
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective sensing of 2,4,6-trinitrophenol (TNP) in aqueous media with “aggregation-induced emission enhancement” (AIEE)-active iridium(III) complexes†
Weilong
Che
a,
Guangfu
Li
a,
Xingman
Liu
a,
Kuizhan
Shao
a,
Dongxia
Zhu
*a,
Zhongmin
Su
 *a and
Martin R.
Bryce
*b
*a and
Martin R.
Bryce
*b
aKey Laboratory of Nanobiosensing and Nanobioanalysis at Universities of Jilin Province, Department of Chemistry, Northeast Normal University, 5268 Renmin Street, Changchun, Jilin Province 130024, P. R. China. E-mail: zhudx047@nenu.edu.cn; zmsu@nenu.edu.cn
bDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 29th January 2018
Abstract
A series of new phosphorescent cyclometalated iridium(III) complexes which possess aggregation-induced emission enhancement (AIEE) detect 2,4,6-trinitrophenol (TNP) selectively with high quenching constants in aqueous media. The sensing mechanism was systematically investigated by mass spectrometry, 1H and 19F NMR spectroscopy. X-ray crystal structure analysis reveals an O–H⋯O interaction between TNP and the ancillary ligand which explains the high selectivity for TNP compared to other nitro-aromatics.
Nitro-aromatics such as 2-nitrotoluene (oNT), nitrobenzene (NB), 2,4,6-trinitrotoluene (TNT) and 2,4,6-trinitrophenol (TNP) (picric acid) are widespread explosive materials that threaten society through terrorist activities. TNP exhibits enhanced explosive power compared to TNT.1 TNP is also a notorious environmental pollutant of irrigation land and ground water supplies due to its high acidity and good solubility in water.2 The selective detection of TNP from mixtures of other nitro-aromatic explosives is very difficult because they all exhibit strong electron affinity.3 Therefore, there is an urgent need for an efficient selective sensor for detecting TNP.4 The current detection methods for organic analytes include Raman spectroscopy, cyclic voltammetry, gas chromatography, mass spectrometry, ion mobility spectrometry, electrochemical sensing, photoluminescence (PL) spectroscopy and others.5 Among these reported methods, PL-based methods have received significant attention due to their relative simplicity, rapid response time, high selectivity, high sensitivity and low cost.6
A variety of PL materials including dual-emission quantum dot hybrids,7 microporous metal–organic frameworks (MOFs),8 conjugated polymers (CPs)9 and fluorescent film sensors,10 have been used to detect of nitro-aromatics. A drawback of this strategy is that due to aggregation-caused quenching (ACQ) the emission intensity decreases when the concentration of the luminophores increases.11 However, Tang's group pioneered a class of compounds which show almost no luminescence in solution and a dramatic enhancement of emission in the aggregated state, named aggregation-induced emission (AIE).12 The similar phenomenon of AIE enhancement (AIEE) was subsequently reported by Park et al.13 Fluorescence-based sensors often suffer from small Stokes shifts and photobleaching effects.14 On this account, the development of phosphorescent iridium(III) complexes as chemosensors represents a breakthrough.15 This is because of the excellent photophysical properties of Ir(III) complexes such as long emissive lifetimes, large Stokes shifts, high quantum yields, and easy tuning of emission wavelength compared to the fluorescent analogues.16 Furthermore, most Ir(III) complexes are thermally and chemically stable and are strongly emissive at room temperature. Ir(III) complexes have high sensitivity and selectivity for the detection of nitro-aromatic explosives and have relatively low detection limits compared to reported fluorescent AIE materials.17 Thus, by combining the desirable features of AIE and phosphorescence, the development of new AIE- or AIEE-active Ir(III)-based sensors is a promising solution for contemporary challenges for today's society.18
AIE occurs due to the restriction of intramolecular motions (RIM) in the aggregated state, thereby blocking the nonradiative pathways and opening up a radiative channel.19 We have previously reported an iridium complex ((ppy)2Ir(oz)) with 2-(2′-hydroxyphenyl)-2-oxazoline (Hoz) as an ancillary ligand, shown in Fig. S1a (ESI†).20 The crystal packing of (ppy)2Ir(oz) has relatively weak C–H⋯π (2.77–2.86 Å) intermolecular interactions which result in an effective pathway for nonradiative decay in the crystalline state (Fig. S1b, ESI†) and (ppy)2Ir(oz) shows strong ACQ (Fig. S5, ESI†). We reasoned that the introduction of electron-withdrawing substituents, such as fluorine, chlorine or a trifluoromethyl group, into the cyclometalated ligands should enhance intermolecular interactions and thereby restrict the molecular motions in the aggregated state and hence allow AIE to occur, instead of ACQ.
Up to now, there are very few studies on the detection of explosives with AIE-active Ir(III) complexes.21 Laskar et al. reported that two Ir(III) complexes detect TNP by a mechanism which combines electron and energy transfer.22 Mukherjee et al. observed that the high acidity of TNP23 caused luminescent MOFs to decompose, making detection of TNP unreliable, whereas other nitro-aromatics were successfully sensed by MOFs.24
We now report that the strongly acidic TNP interacts with the new Ir(III) complexes 1–4 (Fig. 1) and effectively inhibits their AIEE properties. This leads to the desired highly selective and sensitive phosphorescent detection of TNP in aqueous media.
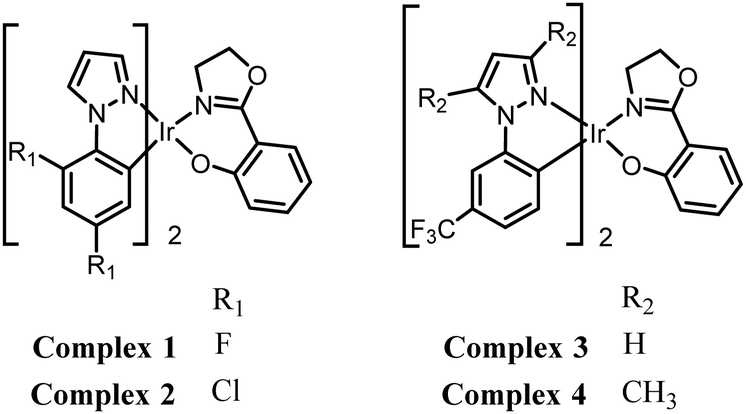 | ||
| Fig. 1 Chemical structures of the complexes 1–4. | ||
Complexes 1–4 possess good solubility in acetonitrile, and the solutions are weakly emissive. A gradual enhancement of PL intensity is observed with increasing the water fraction in the acetonitrile–water mixture, indicating that complex 1 is AIEE active (Fig. 2). Transmission electron microscopy and electron diffraction experiments indicated that crystalline aggregates are formed in the mixtures with a water ratio of 90% (Fig. S6, ESI†). The phosphorescent quantum yield of complex 1 in the solid state is 30% and this relatively strong emission can be attributed to the presence of the fluorine atoms, as the comparable value for the non-fluorinated analogue is only 4%. The packing in the X-ray crystal structure of 1 shows several short C–H⋯F intermolecular interactions with distances of 2.49–2.50 Å (Fig. 3). These contacts can effectively restrict the intramolecular motion of complex 1, thus blocking the nonradiative pathways in the solid state. The complexes 2–4 also show an AIEE effect (Fig. S7–S9, ESI†) and they exhibit short intermolecular contacts, which are π⋯π (complex 2) or C–H⋯F (complexes 3–4) with distances of 3.55 Å (π⋯π) or 2.74 Å (C–H⋯F) or 2.55–2.79 Å (C–H⋯F) in the crystalline state (Fig. S2–S4, ESI†). These interactions could also limit intramolecular motions. The phosphorescence quantum yields of 2–4 in the solid state are 18%, 14% and 8%, respectively.
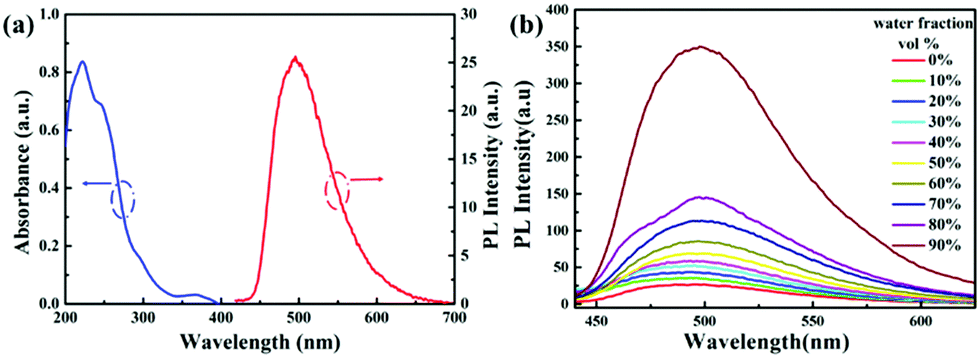 | ||
| Fig. 2 (a) UV-Vis absorption and emission spectra of complex 1 (10 μM) in acetonitrile solution at room temperature. (b) Emission spectra of complex 1 (10 μM) in acetonitrile–water mixtures with different water fractions (0–90% v/v) at room temperature. | ||
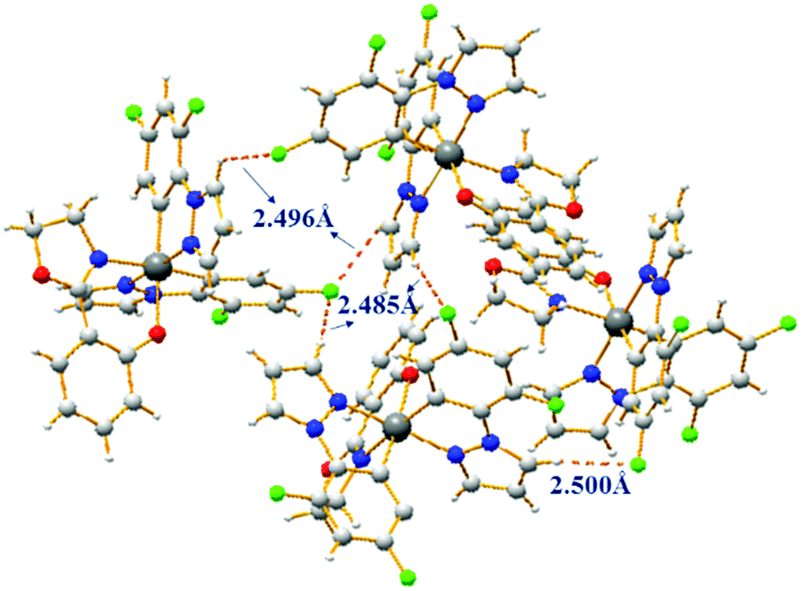 | ||
| Fig. 3 Molecular packing of complex 1 in the crystal with short intermolecular contacts shown as dashed lines. | ||
Complexes 1–4 effectively prevent emission quenching in aqueous media by virtue of their AIEE properties. The phosphorescence quantum yields of 1–4 in acetonitrile–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 v/v) solution are 21%, 15%, 11% and 10%, respectively. Therefore, TNP detection was investigated. As shown in Fig. 4a, complex 1 displays strong phosphorescence at around 500 nm in acetonitrile–water (1:9 v/v) (10 μM) solution without TNP, and a rapid quenching of emission is visible when the concentration of TNP is increased. The quenching is clearly identified when 0.5 ppm of TNP is added and 99% of the emission is quenched when the TNP concentration reached 5 ppm. Similarly, the phosphorescence of complexes 2–4 is also quenched by TNP in acetonitrile–water (1:9 v/v) (10 μM) solutions, with quenching efficiencies of 95%, 97% and 94%, respectively (Fig. S10–S12, ESI†). The emission of 1–4 is immediately quenched when TNP is added to the solutions, indicating that TNP rapidly interacts with the complexes. This facilitates the effective “real-time” detection of TNP.
:9 v/v) solution are 21%, 15%, 11% and 10%, respectively. Therefore, TNP detection was investigated. As shown in Fig. 4a, complex 1 displays strong phosphorescence at around 500 nm in acetonitrile–water (1:9 v/v) (10 μM) solution without TNP, and a rapid quenching of emission is visible when the concentration of TNP is increased. The quenching is clearly identified when 0.5 ppm of TNP is added and 99% of the emission is quenched when the TNP concentration reached 5 ppm. Similarly, the phosphorescence of complexes 2–4 is also quenched by TNP in acetonitrile–water (1:9 v/v) (10 μM) solutions, with quenching efficiencies of 95%, 97% and 94%, respectively (Fig. S10–S12, ESI†). The emission of 1–4 is immediately quenched when TNP is added to the solutions, indicating that TNP rapidly interacts with the complexes. This facilitates the effective “real-time” detection of TNP.
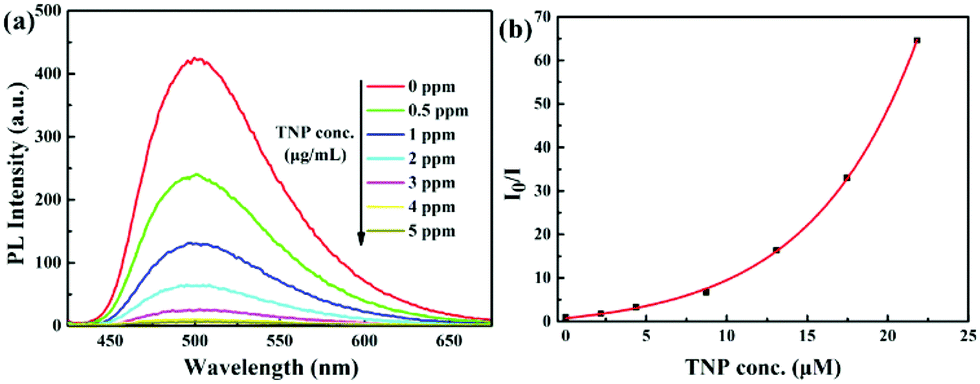 | ||
| Fig. 4 (a) PL spectra of complex 1 (10 μM) in acetonitrile–water (v/v = 1:9) containing different amounts of TNP. (b) Corresponding Stern–Volmer plot of TNP. | ||
The Stern–Volmer (S–V) plot for complex 1 is nonlinear and shows that higher concentrations of TNP lead to a higher efficiency of emission quenching (Fig. 4b). The quenching constant is 1.50 × 105 M−1 for complex 1, which is relatively high compared to the reported Ir(III) complexes.21a,22,25 The S–V curves for complexes 2–4 are also nonlinear, and the quenching constants are 0.32 × 105 M−1, 1.66 × 105 M−1 and 0.86 × 105 M−1, respectively (Fig. S10–S12 and discussion in ESI†). The detection limits for complexes 1–4 as sensors for TNP are calculated to be 0.23, 0.15, 1.05 and 1.65 μM, respectively.26
The emission response of complex 1 to different nitro-aromatic explosives, namely 2,4,6-trinitrotoluene (TNT), 3-nitrotoluene (mNT), nitrobenzene (NB), 2,6-dinitrotoluene (2,6-DNT), 2,4-dinitrotoluene (2,4-DNT), 2-nitrotoluene (oNT) and 1,3-dinitrobenzene (1,3-DNB), established high selectivity for TNP (Fig. 5a). Compared with TNP, other nitro-aromatics have little effect under the same conditions. In addition, the response of different concentrations of nitro-aromatics to the emission quenching of complex 1 was also evaluated (Fig. S13, ESI†). It is observed that with the increase in concentration of the nitro-aromatic species, TNP clearly enhances the quenching efficiency, whereas the others showed no distinct change. It is worth noting that the emission intensity is significantly quenched after the addition of TNP (5 ppm) into the systems containing other nitro-aromatics (Fig. 5b), indicating that the other compounds have little effect on the detection of TNP by complex 1. These results demonstrate the excellent selectivity of complex 1 for TNP. Considering the strong acidity of TNP, we also investigated the response of complex 1 to TNP in CH3CN/aqueous HEPES buffer (1 mM, pH 7.3; 1:4, v/v) solution (Fig. S14, ESI†). The data show that complex 1 has relatively good sensitivity and selectivity for the detection of TNP, indicating that TNP interacts with complex 1 even in HEPES buffer solution. As depicted in Fig. S15–S17 (ESI†), similar phenomena are also observed with complexes 2–4, which are also suitable for selective detection of TNP.
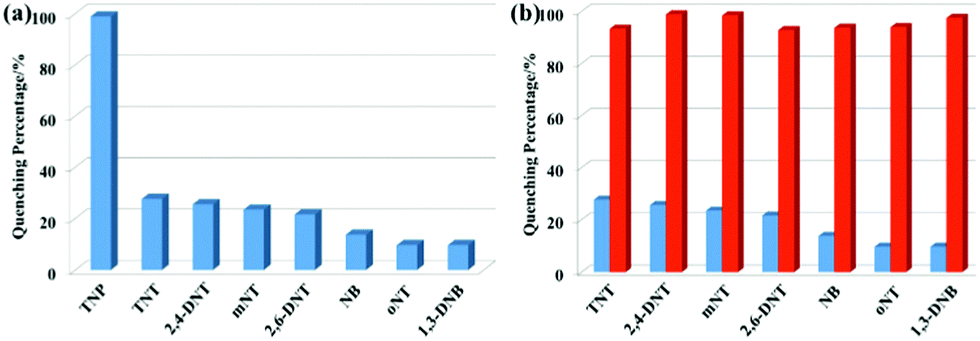 | ||
| Fig. 5 (a) Quenching percentage obtained for different analytes (5 ppm). (b) Quenching percentage of complex 1 (10 μM) with analytes (5 ppm) in acetonitrile–water (v/v = 1:9) mixtures before (blue) and after (red) the addition of 5 ppm TNP. | ||
The origin of the selectivity of complexes 1–4 towards TNP was investigated and the proposed mechanism is shown for complex 1 in Scheme 1. Spectroscopic evidence to support this mechanism is presented in the ESI.† To probe the interaction between complex 1 and TNP, a single crystal of 1·TNP suitable for X-ray analysis was obtained. An O–H⋯O interaction between complex 1 and TNP was clearly observed, and the Ir–O bond lengthened from 2.11 Å in 1 to 2.20 Å in 1·TNP (Fig. 6). This suggests that a specific binding of TNP initiates the fragmentation of complex 1 that is observed spectroscopically, as discussed in the ESI.† The proton of TNP will dissociate more easily in acetonitrile–water medium than in dichloromethane solution used for growing the crystals. We conclude, therefore, that a specific O–H⋯O interaction between TNP and the complexes 1–4 plays a significant role in the sensing system. Under the same conditions, the emission intensities of complexes 1–4 after addition of 5 ppm of 2,4-dinitrophenol (2,4-DNP) are quenched much less compared to TNP (Fig. S27, ESI†). This is due to the increased acidity of 2,4-DNP (pKa = 4.11) and TNP (pKa = 0.38) in aqueous media.27 This accounts for the selective detection of TNP.
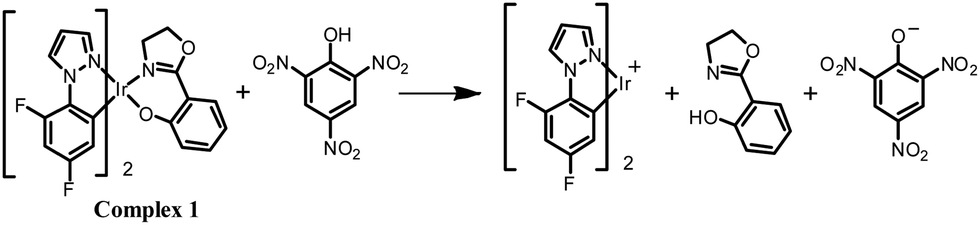 | ||
| Scheme 1 A proposed mechanism for the reaction of complex 1 with TNP. | ||
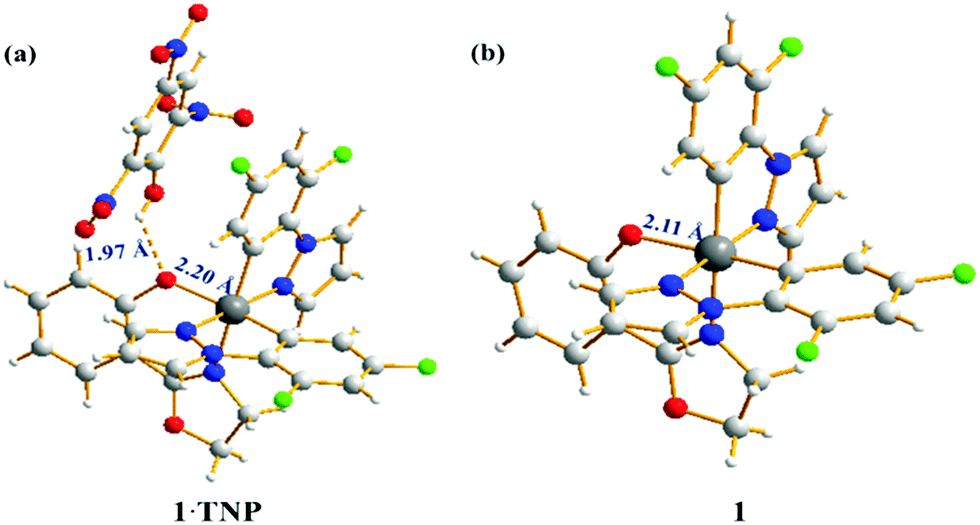 | ||
| Fig. 6 Crystal structure of complex 1·TNP (a) and complex 1 (b). Both single crystals were obtained by slow evaporation of dichloromethane. | ||
Other nitro-aromatic compounds weakly decrease the emission intensity of 1–4. This may be due to the fact that the nitro compounds are electron deficient, and if the lowest unoccupied molecular orbital (LUMO) energies of the complexes are higher than those of the analytes, then photoinduced electron transfer (PET) can occur from complexes to analytes, leading to the emission intensity decrease.21a To confirm this hypothesis, the structures of complexes 1–4 were optimized by referring to the X-ray diffraction data, and their highest occupied molecular orbital (HOMO) and LUMO energy levels in solution were calculated by DFT methods. The higher energy LUMOs of the complexes facilitate the transfer of electrons to the lower LUMOs of the nitro-aromatics, establishing that PET is a lausible mechanism for slightly decreasing the complexes’ emission in response to a range of nitro-aromatic compounds (Fig. S28, ESI†). The PET process may also occur between TNP and the complexes 1–4, but TNP also leads to the decomplexation of the Hoz ligand in aqueous media; and this is the main mechanism for the highly selective sensing of TNP.
To demonstrate a practical application of complex 1 as a sensor for TNP, filter papers that emit strong phosphorescence under 365 nm UV illumination were prepared by their immersion in an acetonitrile solution of complex 1 and then drying at room temperature in air. It can be seen clearly with the naked eye that the phosphorescent paper was stained by adding a drop of a solution of TNP (5 ppm) in acetonitrile–water (1:9 v/v) (Fig. 7). For control experiments, adding solutions of other nitro-aromatic compounds onto the phosphorescent papers led to no obvious change. These results clearly illustrate that complex 1 is suitable for immediate visualization of traces of TNP by a very simple practical method that should be applicable for rapid on-site testing.
 | ||
| Fig. 7 Luminescent photographs of filter papers impregnated with complex 1 against 5 ppm of different nitro-aromatic compounds. All photographs were taken under 365 nm UV illumination. The scale bar is 5 mm. | ||
In conclusion, four new AIEE-active iridium(III) complexes detect TNP selectively over a range of other nitro-aromatic compounds with high efficiency in aqueous medium. The complexes’ emission is quenched by TNP due to a specific O–H⋯O interaction between the TNP and the ancillary ligand of the complexes, which results in the liberation of the Hoz unit. This explains the high selectivity for TNP and is a conceptually different mechanism compared with previous nitro-aromatic sensors. The combined selectivity and sensitivity of complexes 1–4 (especially 1) means they strongly compete with other luminescent materials6,22,24 as viable sensors for detecting TNP. In addition, a test paper demonstrated the naked-eye detection of trace amounts of TNP.
The work was funded by NSFC (No. 51473028), the key scientific and technological project of Jilin province (20150204011GX, 20160307016GX), the development and reform commission of Jilin province (20160058). Work in Durham was funded by EPSRC grant EP/L02621X/1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543 RSC; (b) Y. Salinas, R. M. Máñez, M. D. Marcos, F. Sancenón, A. M. Costero, M. Parraad and S. Gil, Chem. Soc. Rev., 2012, 41, 1261 RSC; (c) K. S. Asha, G. S. Vaisakhan and S. Mandal, Nanoscale, 2016, 8, 11782 RSC.
- (a) E. G. Kayser and N. E. Burlinson, J. Energ. Mater., 1988, 6, 45 CrossRef CAS; (b) J.-F. Xiong, J.-X. Li, G.-Z. Mo, J.-P. Huo, J.-Y. Liu, X.-Y. Chen and Z.-Y. Wang, J. Org. Chem., 2014, 79, 11619 CrossRef CAS PubMed.
- D. Dinda, A. Gupta, B. K. Shaw, S. Sadhu and S. K. Saha, ACS Appl. Mater. Interfaces, 2014, 6, 10722 CAS.
- T.-P. Huynh, A. Wojnarowicz, A. Kelm, P. Woznicki, P. Borowicz, A. Majka, F. D’Souza and W. Kutner, ACS Sens., 2016, 1, 636 CrossRef CAS.
- (a) S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871 RSC; (b) I. A. Popov, H. Chen, O. N. Kharybin, E. N. Nikolaev and R. G. Cooks, Chem. Commun., 2005, 1953 CAS.
- (a) Y. Peng, A.-J. Zhang, M. Dong and Y.-W. Wang, Chem. Commun., 2011, 47, 4505 RSC; (b) M. Dong, Y.-W. Wang, A.-J. Zhang and Y. Peng, Chem. – Asian J., 2013, 8, 1321 CrossRef CAS PubMed; (c) Y. Xu, B. Li, W. Li, J. Zhao, S. Sun and Y. Pang, Chem. Commun., 2013, 49, 4764 RSC; (d) S. Kaur, A. Gupta, V. Bhalla and M. Kumar, J. Mater. Chem. C, 2014, 2, 7356 RSC; (e) V. Bhalla, H. Arora, H. Singh and M. Kumar, Dalton Trans., 2013, 42, 969 RSC; (f) M. Kumar, S. I. Reja and V. Bhalla, Org. Lett., 2012, 14, 6084 CrossRef CAS PubMed; (g) D. Li, J. Liu, R. T. K. Kwok, Z. Liang, B. Z. Tang and J. Yu, Chem. Commun., 2012, 48, 7167 RSC; (h) Z.-H. Fu, Y.-W. Wang and Y. Peng, Chem. Commun., 2017, 53, 10524 RSC.
- E. R. Goldman, I. L. Medintz, J. L. Whitley, A. Hayhurst, A. R. Clapp, H. T. Uyeda, J. R. Deschamps, M. E. Lassman and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 6744 CrossRef CAS PubMed.
- (a) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153 CrossRef CAS PubMed; (b) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881 CrossRef CAS PubMed.
- (a) M. D. Woodka and V. P. Schnee, Anal. Chem., 2010, 82, 9917 CrossRef CAS PubMed; (b) S. Kazunori and T. M. Swager, Bull. Chem. Soc. Jpn., 2007, 80, 2074 CrossRef.
- G. He, G. Zhang, F. Lü and Y. Fang, Chem. Mater., 2009, 21, 1494 CrossRef CAS.
- (a) R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS; (b) K.-Y. Pu and B. Liu, Adv. Funct. Mater., 2009, 19, 277 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740 RSC.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed.
- (a) M. Yu, Q. Zhao, L. Shi, F. Li, Z. Zhou, H. Yang, T. Yi and C. Huang, Chem. Commun., 2008, 2115 RSC; (b) Y. Tao, M. Li, J. Ren and X. Qu, Chem. Soc. Rev., 2015, 44, 8636 RSC; (c) S. Lin, W. Gao, Z. Tian, C. Yang, L. Lu, J.-L. Mergny, C.-H. Leung and D.-L. Ma, Chem. Sci., 2015, 6, 4284 RSC.
- (a) P. Alam, G. Kaur, C. Climent, S. Pasha, D. Casanova, P. Alemany, A. R. Choudhury and I. R. Laskar, Dalton Trans., 2014, 43, 16431 RSC; (b) W. Wang, Z. Mao, M. Wang, L. J. Liu, D. W. Kwong, C. H. Leung and D. L. Ma, Chem. Commun., 2016, 52, 3611 RSC.
- (a) Y. Chi and P. T. Chou, Chem. Soc. Rev., 2010, 39, 638 RSC; (b) Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC; (c) C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418 CrossRef CAS; (d) S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2985 CrossRef CAS.
- L.-L. Wen, X.-G. Hou, G.-G. Shan, W.-L. Song, S.-R. Zhang, H.-Z. Sun and Z.-M. Su, J. Mater. Chem. C, 2017, 5, 10847 RSC.
- (a) G. Li, W. Guan, S. Du, D. Zhu, G. Shan, X. Zhu, L. Yan, Z. Su, M. R. Bryce and A. P. Monkman, Chem. Commun., 2015, 51, 16924 RSC; (b) Z. Mao, M. Wang, J. Liu, L. J. Liu, S. M. Lee, C. H. Leung and D. L. Ma, Chem. Commun., 2016, 52, 4450 RSC; (c) Y. Han, H.-T. Cao, H.-Z. Sun, G.-G. Shan, Y. Wu, Z.-M. Su and Y. Liao, J. Mater. Chem. C, 2015, 3, 2341 RSC.
- (a) J. Zhou, Z. Chang, Y. Jiang, B. He, M. Du, P. Lu, Y. Hong, H. S. Kwok, A. Qin, H. Qiu, Z. Zhao and B. Z. Tang, Chem. Commun., 2013, 49, 2491 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (c) G. Li, Y. Wu, G. Shan, W. Che, D. Zhu, B. Song, L. Yan, Z. Su and M. R. Bryce, Chem. Commun., 2014, 50, 6977 RSC.
- K. Chao, K. Shao, T. Peng, D. Zhu, Y. Wang, Y. Liu, Z. Su and M. R. Bryce, J. Mater. Chem. C, 2013, 1, 6800 RSC.
- (a) X.-G. Hou, Y. Wu, H.-T. Cao, H.-Z. Sun, H.-B. Li, G.-G. Shan and Z.-M. Su, Chem. Commun., 2014, 50, 6031 RSC; (b) K. S. Bejoymohandas, T. M. George, S. Bhattacharya, S. Natarajan and M. L. P. Reddy, J. Mater. Chem. C, 2014, 2, 515 RSC; (c) T. Fei, K. Jiang and T. Zhang, Sens. Actuators, B, 2014, 199, 148 CrossRef CAS.
- P. Alam, G. Kaur, V. Kachwal, A. Gupta, A. R. Choudhury and I. R. Laskar, J. Mater. Chem. C, 2015, 3, 5450 RSC.
- S. S. Nagarkar, A. V. Desai and S. K. Ghosh, CrystEngComm, 2016, 18, 2994 RSC.
- B. Gole, A. K. Bar and P. S. Mukherjee, Chem. – Eur. J., 2014, 20, 13321 CrossRef CAS PubMed.
- Y. Cui, L.-L. Wen, G.-G. Shan, H.-Z. Sun, H.-T. Mao, M. Zhang and Z.-M. Su, Sens. Actuators, B, 2017, 244, 314 CrossRef CAS.
- J. Xu, Y. Zhang, H. Yu, X. Gao and S. Shao, Anal. Chem., 2016, 88, 1455 CrossRef CAS PubMed.
- N. Dey, S. K. Samanta and S. Bhattacharya, ACS Appl. Mater. Interfaces, 2013, 5, 8394 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR spectra, 19F NMR spectra, mass spectra, absorption and emission spectra and procedures for the DFT calculations. CCDC 1573144–1573148. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc08832a |
| This journal is © The Royal Society of Chemistry 2018 |