
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water transport through graphene oxide membranes: the roles of driving forces†
J. Y.
Chong
,
B.
Wang
and
K.
Li
 *
*
Barrer Centre, Department of Chemical Engineering, Imperial College London, London SW7 2AZ, UK. E-mail: kang.li@imperial.ac.uk
First published on 21st February 2018
Abstract
Graphene oxide (GO) membranes have shown excellent selectivities in nanofiltration and pervaporation. However, the water transport mechanisms in the unique membrane laminar structure are still not well understood, especially in pervaporation which involves selective permeation and evaporation. Herein, water transport in GO membranes was tested under two different modes: pressure-driven permeation and pervaporation. The pure water flux was found to be 1–2 orders of magnitude higher in pervaporation due to the large capillary pressure induced by evaporation. The water flux in pervaporation was suggested to be limited by evaporation at room temperature but surface diffusion at high temperature.
Graphene oxide (GO) membranes can be fabricated by assembling two-dimensional GO flakes layer-by-layer on top of each other. The membranes form a uniform laminar structure with a well-defined interlayer space,1–3 and they have been studied extensively for applications such as nanofiltration and pervaporation. GO membranes showed excellent selectivities in rejecting small molecules and multivalent ions in pressure-driven nanofiltration,4–7 and also in dewatering of organic solvents such as alcohol and dimethyl carbonate in pervaporation.8–10 Though the interlayer space of the GO membranes has endowed the membranes with excellent molecular sieving properties,11 the separation mechanisms in these two applications were found to be different. The rejection of small molecules in nanofiltration was mainly through size exclusion,7,12 while the separation of water from organic solvents in pervaporation, which involved selective permeation and evaporation steps in series, was predominantly due to the preferential adsorption of water in GO membranes.10
Solution–diffusion or adsorption–diffusion models have been adopted by many to explain the good performance of GO membranes in pervaporation.10,13,14 In these models, the permeability coefficient of the individual liquid is the product of the sorption coefficient and the diffusivity. GO membranes were found to have a high water adsorption capacity and this was shown by Huang et al. via a direct measurement of water adsorption using the quartz crystal microbalance technique.10 In a recent study by Cho et al., GO membranes showed a 40% weight increase in humid air, and a 120% weight increase when immersed in water.15 The preferential water adsorption was due to the large amount of hydrophilic functional groups in GO membranes and the low water condensation partial pressure resulted due to the fine laminar structure. Some researchers believe that water molecules have a much higher permeation rate in GO membranes compared to other solvents because of the selective adsorption, which subsequently leads to effective water separation from a water–solvent mixture.10,13 However, while the preferential adsorption of water was repeatedly confirmed, the diffusion of water in GO membranes was not much discussed in the adsorption–diffusion model. In the adsorption–diffusion or solution–diffusion models, a phase transition has to occur at the feed-side membrane surface so that water will turn from a continuous bulk liquid phase to individual molecules or clusters.16 An apparent difficulty for the adsorption–diffusion model to explain the pervaporation performance in GO membranes is that molecular diffusion is unlikely to be fast in the long and tortuous transport path in GO membranes,17 which is opposite to the high water flux observed in experimental results.
To explain the high water flux observed, the fast transport hypothesis proposed by Nair et al. has often been adopted by researchers to explain the transport mechanism in the interlayer space.13,18,19 In the hypothesis, water was proposed to pass through the graphitic domains in GO membranes in a low-friction flow due to the hydrophobic pore walls. However, the hypothesis suggested that water molecules condensed and permeated through the interlayer space in a bulk phase with one to three layers of water. If this hypothesis is adopted, the adsorption–diffusion model will no longer be suitable, as the transport of water in the interlayer space will follow a pore flow model instead. Different from the solution–diffusion or adsorption–diffusion models, the driving force for water permeation in the pore flow model is not the concentration gradient but the pressure difference in the membranes.16 Therefore, two important questions are raised if the pore flow model is applied to explain the transport mechanism in pervaporation: (i) what is the origin of the pressure difference in the nanochannels that leads to a high water flux; (ii) where and how does the liquid phase water in the nanochannels turn to the vapour phase?
To answer the first question, the pure water flux of GO membranes was tested in this study using two different modes: pressure-driven permeation and pervaporation. The comparison made between the two modes enabled us to define the difference in driving forces in these modes, and to identify the origins of the driving force particularly in pervaporation. For the second question, we compared the pure water flux of GO membranes under the pervaporation mode to the evaporation rate of free water. By doing so, the effective evaporating area of GO membranes could be assessed, which allowed us to understand the possible steps involved in evaporation. The use of pure water rather than solvent–water mixtures in this study simplified the analysis so that we could focus on the transport steps on the permeate side, without complications on the feed side. The experimental results suggested that the driving force for water permeation through GO membranes was much larger in pervaporation due to the high capillary pressure maintained by evaporation, and the rate-determining step should be the mass transfer steps on the permeate side surface.
In this study, flat-sheet GO membranes were prepared on polyethersulfone (PES) substrates by using a filtration method. After drying at room temperature for at least 24 hours, the GO membranes were sealed in a dead-end filtration cell for permeation tests. A detailed description of the synthesis and testing methods can be found in the ESI.† First, the membranes were tested with pure water for pressure-driven permeation. A transmembrane pressure of 6 bar was applied for at least 24 hours to ensure that the membranes were compacted and a steady-state permeance was obtained.20 Then, the same wet membrane samples were used for the pervaporation test. This was to ensure the same membrane structure and transport passages for both pressure-driven permeation and pervaporation tests, and thus the fairness of the comparison. A vacuum system was connected on the permeate side of the membranes during pervaporation, and the water vapour permeated was condensed using a liquid nitrogen cold trap. The experimental set-up is shown in the ESI.†
Fig. 1(a–c) show the photo of a GO membrane and the SEM images of the surface and the cross-section of the GO membrane on a PES substrate. The GO membrane had a smooth shiny coating on the PES substrate and some wrinkles were observed on the membrane surface under the SEM. The thickness of the GO membrane was about 350 nm and the GO layer was located nicely on the PES substrate. Fig. 1d shows the water permeance of the GO membranes in the pressure-driven permeation test. The membrane showed a permeance of about 1.5 L m−2 h−1 bar−1 (LMH bar−1) in the beginning of the permeation test. However, the permeance dropped significantly over time and reached a steady state permeance of 0.05 LMH bar−1. As reported in our previous study, the high water permeance in the beginning of the permeation tests could be due to the loosely packed microstructure.20 Because of the membrane formation mechanism, a hierarchical structure was normally obtained by the current GO membrane synthesis methods.21 The imperfect stacking of GO flakes created extra large transport channels for water to pass through the membranes in the beginning of the permeation test. However, the loosely packed microstructure was compacted under pressure during the permeation test, and the extra transport channels were reduced over time, leading to a lower permeation flux. GO membranes showed a low steady state water permeance eventually due to the long transport path in the membranes, as a result of the high aspect ratio of the GO flakes.
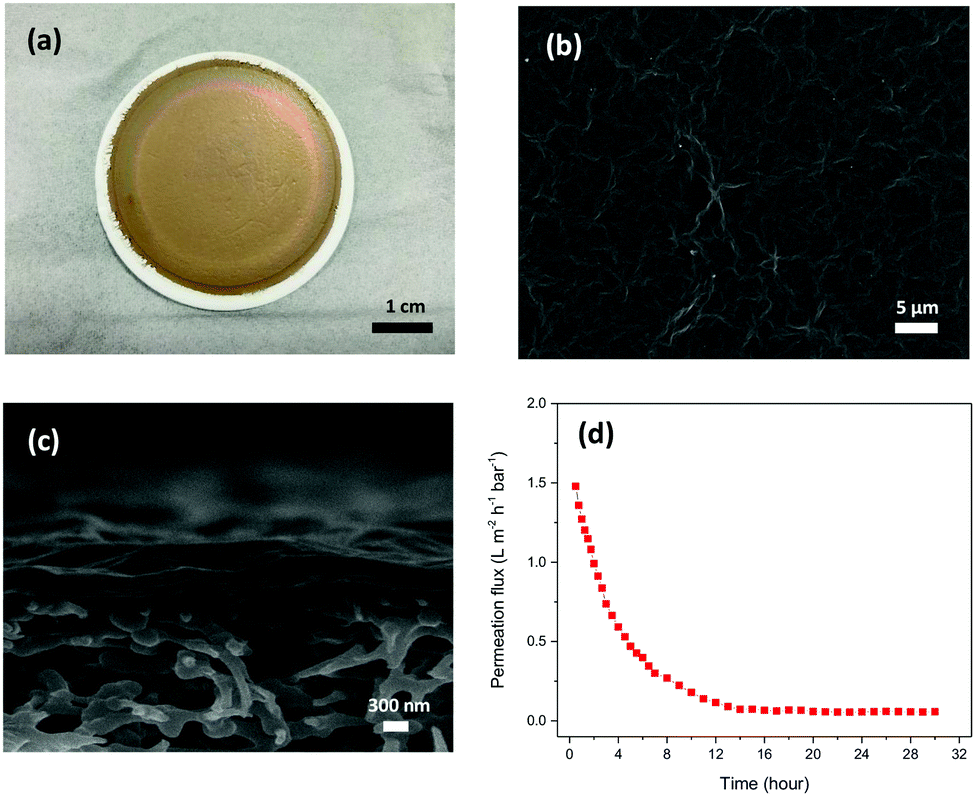 | ||
| Fig. 1 (a) Pictures of flat sheet GO membranes on a PES substrate. SEM images of the surfaces (b) and the cross-sections (c) of GO membranes. (d) Water permeance of GO membranes during pressure-driven permeation. | ||
Fig. 2 shows the pure water permeance of GO membranes under both pressure-driven and pervaporation conditions. GO membranes showed a very low steady-state permeance, 0.05 LMH bar−1, when tested under the pressure-driven mode. However, when tested under the pervaporation mode at room temperature (RT), the pure water flux of GO membranes was 1.62 LMH, with an apparent pressure difference of 1 bar. The flux was about 30 times higher than the permeance when tested under pressure-driven permeation. When vacuum is applied on the permeate side in pervaporation, water on the permeate side surface is evaporated quickly due to a low vapour partial pressure. Whenever the evaporation is faster than the permeation of liquid water, the liquid–vapour interface may move and locate at the entrance or even inside the nanochannels in the GO membrane. When the interface reaches the nanochannels, a large capillary pressure is generated as a result of the extremely small pore size, and will drag water from the feed side to the permeate side swiftly. Our estimation shows that the capillary pressure in GO membranes could be as large as 1440 bar (see ESI† for the calculation), a similar estimation was also reported by Nair et al.18 However, the capillary pressure does not exist under the pressure-driven permeation mode as there is no phase change on the permeate side. Fig. 3 illustrates the water transport in the interlayer space of GO membranes where the real tortuous transport path is simplified to a long nanochannel. As illustrated in Fig. 3(a), a liquid–vapour interface does not form in the interlayer space as the water permeated through the membrane is still in the liquid form. In this case, the water permeation only relies on the hydraulic pressure applied (6 bar in this study), which is much smaller than the capillary pressure.
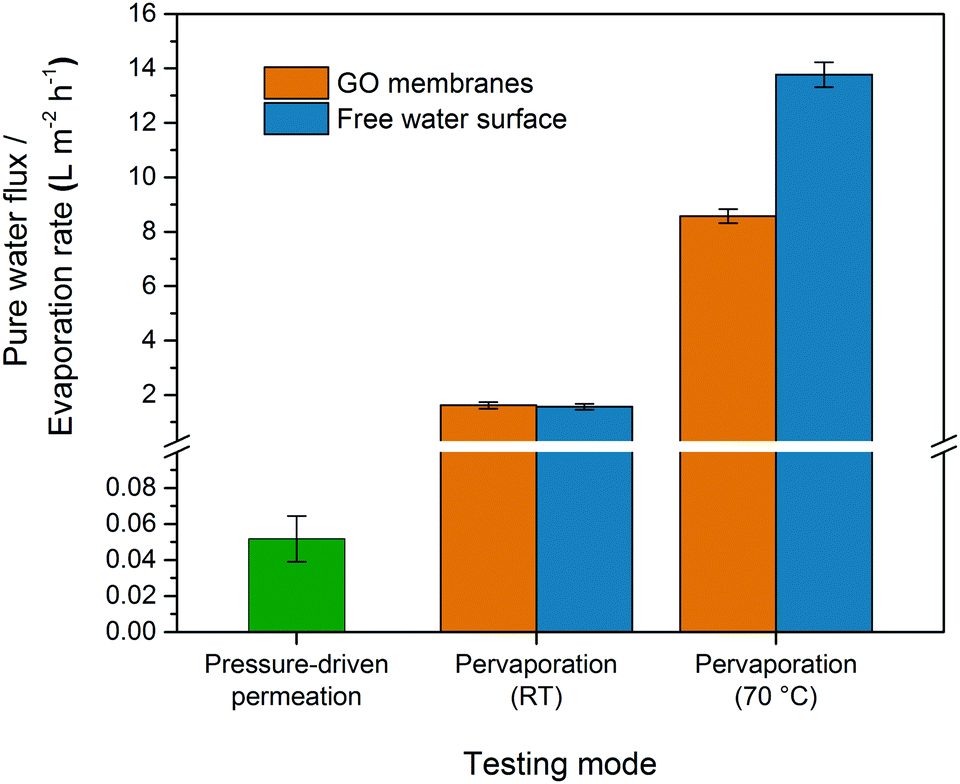 | ||
| Fig. 2 Pure water flux of GO membranes tested using pressure-driven permeation (L m−2 h−1 bar−1) and pervaporation at room temperature (RT) and at 70 °C. | ||
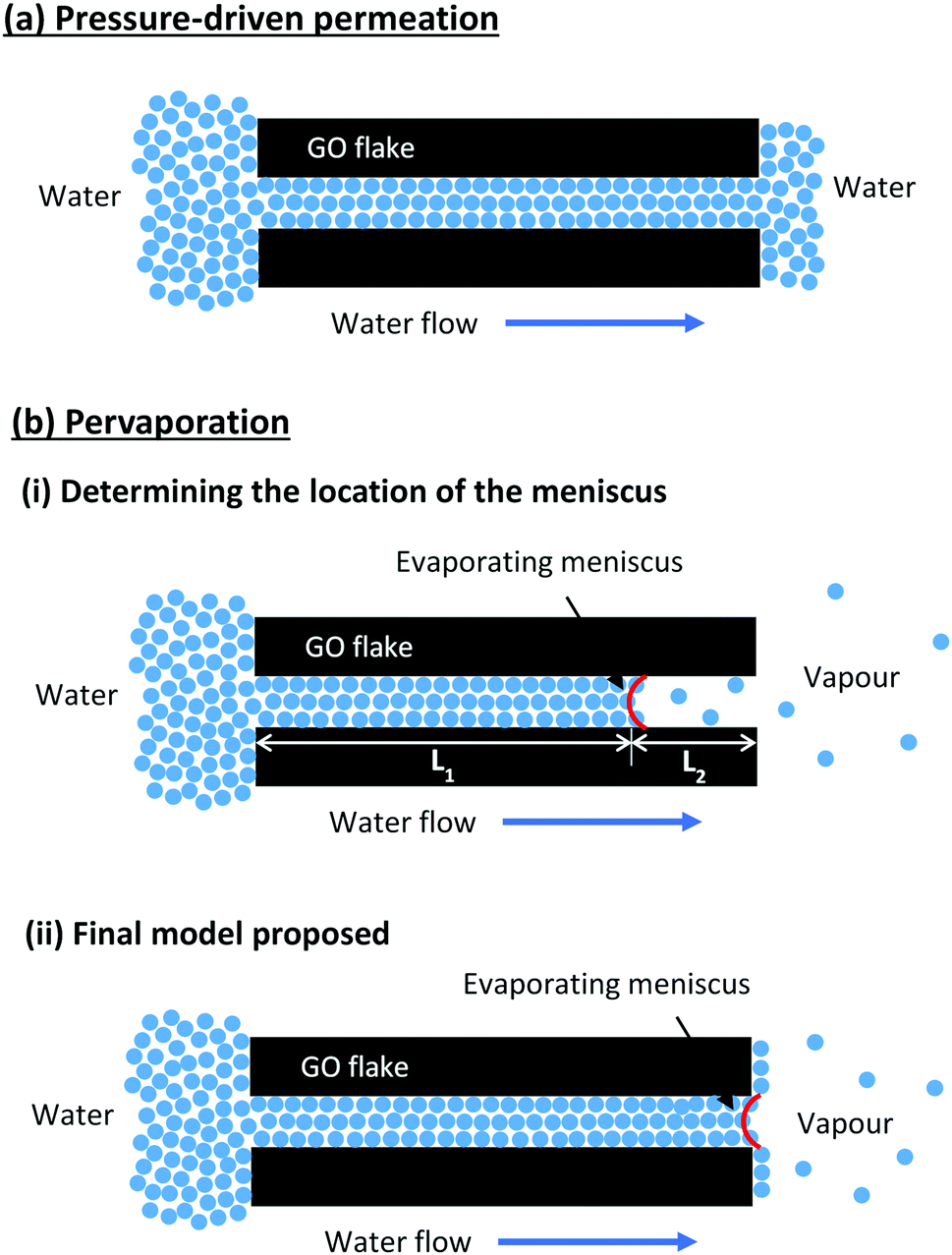 | ||
| Fig. 3 Illustration of the water transport mechanisms in the nanochannels in GO membranes under the (a) pressure-driven permeation and (b) pervaporation modes. | ||
Fig. 3(b) illustrates the water transport in the interlayer space of GO membranes during pure water pervaporation. Water enters the nanochannels due to the hydrophilic nature of the membranes, and then permeates through the interlayer space driven by the capillary pressure generated by the meniscus on the other side. The permeation flux in the interlayer space before the evaporating meniscus (Q1) is proportional to the capillary pressure (Pc) but inversely proportional to the passage length (L1):
| Q1 ∝ Pc/L1 | (1) |
At the evaporating interface, water molecules evaporate and diffuse through the interlayer space if the interface is in the membrane. The vapour pressure at the interface could be assumed to be the saturated pressure and the permeation rate (Q2) could be determined by the diffusivity of water in the interlayer space (D), the difference between the vapour pressure at the meniscus (PI) and the vacuum pressure (PV), and the passage length of the water vapour L2:
| Q2 ∝ D × (PI − PV)/L2 | (2) |
In a steady state, Q1 = Q2, and the position of the evaporating meniscus moves to attain the steady state permeation flux, which will then determine the L1 and L2. Since the transport of the condensed water through L1 is much faster due to the high capillary pressure, it is reasonable to predict that the evaporating meniscus would be close to the membrane downstream surface. In that case, the evaporation steps determine the overall water flux. To verify this, we carried out a simple experiment to measure the evaporation rate of a free water surface in vacuum at room temperature. A cylindrical glass bottle half filled with water was connected to a vacuum system and the evaporation rate of water was determined from the change in the mass of the bottle. The water evaporation rate at room temperature was found to be 1.57 LMH (Fig. 2), which was essentially the same as the pure water flux of GO membranes in pervaporation. Similar results have also been reported by Nair et al., where the water vapour permeation rate through GO membranes was close to the evaporation rate of a free water surface with the same area.18 However, the experimental results from pervaporation would be difficult to explain if the water transport only followed the model illustrated in Fig. 3(b)(i). As the surface porosity of GO membranes was very low (<0.1%),18 the water evaporation would be very slow if water evaporated only via the liquid–vapour interface in the nanochannels.
To match the experimental results, the evaporation area in pervaporation has to be extended to the whole membrane surface rather than staying inside the pores. Such extension of the evaporation area can be realised due to the high surface hydrophilicity of GO membranes, where water can spread on the surface and turn into adsorbed molecules. The adsorbed water molecules can migrate to the whole permeate side membrane surface via surface diffusion before desorption, as schematically shown in Fig. 3(b)(ii). Such full coverage by surface diffusion is very likely since the required diffusion distance from the exits of the nanochannels to the centre of each GO flake is only a few micrometres, which can be easily covered if the desorption is relatively slow. Since water molecules on the GO surface are bonded by hydrogen bonds,22 their desorption from the membrane surface would have a similar energy requirement to evaporation from bulk water, which is also to break the hydrogen bond. Therefore, the water flux through GO membranes in pervaporation has a similar value to that in free evaporation.
To further study the effect of the evaporation rate on the water flux of GO membranes, the pervaporation test was also carried out at 70 °C. The pure water flux increased to about 8.57 LMH (Fig. 2), which was 5.3 times higher than the flux at room temperature. However, the evaporation rate of the free water surface increased to 13.8 LMH at 70 °C, which was 8 to 9 times higher than that at room temperature. The different rates between pervaporation and free evaporation indicated the change in the rate-determining step at higher temperatures. It is known that surface diffusion of small molecules like water normally has a very low activation energy (<20 kJ mol−1),23,24 while the activation energy for desorption could be higher (the latent heat of vaporization of water is higher than 40 kJ mol−1 between room temperature and 70 °C).25,26 When the temperature increases, the rate of desorption increases more than that of surface diffusion. Consequently, the centre of the GO flakes will not be covered by water molecules as they leave the surface before reaching the centre. This leads to the reduction of the effective evaporation area and hence the water flux of pervaporation will be lower than in free evaporation. Besides the competition between surface diffusion and desorption, another possible reason for the lower water flux could be the relatively slow permeation in the nanochannels compared to the much enhanced evaporation rate at high temperature. However this is not very likely considering the huge capillary pressure that could be generated in the nanochannels (>1000 bar).
In conclusion, GO membranes showed a much higher pure water flux in pervaporation compared to pressure-driven permeation due to the difference in driving forces. A two-step pore flow–evaporation model has been proposed to understand the high water flux in pervaporation. Water permeated through GO membranes at a high speed due to the high capillary pressure in the membranes. However, the overall water flux was limited by the evaporation rate at room temperature, but by surface diffusion on the permeate side membrane surface at high temperature. The performance of GO membranes in pervaporation can be further enhanced by increasing the effective surface area or by surface modification to improve the surface diffusion on the permeate side surface.
The authors gratefully acknowledge the research funding provided by the EPSRC in the United Kingdom (Grant no. EP/M022250/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- N. V. Medhekar, A. Ramasubramaniam, R. S. Ruoff and V. B. Shenoy, ACS Nano, 2010, 4, 2300–2306 CrossRef CAS PubMed.
- G. Liu, W. Jin and N. Xu, Chem. Soc. Rev., 2015, 44, 5016–5030 RSC.
- H. Huang, Y. Mao, Y. Ying, Y. Liu, L. Sun and X. Peng, Chem. Commun., 2013, 49, 5963–5965 RSC.
- N. F. D. Aba, J. Y. Chong, B. Wang, C. Mattevi and K. Li, J. Membr. Sci., 2015, 484, 87–94 CrossRef CAS.
- J. Y. Chong, N. F. D. Aba, B. Wang, C. Mattevi and K. Li, Sci. Rep., 2015, 5, 15799 CrossRef CAS PubMed.
- A. Akbari, P. Sheath, S. T. Martin, D. B. Shinde, M. Shaibani, P. C. Banerjee, R. Tkacz, D. Bhattacharyya and M. Majumder, Nat. Commun., 2016, 7, 10891 CrossRef CAS PubMed.
- Y. P. Tang, D. R. Paul and T. S. Chung, J. Membr. Sci., 2014, 458, 199–208 CrossRef CAS.
- G. Li, L. Shi, G. Zeng, Y. Zhang and Y. Sun, RSC Adv., 2014, 4, 52012–52015 RSC.
- K. Huang, G. Liu, Y. Lou, Z. Dong, J. Shen and W. Jin, Angew. Chem., Int. Ed., 2014, 53, 6929–6932 CrossRef CAS PubMed.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS PubMed.
- J. Abraham, K. S. Vasu, C. D. Williams, K. Gopinadhan, Y. Su, C. T. Cherian, J. Dix, E. Prestat, S. J. Haigh, I. V. Grigorieva, P. Carbone, A. K. Geim and R. R. Nair, Nat. Nanotechnol., 2017, 12, 546–550 CrossRef CAS PubMed.
- W.-S. Hung, Q.-F. An, M. De Guzman, H.-Y. Lin, S.-H. Huang, W.-R. Liu, C.-C. Hu, K.-R. Lee and J.-Y. Lai, Carbon, 2014, 68, 670–677 CrossRef CAS.
- W.-S. Hung, C.-H. Tsou, M. De Guzman, Q.-F. An, Y.-L. Liu, Y.-M. Zhang, C.-C. Hu, K.-R. Lee and J.-Y. Lai, Chem. Mater., 2014, 26, 2983–2990 CrossRef CAS.
- Y. H. Cho, H. W. Kim, H. D. Lee, J. E. Shin, B. M. Yoo and H. B. Park, J. Membr. Sci., 2017, 544, 425–435 CrossRef CAS.
- J. G. Wijmans and R. W. Baker, J. Membr. Sci., 1995, 107, 1–21 CrossRef CAS.
- R. Devanathan, D. Chase-Woods, Y. Shin and D. W. Gotthold, Sci. Rep., 2016, 6, 29484 CrossRef PubMed.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS PubMed.
- C.-H. Tsou, Q.-F. An, S.-C. Lo, M. De Guzman, W.-S. Hung, C.-C. Hu, K.-R. Lee and J.-Y. Lai, J. Membr. Sci., 2015, 477, 93–100 CrossRef CAS.
- J. Y. Chong, B. Wang, C. Mattevi and K. Li, J. Membr. Sci., 2018, 549, 385–392 CrossRef CAS.
- K. Nandy, M. J. Palmeri, C. M. Burke, Z. An, S. T. Nguyen, K. W. Putz and L. C. Brinson, Adv. Mater. Interfaces, 2016, 3, 1500666 CrossRef.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- T. Mitsui, M. K. Rose, E. Fomin, D. F. Ogletree and M. Salmeron, Science, 2002, 297, 1850–1852 CrossRef CAS PubMed.
- P. C. Carman and F. A. Raal, Proc. R. Soc. London, Ser. A, 1951, 209, 38–58 CrossRef CAS.
- P. Datt, in Encyclopedia of Snow, Ice and Glaciers, ed. V. P. Singh, P. Singh and U. K. Haritashya, Springer, Netherlands, Dordrecht, 2011 Search PubMed.
- X. Li, X. Chen and Z. Li, J. Chem. Eng. Data, 2010, 55, 3164–3169 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures. See DOI: 10.1039/c7cc09120f |
| This journal is © The Royal Society of Chemistry 2018 |