer et al. have reported nanorheology probes based on magneto-plasmonic Au–Fe nanohelices (Fig. 10b).111 The active rheology probe can be actuated using a weak magnetic field and operates using a novel optical sensing scheme based on circular dichroism (CD).112 Owing to the intense chiroptical properties of the nanohelices, this sensing scheme is practically background-free and allows optical signal identification in optically dense media (OD as high as 3). By analysing the time- and angle-dependent CD signal of the actively actuating nanohelices, direct viscosity measurement in full blood (50% v/v red blood cells) samples has been achieved. Interestingly, the measured dynamic viscosity is insensitive to the concentration of red blood cells and matches that of blood plasma because the nanohelices are almost two orders of magnitude smaller than the red blood cells that are responsible for the shear thinning properties of blood.
In addition to actively powered nanomachines, hybrid nanosystems also offer facile routes to mediate cross-scale interactions (e.g. between small molecules and plasmonic NPs) through the formation of hierarchical nanostructures. For instance, Lee and Scherman utilised cucurbiturils (CBs), a family of rigid macrocyclic molecules, to guide the formation of precise plasmonic nanojunctions through an aqueous self-assembly approach (Fig. 1 top row, middle panel).113,114 The well-defined spacing between plasmonic NPs gives rise to reproducible and strong localised surface plasmon resonance (LSPR) signals, as well as surface-enhanced Raman scattering (SERS) activity.115 Notably, CB can position small molecules right at the centre of plasmonic hotspots via host–guest complexation, enabling ultrasensitive and selective detection of a range of analytes,116 including neurotransmitters.117 Meanwhile, Klajn et al. reported the synthesis and dynamic assembly of nano-flasks using azobenzene-functionalised inorganic NPs (Fig. 1 top row, left panel).118 The nano-flasks can be reversibly self-assembled and disassembled upon UV and visible light irradiation, allowing the encapsulated molecules within the nano-flasks to be trapped and released dynamically. The selectivity of molecules encapsulated in the nano-flasks can be optimised by functionalising the NPs with ligands of different functional groups and multi-wall layers. They also showed that the encapsulated molecules can undergo various chemical reactions and can be potentially used as nanoreactors.
Nanoscale encapsulation is mostly limited to molecular systems (e.g. macrocyclic molecules, micelles and vesicles) while nanocolloidal analogues are rare mainly because it remains a challenge to make NPs with concave surfaces, let alone a deep cavity. Driven by scientific curiosity, Lee and Fischer et al. reported a scheme for making 1:1 host–guest complexes of inorganic NPs (Fig. 1 top row, right panel).119 In particular, a Au NP can be specifically encapsulated deep inside the cavity of a SiO2 nanocup with high yield (>70%) and regiospecificity (>90%) via a novel intermediate of reactive double Janus nanoparticle. These complexes are formed by a kinetically controlled mechanism involving a delicate interplay between bipolar galvanic corrosion and alloying–dealloying oxidation. Release of the NP guest from the nanocups can be efficiently triggered by an external stimulus.
5. Discussion and outlook
This Feature Article highlighted and discussed a range of artificial molecular and nanostructures that show promise for making next-generation nanomachinery – artificial nanosystems capable of performing useful function, very often, by means of doing mechanical work at the nanoscale. The artificial nanoscale structures are classified into three major groups, namely molecular machines, self-assembled nanomachines and hybrid inorganic nanomachines, yet the actual boundary between these groups can sometimes be vague and subject to a specific context (Fig. 1). Many of these structures are made by bottom-up synthesis and self-assembly approaches, which therefore show higher compatibility to scale-up production and solution-based applications.It is interesting, and perhaps useful, to note that despite the breadth in length scale and materials, common structural features that are critical to the functioning of nanomachines can be identified. Many of these design rules and operating principles reflect those found in living systems, which consist of molecular and nanomotors from billions of years of evolution in nature.
(a) The existence of kinetically stable far-from-equilibrium states is crucial to nanosystems that extract mechanical work from other forms of energy, e.g. light and chemical. In the case of molecular rotors (Section 2),14 the less thermodynamically stable cis-isomer generated from photo-isomerisation should be kinetically stable to allow thermally driven helix inversion to occur in the next step. Otherwise, reverse photo-isomerisation or photo-degradation of the molecule may result. The same rule applies to soft (Section 3.1) and inorganic and hybrid (Section 4) nanomotors. Here, a chemical potential gradient or a thermal gradient has to be maintained across the nanomotor, which is typically achieved by chemical catalysis and plasmonic heating, respectively. Throughout the entire process, the structure of the nanomotor should be kinetically stable and remain intact. In a nutshell, molecular and nanomotors operate in far-from-equilibrium states120 which are maintained by constant dissipation of energy. In this process, work (very often mechanical) can be extracted to perform useful tasks. In order to be functional, the motors are required to remain intact without degradation.
(b) Compartmentalised and hybrid structures are often employed to enhance the kinetic stability of the nanosystems meanwhile allowing selective or on-demand exchange of molecular species across different parts of the nanomachine or with the surroundings. At the molecular level, the formation of mechanically interlocked supramolecular complexes significantly increases the kinetic stability of the system. This can be illustrated by Stoddart's molecular shuttle13 (Section 2.1), which is made of a rotaxane structure. The molecular wheel is threaded on an axle with both ends capped by a stopper, so that the wheel can slide freely along the axle without falling out. If the capping group at the end of the axle is removed, however, it becomes physically possible for the wheel to slide off the axle, and this structure is known as a pseudorotaxane. At the nanoscale, compartmentalised structures are also useful in mediating on-demand release of cargo, illustrated by smart nanocarriers based on DNA origami78 (Section 3.2), and in acting as a barrier for sustaining a concentration gradient across the soft nanomotors61,64 (Section 3.1).
(c) Asymmetry or anisotropy in molecular and nanostructures are commonly utilised to act as asymmetric kinetic barriers and to generate gradients of chemical potential for directed motion of molecules or nanostructures, respectively. For instance, the direction and type of motion of the electrically driven four-wheeled nanocar25 (Section 2.2) can be delicately controlled by the rotation direction of the four rotor wheels, which is determined by the chirality of the individual molecular rotor. Meanwhile, structural anisotropy is also a critical requirement for active propulsion at the nanoscale. For self-propelling nanomotors (Sections 3.1 and 4.1), breaking the plane of symmetry by formation of Janus-like structures is typically essential to the generation of a concentration/thermal gradient that powers the propulsion. For propulsion by mechanical rotation, as in the case of nanopropellers (Section 4.2), chiral structures, in particular helical structures,106 are required to enable the break of time-reversal symmetry and the strong coupling between rotational and translational motion.
(d) Information-rich and programmable constructs are often found in sophisticated molecular and nanomachines. When designing and constructing artificial nanomachines, one typical approach is to specify the function of each individual component and then build precisely what has been designed. Although it may seem easy for molecular machines, which have atomistically precise molecular structure, the actual synthesis could be challenging because of the required stereochemistry. For self-assembled systems, DNA origami and related approaches (Section 3.2) are outstanding platforms for making nanomachines because of their programmable and dynamic constructs. Indeed DNA origami nanomachines have been shown to assemble NPs in a stepwise and programmable manner,77 as well as to perform molecular computation in living insects.82 Regarding inorganic nanomachines, sophisticated programmable nanostructures are less available because neither top-down nor bottom-up techniques offer atomistic precision and the surface of inorganic NPs often remains uncharacterised. Yet, the recent development of bottom-up fabrication techniques, e.g. nano-GLAD,104 offer promising avenue towards programmable hybrid nanostructures.
(e) Dynamic structures with well-defined movable components are commonly exploited to mediate the stimuli-responsiveness and the information processing in many molecular and nanomachines. This type of structures is generally designed by engineering the reversible supramolecular interactions between different components within a nanomachine. Mechanically interlocked rotaxanes used in molecular shuttles13 represent a classical example where the position of the shuttle can be controlled by the switchable binding constant of different stations on the axle. Other examples include the use of a photoswitchable double bond as a molecular switch. At the nanoscale, in addition to more localised supramolecular interactions (e.g. hydrogen bonding, halogen bonding, ion–dipole, metal coordination and π–π stacking), wider diversity of supramolecular forces that rely on ensemble effects comes into play, including hydrophobic forces, entropic coiling of polymers and depletion interactions. The advantage of supramolecular interactions is that they are strong enough to hold a particular structure together, yet their dynamic nature allows them to be “switched off” upon a specific stimulus. For instance, Klajn et al. recently reported the dynamic trapping and release of molecules can be achieved using azobenzene-functionalised inorganic NPs that self-assemble or dissociate in response to light (Section 4.3).118
The next-generation nanomachines are likely to consist of multiple functional components, taking advantage of the physiochemical properties across a range of length scales and material systems, for instance the atomistic precision of molecular components, the dynamic nature of self-assemble nanostructures and the outstanding photonic properties of inorganic NPs. Different components will be synthesised or fabricated predominantly by bottom-up techniques and brought together by hierarchical self-assembly approaches. Research motivation will gradually shift from curiosity-driven to application-driven. Given the interdisciplinary nature, future development of the nanomachines calls for close collaboration between experts of a wide range of disciplines, including synthetic and supramolecular chemistry, system chemistry, biophysics, molecular modelling and nanoscience in general.
The investigation of complex dynamics and functioning mechanisms of nanomachines will motivate new development of in situ characterisation techniques with improved sensitivity and spatiotemporal resolution. Current measurement techniques based on optical scattering, such as dynamic light scattering and dark-field optical microscopy, generally suffer from poor signal-to-noise ratio for small NPs which appear to be “dimmer” (scattering intensity scales with r6) and at the same time moving faster (translational diffusion scales with 1/r). Microscopy based on non-linear optics, e.g. pump–probe four-wave mixing imaging,121 offers a promising avenue towards in situ 3-D tracking of single NPs with impressive spatiotemporal resolution. Meanwhile, liquid-cell transmission electron microscopy (LC TEM),122 which allows real-time imaging of NPs in liquid media, emerges as a powerful technique for studying dynamics of nanosystems. When coupled to an aberration-corrected setup and an electron-counting direct detection camera, LC TEM can potentially achieve atomic resolution with ∼ms time resolution. It is also noted that next generation characterisation techniques will likely to take advantage of advanced computer algorithms and modelling for real-time data interpretation and visualisation.
Another potential future direction of nanomachine research could be about the interaction and cooperation of molecular and nanomachines, similar but not limited to metabolic channelling123 between multiple enzymes or formation of microbial colonies in biological systems. For instance, Gibbs et al. recently reported strategies for engineering long-range magnetic interaction between artificial micromotors,124 whereas Rurack et al. have demonstrated hierarchical chemical communication between groups of nanoparticles.125 These and similar approaches can be extended to designing molecular and nanomachines that can “talk” and “collaborate” with each other. This type of systems can consist of one or more kinds of machines, each with a specific role assigned. The interaction can be mediated by chemical signals or other physical cues, e.g. magnetic field and local temperature gradient. At the system scale, interacting nanomachines will give rise to novel emergent properties, which potentially enable intelligent and self-evolving nanosystems, as well as offer new prospects in drug discovery, catalysis and understanding the origin of life.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
SM and TCL are grateful to the Research Project Grant (RPG-2016-393) funded by the Leverhulme Trust. EE, WIKC and TCL are grateful to the Studentship funded by the A*STAR-UCL Research Attachment Programme through the EPSRC Centre for Doctoral Training in Molecular Modelling and Materials Science (EP/L015862/1).Notes and references
- R. P. Feynman, Eng. Sci., 1960, 23(5), 22–36 Search PubMed.
- L. Soler and S. Sanchez, Nanoscale, 2014, 6, 7175–7182 RSC.
- W. Gao and J. Wang, ACS Nano, 2014, 8, 3170–3180 CrossRef CAS PubMed.
- M. N. Stojanovic, D. Stefanovic and S. Rudchenko, Acc. Chem. Res., 2014, 47, 1845–1852 CrossRef CAS PubMed.
- V. Richards, Nat. Chem., 2016, 8, 1090 CrossRef CAS PubMed.
- C. Cheng and J. F. Stoddart, ChemPhysChem, 2016, 17, 1780–1793 CrossRef CAS PubMed.
- Y.-J. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS PubMed.
- F. Zhang, J. Nangreave, Y. Liu and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198–11211 CrossRef CAS PubMed.
- C. Angell, S. Xie, L. Zhang and Y. Chen, Small, 2016, 12, 1117–1132 CrossRef CAS PubMed.
- V. Balzani, A. Credi, B. Ferrer, S. Silvi and M. Venturi, in Molecular Machines, ed. T. R. Kelly, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 1–27 Search PubMed.
- D. A. Leigh, Angew. Chem., Int. Ed., 2016, 55, 14506–14508 CrossRef CAS PubMed.
- C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS.
- P. L. Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5133 CrossRef CAS PubMed.
- B. L. Feringa, Acc. Chem. Res., 2001, 34, 504–513 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- P. Thordarson, E. J. Bijsterveld, A. E. Rowan and R. J. Nolte, Nature, 2003, 424, 915–918 CrossRef CAS PubMed.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, J. Org. Chem., 1983, 48, 3619–3620 CrossRef CAS.
- D. Tuncel, H. B. Tiftik and B. Salih, J. Mater. Chem., 2006, 16, 3291–3296 RSC.
- T.-C. Lee, E. Kalenius, A. I. Lazar, K. I. Assaf, N. Kuhnert, C. H. Grün, J. Jänis, O. A. Scherman and W. M. Nau, Nat. Chem., 2013, 5, 376 CrossRef CAS PubMed.
- S. Kassem, A. T. L. Lee, D. A. Leigh, V. Marcos, L. I. Palmer and S. Pisano, Nature, 2017, 549, 374–378 CrossRef CAS PubMed.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D’Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- R. Klajn, Pure Appl. Chem., 2010, 82, 2247 CrossRef.
- N. Katsonis, M. Lubomska, M. M. Pollard, B. L. Feringa and P. Rudolf, Prog. Surf. Sci., 2007, 82, 407–434 CrossRef CAS.
- R. A. van Delden, M. K. J. ter Wiel, M. M. Pollard, J. Vicario, N. Koumura and B. L. Feringa, Nature, 2005, 437, 1337–1340 CrossRef CAS PubMed.
- T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Macia, N. Katsonis, S. R. Harutyunyan, K.-H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed.
- K. Ariga, Y. Terasaka, D. Sakai, H. Tsuji and J.-i. Kikuchi, J. Am. Chem. Soc., 2000, 122, 7835–7836 CrossRef CAS.
- X. Huang, C. Li, S. Jiang, X. Wang, B. Zhang and M. Liu, J. Am. Chem. Soc., 2004, 126, 1322–1323 CrossRef CAS PubMed.
- T. D. Nguyen, H.-R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. F. Stoddart and J. I. Zink, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029–10034 CrossRef CAS PubMed.
- T. D. Nguyen, Y. Liu, S. Saha, K. C. F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS PubMed.
- S. Angelos, Y.-W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344–11346 CrossRef CAS PubMed.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC.
- M. Yamada, M. Kondo, J.-i. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed.
- M. Yamada, M. Kondo, R. Miyasato, Y. Naka, J.-i. Mamiya, M. Kinoshita, A. Shishido, Y. Yu, C. J. Barrett and T. Ikeda, J. Mater. Chem., 2009, 19, 60–62 RSC.
- S. Palagi, A. G. Mark, S. Y. Reigh, K. Melde, T. Qiu, H. Zeng, C. Parmeggiani, D. Martella, A. Sanchez-Castillo, N. Kapernaum, F. Giesselmann, D. S. Wiersma, E. Lauga and P. Fischer, Nat. Mater., 2016, 15, 647–653 CrossRef CAS PubMed.
- R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325–1349 CrossRef CAS.
- N. Stephanopoulos, J. H. Ortony and S. I. Stupp, Acta Mater., 2013, 61, 912–930 CrossRef CAS PubMed.
- B. K. Anatoly, J. Phys.: Condens. Matter, 2013, 25, 463101 CrossRef PubMed.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- O. Garbuzenko, J. Winkler, M. Tomassone and T. Minko, Langmuir, 2014, 30, 12941–12949 CrossRef CAS PubMed.
- D. Jiao, J. Geng, X. J. Loh, D. Das, T.-C. Lee and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 9633–9637 CrossRef CAS PubMed.
- X. J. Loh, M.-H. Tsai, J. d. Barrio, E. A. Appel, T.-C. Lee and O. A. Scherman, Polym. Chem., 2012, 3, 3180–3188 RSC.
- X. J. Loh, J. del Barrio, T.-C. Lee and O. A. Scherman, Chem. Commun., 2014, 50, 3033–3035 RSC.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- X. J. Loh, J. del Barrio, P. P. C. Toh, T.-C. Lee, D. Jiao, U. Rauwald, E. A. Appel and O. A. Scherman, Biomacromolecules, 2012, 13, 84–91 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- E. Ellis, K. Zhang, Q. Lin, E. Ye, A. Poma, G. Battaglia, X. J. Loh and T.-C. Lee, J. Mater. Chem. B, 2017, 5, 4421–4425 RSC.
- W. Gao and J. Wang, Nanoscale, 2014, 6, 10486–10494 RSC.
- F. Wong, K. K. Dey and A. Sen, Annu. Rev. Mater. Res., 2016, 46, 407–432 CrossRef CAS.
- S. Sanchez, L. Soler and J. Katuri, Angew. Chem., Int. Ed., 2015, 54, 1414–1444 CrossRef CAS PubMed.
- S. Forster and M. Konrad, J. Mater. Chem., 2003, 13, 2671–2688 RSC.
- R. Deng, F. Liang, J. Zhu and Z. Yang, Mater. Chem. Front., 2017, 1, 431–443 RSC.
- X. Li, H. Yang, L. Xu, X. Fu, H. Guo and X. Zhang, Macromol. Chem. Phys., 2010, 211, 297–302 CrossRef CAS.
- I. Voets, R. Fokkink, T. Hellweg, S. King, P. de Waard, A. de Keizer and M. Stuart, Soft Matter, 2009, 5, 999–1005 RSC.
- J. Dupont and G. Liu, Soft Matter, 2010, 6, 3654–3661 RSC.
- J. Marsat, M. Heydenreich, E. Kleinpeter, H. Berlepsch, C. Bottcher and A. Laschewsky, Macromolecules, 2011, 44, 2092–2105 CrossRef CAS.
- A. Groschel, A. Walther, T. Lobling, J. Schmelz, A. Hanisch, H. Schmalz and A. Muller, J. Am. Chem. Soc., 2012, 134, 13850–13860 CrossRef PubMed.
- A. Groschel, F. Schacher, H. Schmalz, O. Borisov, E. Zhulina, A. Walther and A. Muller, Nat. Commun., 2012, 3, 710 CrossRef PubMed.
- B. Pham, C. Such and B. Hawkett, Polym. Chem., 2015, 6, 426–435 RSC.
- A. Joseph, C. Contini, D. Cecchin, S. Nyberg, L. Ruiz-Perez, J. Gaitzsch, G. Fullstone, X. Tian, J. Azizi, J. Preston, G. Volpe and G. Battaglia, Sci. Adv., 2017, 3, e1700362 CrossRef PubMed.
- D. A. Wilson, R. J. M. Nolte and J. C. M. van Hest, Nat. Chem., 2012, 4, 268 CrossRef CAS PubMed.
- Y. Tu, F. Peng, A. A. M. André, Y. Men, M. Srinivas and D. A. Wilson, ACS Nano, 2017, 11, 1957–1963 CrossRef CAS PubMed.
- F. Peng, Y. Tu, J. C. M. van Hest and D. A. Wilson, Angew. Chem., 2015, 127, 11828–11831 CrossRef.
- L. K. E. A. Abdelmohsen, M. Nijemeisland, G. M. Pawar, G.-J. A. Janssen, R. J. M. Nolte, J. C. M. van Hest and D. A. Wilson, ACS Nano, 2016, 10, 2652–2660 CrossRef CAS PubMed.
- F. Peng, Y. Tu, A. Adhikari, J. C. J. Hintzen, D. W. P. M. Lowik and D. A. Wilson, Chem. Commun., 2017, 53, 1088–1091 RSC.
- N. Lapique and Y. Benenson, Nat. Nanotechnol., 2017 DOI:10.1038/s41565-017-0004-z.
- J. Pan, F. Li, T.-G. Cha, H. Chen and J. H. Choi, Curr. Opin. Biotechnol., 2015, 34, 56–64 CrossRef CAS PubMed.
- T. Omabegho, R. Sha and N. C. Seeman, Science, 2009, 324, 67 CrossRef CAS PubMed.
- R. A. Muscat, J. Bath and A. J. Turberfield, Nano Lett., 2011, 11, 982–987 CrossRef CAS PubMed.
- T. Torring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646 RSC.
- A. H. Okholm and J. Kjems, Adv. Drug Delivery Rev., 2016, 106, 183–191 CrossRef CAS PubMed.
- B. Wei, M. Dai and P. Yin, Nature, 2012, 485, 623 CrossRef CAS PubMed.
- C. E. Castro, F. Kilchherr, D.-N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297 CrossRef CAS PubMed.
- K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, S. Taylor, R. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206–210 CrossRef CAS PubMed.
- H. Gu, J. Chao, S.-J. Xiao and N. C. Seeman, Nature, 2010, 465, 202–205 CrossRef CAS PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed.
- A. Idili, A. Vallée-Bélisle and F. Ricci, J. Am. Chem. Soc., 2014, 136, 5836–5839 CrossRef CAS PubMed.
- J. Xu, Z.-S. Wu, Z. Wang, H. Li, J. Le and L. Jia, Biomaterials, 2016, 100, 110–117 CrossRef CAS PubMed.
- A. E. Marras, L. Zhou, H.-J. Su and C. E. Castro, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 713–718 CrossRef CAS PubMed.
- Y. Amir, E. Ben-Ishay, D. Levner, S. Ittah, A. Abu-Horowitz and I. Bachelet, Nat. Nanotechnol., 2014, 9, 353–357 CrossRef CAS PubMed.
- S. Arnon, N. Dahan, A. Koren, O. Radiano, M. Ronen, T. Yannay, J. Giron, L. Ben-Ami, Y. Amir, Y. Hel-Or, D. Friedman and I. Bachelet, PLoS One, 2016, 11, e0161227 Search PubMed.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- D. Mandal, A. Nasrolahi Shirazi and K. Parang, Org. Biomol. Chem., 2014, 12, 3544–3561 CAS.
- A. Ljubetic, F. Lapenta, H. Gradisar, I. Drobnak, J. Aupic, Z. Strmsek, D. Lainscek, I. Hafner-Bratkovic, A. Majerle, N. Krivec, M. Bencina, T. Pisanski, T. C. Velickovic, A. Round, J. M. Carazo, R. Melero and R. Jerala, Nat. Biotechnol., 2017, 35, 1094–1101 CAS.
- S. Sengupta, K. K. Dey, H. S. Muddana, T. Tabouillot, M. E. Ibele, P. J. Butler and A. Sen, J. Am. Chem. Soc., 2013, 135, 1406–1414 CrossRef CAS PubMed.
- C. Riedel, R. Gabizon, C. A. M. Wilson, K. Hamadani, K. Tsekouras, S. Marqusee, S. Presse and C. Bustamante, Nature, 2015, 517, 227–230 CrossRef CAS PubMed.
- R. Golestanian, Phys. Rev. Lett., 2015, 115, 108102 CrossRef PubMed.
- S. Sengupta, D. Patra, I. Ortiz-Rivera, A. Agrawal, S. Shklyaev, K. K. Dey, U. Córdova-Figueroa, T. E. Mallouk and A. Sen, Nat. Chem., 2014, 6, 415 CrossRef CAS PubMed.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2016, 37, 29–46 CrossRef CAS PubMed.
- H. Wang and M. Pumera, Chem. Rev., 2015, 115, 8704–8735 CrossRef CAS PubMed.
- X. Ma, A. C. Hortelao, A. Miguel-López and S. Sánchez, J. Am. Chem. Soc., 2016, 138, 13782–13785 CrossRef CAS PubMed.
- T.-C. Lee, M. Alarcon-Correa, C. Miksch, K. Hahn, J. G. Gibbs and P. Fischer, Nano Lett., 2014, 14, 2407–2412 CrossRef CAS PubMed.
- W. F. Paxton, K. C. Kistler, C. C. Olmeda, A. Sen, S. K. Angelo St., Y. Cao, T. E. Mallouk, P. E. Lammert and V. H. Crespi, J. Am. Chem. Soc., 2004, 126, 13424–13431 CrossRef CAS PubMed.
- P. H. Colberg, S. Y. Reigh, B. Robertson and R. Kapral, Acc. Chem. Res., 2014, 47, 3504–3511 CrossRef CAS PubMed.
- P. Kreissl, C. Holm and J. de Graaf, J. Chem. Phys., 2016, 144, 204902 CrossRef PubMed.
- M. Xuan, J. Shao, X. Lin, L. Dai and Q. He, ChemPhysChem, 2014, 15, 2255–2260 CrossRef CAS PubMed.
- X. Ma, K. Hahn and S. Sanchez, J. Am. Chem. Soc., 2015, 137, 4976–4979 CrossRef CAS PubMed.
- M. Xuan, Z. Wu, J. Shao, L. Dai, T. Si and Q. He, J. Am. Chem. Soc., 2016, 138, 6492–6497 CrossRef CAS PubMed.
- W. Qin, T. Peng, Y. Gao, F. Wang, X. Hu, K. Wang, J. Shi, D. Li, J. Ren and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 515–518 CrossRef CAS PubMed.
- J. G. Gibbs, A. G. Mark, T.-C. Lee, S. Eslami, D. Schamel and P. Fischer, Nanoscale, 2014, 6, 9457–9466 RSC.
- S. Eslami, J. G. Gibbs, Y. Rechkemmer, J. van Slageren, M. Alarcón-Correa, T.-C. Lee, A. G. Mark, G. L. J. A. Rikken and P. Fischer, ACS Photonics, 2014, 1, 1231–1236 CrossRef CAS.
- A. G. Mark, J. G. Gibbs, T.-C. Lee and P. Fischer, Nat. Mater., 2013, 12, 802 CrossRef CAS PubMed.
- H.-H. Jeong, M. Alarcón-Correa, A. G. Mark, K. Son, T.-C. Lee and P. Fischer, Adv. Sci., 2017, 4, 1700234 CrossRef PubMed.
- D. Schamel, A. G. Mark, J. G. Gibbs, C. Miksch, K. I. Morozov, A. M. Leshansky and P. Fischer, ACS Nano, 2014, 8, 8794–8801 CrossRef CAS PubMed.
- X. Ma, A. Jannasch, U.-R. Albrecht, K. Hahn, A. Miguel-López, E. Schäffer and S. Sánchez, Nano Lett., 2015, 15, 7043–7050 CrossRef PubMed.
- X. Ma, A. C. Hortelão, T. Patiño and S. Sánchez, ACS Nano, 2016, 10, 9111–9122 CrossRef CAS PubMed.
- Q. Wei, H.-M. Song, A. P. Leonov, J. A. Hale, D. Oh, Q. K. Ong, K. Ritchie and A. Wei, J. Am. Chem. Soc., 2009, 131, 9728–9734 CrossRef CAS PubMed.
- Q. Wei, T. C. Lee, E. Appel, H.-m. Song, O. A. Scherman and A. Wei, Presented in part at 246th ACS National Meeting and Exposition, Indianapolis, September, 2013.
- H.-H. Jeong, A. G. Mark, T.-C. Lee, M. Alarcón-Correa, S. Eslami, T. Qiu, J. G. Gibbs and P. Fischer, Nano Lett., 2016, 16, 4887–4894 CrossRef CAS PubMed.
- H.-H. Jeong, A. G. Mark, M. Alarcón-Correa, I. Kim, P. Oswald, T.-C. Lee and P. Fischer, Nat. Commun., 2016, 7, 11331 CrossRef CAS PubMed.
- T.-C. Lee and O. A. Scherman, Chem. Commun., 2010, 46, 2438–2440 RSC.
- T.-C. Lee and O. A. Scherman, Chem. – Eur. J., 2012, 18, 1628–1633 CrossRef CAS PubMed.
- S. Mahajan, T.-C. Lee, F. Biedermann, J. T. Hugall, J. J. Baumberg and O. A. Scherman, Phys. Chem. Chem. Phys., 2010, 12, 10429–10433 RSC.
- R. W. Taylor, T.-C. Lee, O. A. Scherman, R. Esteban, J. Aizpurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878–3887 CrossRef CAS PubMed.
- S. Kasera, L. O. Herrmann, J. d. Barrio, J. J. Baumberg and O. A. Scherman, Sci. Rep., 2014, 4, 6785 CrossRef CAS PubMed.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J.-W. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2015, 11, 82 CrossRef PubMed.
- M. Alarcón-Correa, T.-C. Lee and P. Fischer, Angew. Chem., Int. Ed., 2015, 54, 6730–6734 CrossRef PubMed.
- J. H. van Esch, R. Klajn and S. Otto, Chem. Soc. Rev., 2017, 46, 5474–5475 RSC.
- V. Kravtsov, R. Ulbricht, J. M. Atkin and M. B. Raschke, Nat. Nanotechnol., 2016, 11, 459 CrossRef CAS PubMed.
- N. de Jonge and F. M. Ross, Nat. Nanotechnol., 2011, 6, 695 CrossRef CAS PubMed.
- S. Schoffelen and J. C. M. van Hest, Soft Matter, 2012, 8, 1736–1746 RSC.
- A. Nourhani, D. Brown, N. Pletzer and J. G. Gibbs, Adv. Mater., 2017, 29, 1703910 CrossRef PubMed.
- C. Giménez, E. Climent, E. Aznar, R. Martínez-Máñez, F. Sancenón, M. D. Marcos, P. Amorós and K. Rurack, Angew. Chem., Int. Ed., 2014, 53, 12629–12633 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |