
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrogenation of nitroarenes using an electrogenerated polyoxometalate redox mediator†
Lewis
MacDonald
 ,
Benjamin
Rausch
,
Mark D.
Symes
and
Leroy
Cronin
*
,
Benjamin
Rausch
,
Mark D.
Symes
and
Leroy
Cronin
*
WestCHEM, School of Chemistry, University of Glasgow, University Avenue, Glasgow, G12 8QQ, UK. E-mail: Lee.Cronin@glasgow.ac.uk
First published on 4th January 2018
Abstract
The 2-electron reduced form of the polyoxometalate silicotungstic acid (H4[SiW12O4]) is shown to be an effective and selective hydrogenation agent for a range of nitroarenes without the need for any co-catalyst. The ease of generation of the active species and its recyclability suggest that a new approach to this important class of chemical conversions is possible.
Aromatic amines are a key ingredient in the synthesis of many industrial fine chemicals, including pharmaceuticals, dyes, polymers and agrochemicals.1–4 A common route to such amines is via the reduction of the corresponding nitro-compounds,5 with the most widely-used procedures relying on the use of catalytic hydrogenation using hydrogen gas and catalysts based on Pt/C, PtO2 and RANEY® Ni.6 However, whilst these routes tend to be effective, it is not always practical or desirable to employ molecular hydrogen as the hydrogenation agent. Accordingly, there has been considerable interest in developing systems for the selective reduction of nitro aromatics to their aniline derivatives using soluble hydrogenation agents. One such approach has been to use transfer hydrogenation, whereby a reducing agent is paired with an appropriate catalyst,7,8 which allows hydrogenation to proceed without the need to handle gaseous H2.6,9–11 Amongst the reducing agents used in such transfer hydrogenations are sodium borohydride, silyl hydrides, formate, oxalic acid and hydrazine.6,12–14 However, all of these reducing agents are in a sense sacrificial, inasmuch as they are irreversibly consumed during the hydrogenation process.
Recently, we developed the concept of the electron-coupled-proton buffer, whereby electrochemical water oxidation at the anode leads to reversible reduction and protonation of a suitable redox mediator at the cathode.15–19 Previously, we applied this concept to the spatially- and temporally-separated half reactions of water splitting by using the reduced and protonated mediator to generate hydrogen. However, it also seemed to us that the reduced and protonated mediator would also be an ideal hydrogenation agent for organic synthesis, with the key advantages of avoiding the handling of hydrogen gas and the fact that the hydrogen equivalents are obtained from water, rather than a potentially unsustainable sacrificial source.
Herein, we demonstrate the feasibility of this system and show that reduction of silicotungstic acid by two electrons (giving H6[SiW12O40]) produces a species that is capable of reducing nitroarenes to their corresponding aniline derivatives at room temperature with high selectivity in good to excellent yields. The reactions are conducted in aqueous medium, and reduction is possible even in the case of nitroarenes that are essentially insoluble in water. The resulting anilines are readily isolated in pure form from the reaction medium by simply adjusting the pH followed by extraction into organic solvents. Meanwhile, the redox mediator itself can be recycled from the reaction medium and used to perform subsequent hydrogenation reactions without any apparent loss of activity. On account of the selectivity of this process, the fact that it allows hydrogenations without the need for hydrogen gas, high pressures or any co-catalysts, and because of its potential as a sustainable route for the hydrogenation of nitroarenes, we foresee the development of this system to a range of allied transformations in the future.
In a typical procedure (see Fig. 1), H4[SiW12O40] was dissolved in water to a concentration of 0.5 M and reduced by two electrons to H6[SiW12O40] under an inert atmosphere in a manner similar to that we reported previously (see ESI† for full experimental details).17 To aliquots of this reduced polyoxometalate were then added the nitroarene compounds listed in Table 1 under an inert argon atmosphere, and the reactants were allowed to stir at room temperature for 18 hours. After this time, the pH of the reaction medium was brought above the pKa value of the anticipated aniline product and then the reaction mixture was extracted using either ether or chloroform. The aqueous phase was kept aside for subsequent mediator recovery (see below).
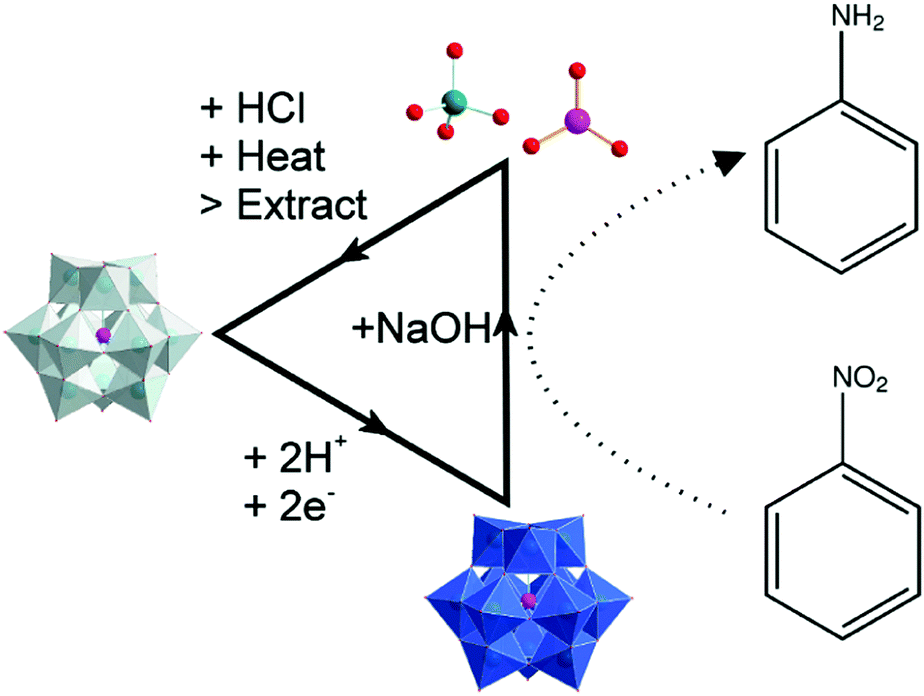 | ||
| Fig. 1 Schematic detailing the process of hydrogenating nitro-containing arene compounds and recycling the polyoxometalate. Colour code – grey polyhedral: oxidized form of the Keggin ion (H4[SiW12O40]), blue polyhedral: reduced form of the Keggin ion (H6[SiW12O40]). Ball and stick diagram: Si = pink, W = green, O = red. | ||
|
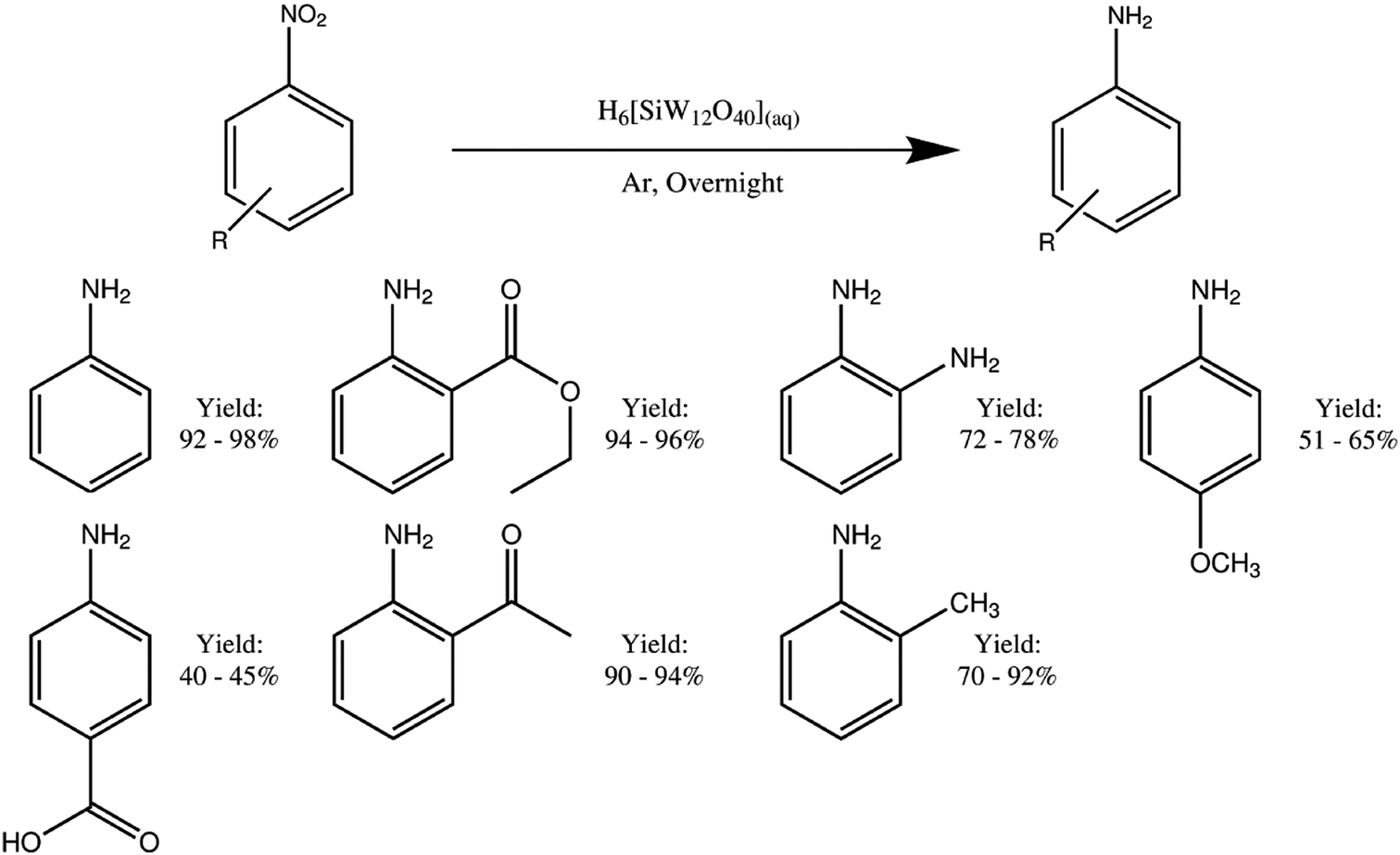
The isolated yields of all the aniline products are shown in Table 1, and their purity was confirmed by 1H and 13C NMR analysis. Comparison to both the starting material and a genuine sample of the anticipated product was made in each case (an example is shown in Fig. 2, and data for the remaining compounds are shown in the ESI†). In Fig. 2, the lack of starting material indicates that all the nitrobenzene has reacted with the reduced mediator, whilst the identical peak pattern with respect to an authentic sample of the anticipated product (aniline) confirms that the selectivity of the reaction is essentially 100%.
Varying isolated yields were obtained for the products shown in Table 1, with the isolated yields for aniline (92–98%), ethyl 2-aminobenzoate (94–96%) and 2′-aminoacetophenone (90–94%) being excellent. The yield of 2-toluidine was also high (70–92%). 2-Phenylenediamine (72–78%) was isolated in good yield, whilst 4-ansidine (51–65%) and 4-aminobenzoic acid (40–45%) were isolatable in more modest yields. One possible explanation for the lower yields in these two latter cases relates to the solubility of the starting materials. This appears to dictate the yield of hydrogenated product obtained, with the more water-soluble compounds (nitrobenzene, ethyl 2-nitrobenzene and 2′-nitroacetophenone) showing markedly higher yields that the insoluble organics (4-nitroanisole and 4-nitrobenzoic acid). However, it is notable that even starting materials with very low solubility in water still showed appreciable conversion to the hydrogenated species. For example, 4-nitroanisole has a solubility of 5.9 mg per 10 mL water,20 yet an isolated yield of between 51 and 65% was obtained when 0.2 g of 4-nitroanisole was present in 10 mL of 0.5 M (reduced) silicotungstic acid. Hence it seems that even largely insoluble species can be effectively hydrogenated by this procedure. An alternative explanation for the lower yield in the case of 4-aminobenzoic acid is the fact that the product is rather water-soluble and hence may not be fully recovered during the extraction process. Likewise, if the methyl group of 4-ansidine is cleaved under these hydrogenation-like conditions, then that would lead to p-nitrophenol, which again would be somewhat water-soluble.
The H4[SiW12O40] used in the hydrogenation process could be recycled by employing the cycle shown in Fig. 1. When the pH of the reaction medium is raised (to allow extraction of the aniline products), the polyoxometalate is subject to partial disassembly into smaller metal–oxo building blocks. However, upon re-acidification and exposure to oxygen, it is possible to re-form H4[SiW12O40]. Hence the (dark blue) aqueous layers from the extraction process (typical pH ≈ 5.5) were treated with concentrated HCl followed by heating to 95 °C and left to stir overnight. During this time the dark blue solution (indicating reduced mediator) would turn yellow (indicating a fully oxidised species). This solution was then filtered through activated carbon, producing a clear, colourless filtrate.21–23
Following literature procedures, H4[SiW12O40] was then isolated from this solution by extraction with diethyl ether and subsequent addition of a mineral acid (in this case concentrated HCl).24 Crystals of pure H4[SiW12O40] were then collected from this extract (see full details in the ESI† and CHN analyses of fresh and recycled H4[SiW12O40] in Table S1). Fig. 3 compares the powder XRD patterns obtained from recycled H4[SiW12O40] (after one use as a hydrogenation mediator) and H4[SiW12O40] after a second use as a hydrogenation mediator. Both species give similar peak patterns. The starred peaks agree with previous literature measurements of H4[SiW12O40] using powder XRD.25 The yield of each recovery cycle was calculated to be ∼70%.
To show the reusability of the recovered H4[SiW12O40], the recycled compound was reduced electrochemically by two electrons as before and the hydrogenation procedure was repeated using nitrobenzene. Fig. 2 shows a comparison of the starting material, an authentic sample of the anticipated product as a standard and the products obtained in the two hydrogenation cycles. There is no distinct variation between the product obtained from electrochemically reduced fresh H4[SiW12O40] and the electrochemically reduced recycled H4[SiW12O40]. A 97% yield was obtained using the recycled H4[SiW12O40] and nitrobenzene. Indeed, the same sample of H4[SiW12O40] could be recycled up to four times (with at least 70% recovery of the H4[SiW12O40] after each step) and subsequently re-used in the reduction of nitrobenzene (delivering a yield of aniline of at least 92% in each cycle). The fact that the recovery of the H4[SiW12O40] after each step is less than 100% is due to its solubility in water and hence the difficulty in obtaining a 100% recovery from its recrystallization.
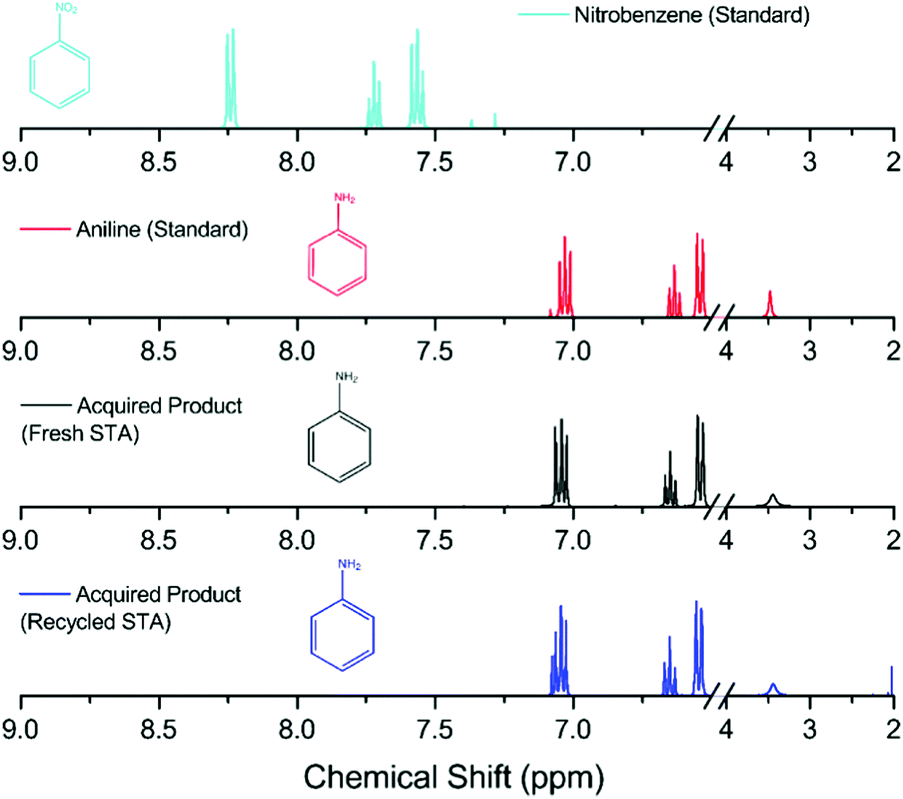 | ||
| Fig. 2 1H NMR spectra of the obtained product compared with the starting material (nitrobenzene) and the anticipated product (aniline) using fresh and recycled H6[SiW12O40] as the hydrogenating agent. All analysis was conducted in CDCl3 (axis has been compressed to show detail of peaks, higher resolution images are shown in the ESI†). Colour code – nitrobenzene = light blue, aniline = red, acquired product (using fresh H6[SiW12O40]) = black, acquired product (using used H6[SiW12O40]) = dark blue. | ||
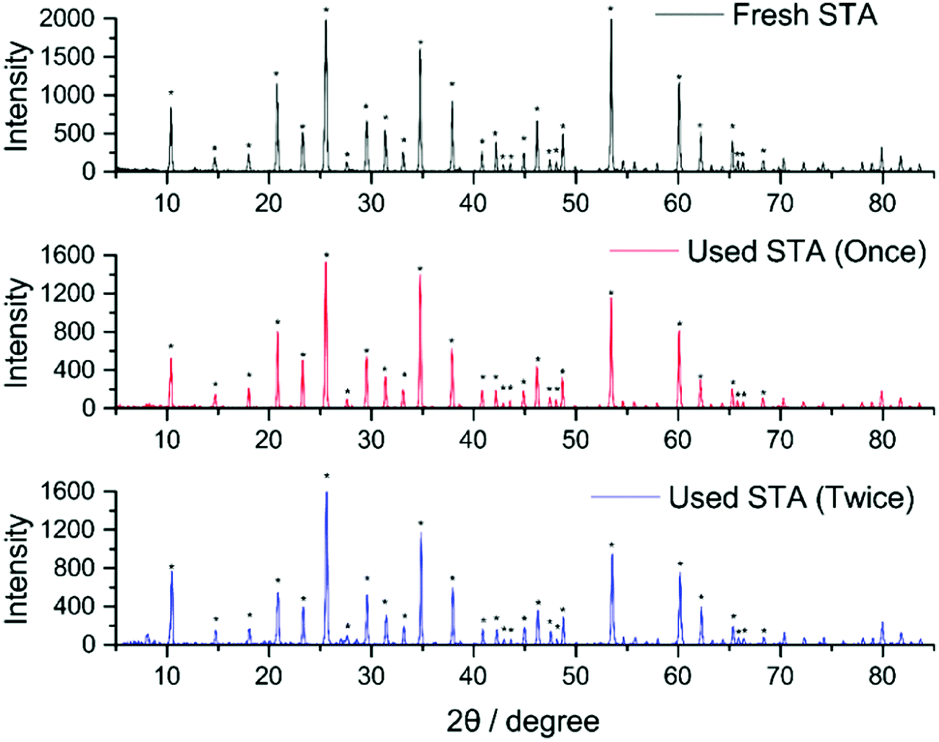 | ||
| Fig. 3 Powder XRD patterns for fresh and the recycled H4[SiW12O40]. (* = peaks confirmed as H4[SiW12O40] from the literature)25 Colour code – H4[SiW12O40] (fresh) = Black, H4[SiW12O40] after one use as a hydrogenation mediator = Red, H4[SiW12O40] after two uses as a hydrogenation mediator = dark blue. | ||
In addition, we also performed the reduction of ethyl 2-nitrobenzoate to ethyl 2-aminobenzoate using recycled H4[SiW12O40] (yield = 89%) and the reduction of 2′-nitroacetophenone to 2′-aminoacetophenone using recycled H4[SiW12O40] (yield = 96%). These yields compare well with those obtained using fresh H4[SiW12O40] (see Table 1). Taken together, these results suggest that the H4[SiW12O40] can be recovered and re-used multiple times for mediated electroreduction of various nitroarene substrates.
In conclusion, we have shown that an electrochemically reduced polyoxometalate, in the form of silicotungstic acid, can selectively hydrogenate the nitro group of a number of nitroarenes in the presence of other functional groups. The procedure is simple and sustainable in that it requires no pressurised hydrogen atmospheres, elevated temperatures, co-catalysts or sacrificial reagents. The mediator is recoverable and recyclable using well known procedures and commonly-available chemicals. Moreover, the recycled mediator can be electrochemically re-reduced and can perform the hydrogenation reaction without an appreciable drop in performance compared with fresh H4[SiW12O40].
We thank the following funders: EPSRC (Grant No. EP/H024107/1, EP/J015156/1, EP/K023004/1, EP/L023652/1), the EC (318671 MICREAGENTS), ERC (project 670467 SMART-POM). The authors would like to thank the BBSRC, Dr Guillaume Marie and Prof Richard Cogdell for their support. MDS thanks the Royal Society for a University Research Fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- X.-A. Ning, J.-Y. Liang, R.-J. Li, Z. Hong, Y.-J. Wang, K.-L. Chang, Y.-P. Zhang and Z.-Y. Yang, Chemosphere, 2015, 134, 367–373 CrossRef CAS PubMed.
- A. P. Harding, P. L. A. Popelier, J. Harvey, A. Giddings, G. Foster and M. Kranz, Regul. Toxicol. Pharmacol., 2015, 71, 244–250 CrossRef CAS PubMed.
- B. Jurado-Sánchez, E. Ballesteros and M. Gallego, Sci. Total Environ, 2013, 463–464, 293–301 CrossRef PubMed.
- A. M. Al-Sabagh, N. M. Nasser, E. A. Khamis and M. Abd-El-Raouf, Egypt. J. Pet., 2015, 24, 363–374 CrossRef.
- A. S. Travis, Manufacture and Uses of the Anilines: A Vast Array of Processes and Products, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
- H. K. Kadam and S. G. Tilve, RSC Adv., 2015, 5, 83391–83407 RSC.
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- H. Imai, T. Nishiguchi and K. Fukuzumi, Chem. Lett., 1976, 655–656 CrossRef CAS.
- Y. Watanabe, T. Ohta, Y. Tsuji, T. Hiyoshi and Y. Tsuji, Bull. Chem. Soc. Jpn., 1984, 57, 2440–2444 CrossRef CAS.
- G. Wienhöfer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875–12879 CrossRef PubMed.
- K. Imamura, K. Hashimoto, H. Kominami, M. Matsumura, B. Ohtani, H. Kominami and B. Ohtani, Chem. Commun., 2012, 48, 4356 RSC.
- E. Garcia-Verdugo, Z. Liu, E. Ramirez, J. Garcia-Serna, J. Fraga-Dubreuil, J. R. Hyde, P. A. Hamley and M. Poliakoff, Green Chem., 2006, 8, 359–364 RSC.
- M. V. Joshi and D. Mukesh, J. Catal., 1997, 168, 273–277 CrossRef CAS.
- M. D. Symes and L. Cronin, Nat. Chem., 2013, 5, 403–409 CrossRef CAS PubMed.
- B. Rausch, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2013, 135, 13656–13659 CrossRef CAS PubMed.
- B. Rausch, M. D. Symes, G. Chisholm and L. Cronin, Science, 2014, 345, 1326–1330 CrossRef CAS PubMed.
- L. G. Bloor, R. Solarska, K. Bienkowski, P. J. Kulesza, J. Augustynski, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2016, 138, 6707–6710 CrossRef CAS PubMed.
- L. MacDonald, J. C. McGlynn, N. Irvine, I. Alshibane, L. G. Bloor, B. Rausch, J. S. J. Hargreaves and L. Cronin, Sustainable Energy Fuels, 2017, 1, 1782–1787 CAS.
- P. M. Gross and J. H. Saylor, J. Am. Chem. Soc., 1929, 53, 1744–1751 CrossRef.
- A. G. Scroggie, J. Am. Chem. Soc., 1929, 51, 1057–1062 CrossRef CAS.
- J. Georgin, G. L. Dotto, M. A. Mazutti and E. L. Foletto, J. Environ. Chem. Eng., 2016, 4, 266–275 CrossRef CAS.
- S. Cheng, L. Zhang, A. Ma, H. Xia, J. Peng, C. Li and J. Shu, J. Environ. Sci. DOI:10.1016/j.jes.2016.12.027.
- E. O. North and G. D. Beal, J. Am. Pharm. Assoc., 1924, 13, 889–898 CAS.
- G. Chen, J. Li, X. Yang and Y. Wu, Appl. Catal., A, 2006, 310, 16–23 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc09036f |
| This journal is © The Royal Society of Chemistry 2018 |