
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aromaticity switching via azulene transformations in azulene-bridged A,D-dithiahexaphyrin†
Michał J.
Białek
 and
Lechosław
Latos-Grażyński
*
and
Lechosław
Latos-Grażyński
*
Department of Chemistry, University of Wrocław, F. Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: lechoslaw.latos-grazynski@chem.uni.wroc.pl; Web: http://llg.chem.uni.wroc.pl/
First published on 19th December 2017
Abstract
The incorporation of an azulene bridge into an aromatic hexaphyrin framework allows manipulating π-electron delocalization pathways. The palladium(II) complex undergoes hydroxyl-triggered azulene contraction or isomerization to an oxynaphthalene unit, transforming the hexaphyrin framework into meso-linked carbaporphyrins. This converts the 26π-electron pathway into the 18π one.
The specific conformational and electronic flexibilities of porphyrinoids are regarded as essential factors in the design of their macrocyclic frames.1–4 The stabilization of appropriate conformations of flexible expanded porphyrins, e.g. hexaphyrins(1.1.1.1.1.1), can be achieved by the incorporation of an appropriate internal linker that binds two opposite meso positions, eventually favouring a flattened macrocyclic framework with a well-defined π-conjugation.5–9
The choice of the linker strongly influences the electronic properties of the macrocycle. It has been established that meso-bridging units participate differently in π-electron delocalization, thus promoting 18π- or 26π-electron macrocyclic aromaticity as reflected by significantly altered spectroscopic and photophysical properties.5–9
In our strategy, the hexaphyrin scaffold was initially bridged with a hydrocarbon exhibiting a dipolar character, i.e. azulene, potentially expanding the π-electron delocalization framework in the third dimension. In addition, such a modification allows for peculiar azulene coordination and eventually for appropriately triggered, profound azulene transformations.10–12
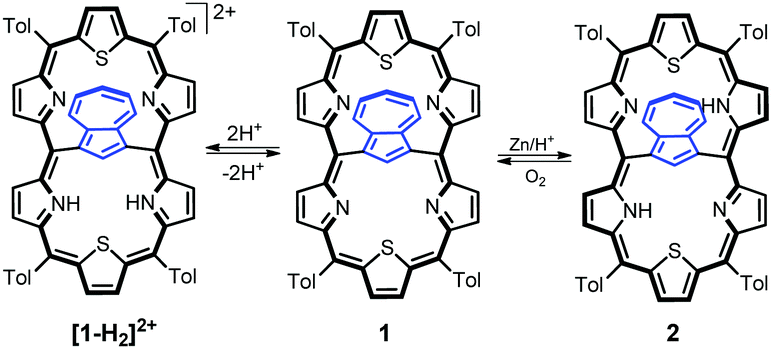
Azulene-1,3-diyl-strapped A,D-dithia[26]hexaphyrin 1 was obtained through a simple modification of the [3 + 1 + 3] approach (Scheme 1) described previously for thiophene-2,5-diyl-strapped core-modified meso-aryl hexaphyrins5,7 and for 1,3-phenylene-,6 thiophene-2,5-diyl- or pyrrole-2,5-diyl-strapped8 hexaphyrins. 1 was isolated in 31% yield. The alternative 23-thiaazuliporphyrin product could be detected if the 16-thiatripyrrane excess was insufficient, analogically to the already known synthesis of 10,15-dimesitil-23-thiaazuliporphyrin.13
 | ||
| Scheme 1 Synthesis of the azulene-1,3-diyl-strapped A,D-dithia[26]hexaphyrin. | ||
1 adopts a folded conformation, as determined by an X-ray diffraction study (Fig. 1). The dihedral angle between the C4meso(I) (atoms C5, C10, C25, C30) and C4meso(II) (atoms C10, C15, C20, C25) planes equals 16.14(5)°.
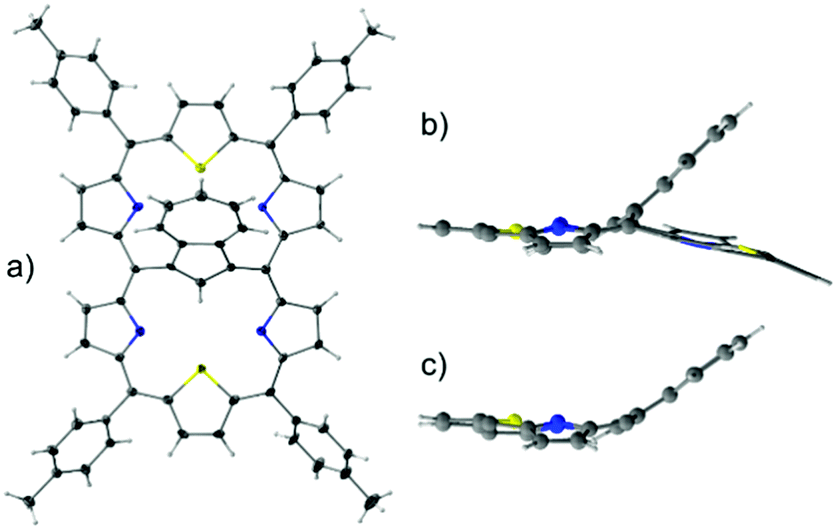 | ||
| Fig. 1 The molecular structure of 1: (a) perspective view and (b) side view, and (c) for comparison the side projection of meso-tetra(p-tolyl)-23-thiaazuliporphyrin10 is shown. | ||
The azulene ring is bent towards one of the porphyrin-like cavities as reflected by the dihedral angle of 39.60(6)° between the C10Az (atoms C37 to C46) and C4meso(I) planes. This angle is, in fact, close to the one observed for meso-tetra(p-tolyl)-23-thiaazuliporphyrin (33.18(6)°; Fig. 1b and c).10
When dissolved in chlorinated solvents, 1 forms a violet solution. The electronic spectrum of 1 shows intensive bands at 455 and 565 nm, and less intensive bands at shorter wavelengths (Fig. 2). The spectrum is supplemented by NIR bands at 1038 nm.
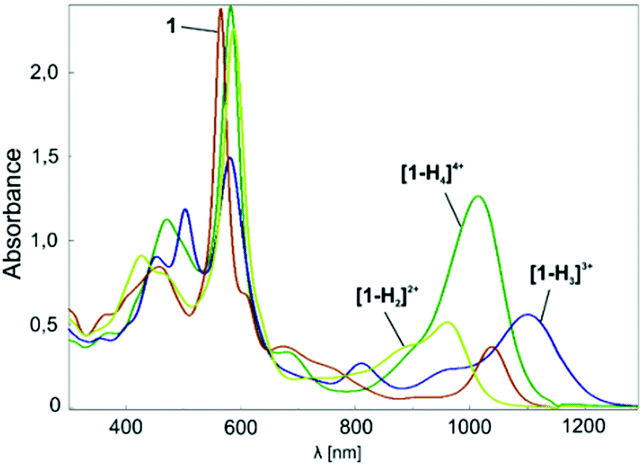 | ||
| Fig. 2 Electronic spectra of 1 in chloroform obtained in the course of titration with TFA at 293 K. | ||
The 1H NMR spectrum of 1 is consistent with the macrocyclic structure as constructed from a 31,34-dithiahexaphyrin frame symmetrically strapped by the azulene-1,3-diyl unit (Fig. 3a). The effective high symmetry of 1 (C2v) inferred from 1H NMR implies a fast conformational rearrangement in the whole investigated temperature range (190–300 K).
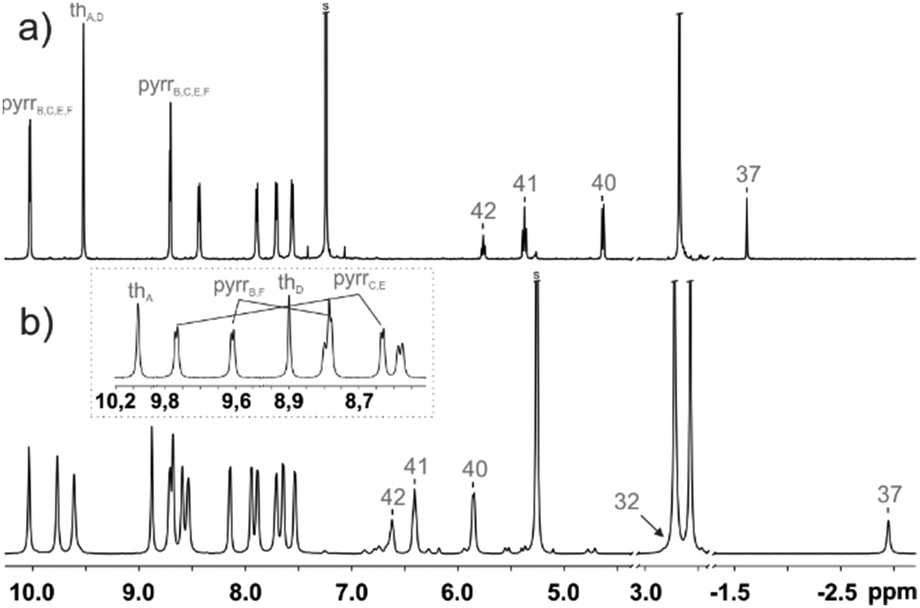 | ||
| Fig. 3 1H NMR spectra of (a) 1 (600 MHz, CDCl3, 250 K) and (b) [1-H2]2+ (600 MHz, CD2Cl2, 190 K). The inset in trace (b) shows the assignment of two pyrrole and thiophene subsets. | ||
The equilibrium involves two energetically identical conformers 1-L and 1-R (Scheme S1, ESI†), corresponding to the molecular structure determined in the solid state, solely differentiated by the orientation of the canted azulene.
The 1H chemical shifts of the thiophene and pyrrole hydrogens of 1 are in the characteristic range determined for aromatic hexaphyrinoids.7,8 The bridging azulene fragment is exposed to a strong macrocyclic ring current effect as reflected by the upfield relocation of all azulene resonances.
Upon addition of TFA, the violet solution of 1 turns blue-green. The titration of 1 with TFA observed using electron spectroscopy is presented in Fig. 2. This yielded three stages of protonation: the dicationic [1-H2]2+, tricationic [1-H3]3+, and tetracationic [1-H4]4+ species.
The addition of two protons removes the equivalency of the two porphyrin-like pockets (Scheme 2). The spectroscopic pattern is unambiguously consistent with the (N32–H, N33, N35, N36–H) distribution, in which two protons are located in one cavity (Fig. 3b). Presumably they are stabilized by the hydrogen bonding to a TFA anion (Fig. S12, ESI†),14 in spite of enforced steric hindrance.
 | ||
| Scheme 2 Diprotonation of 1 and its two-electron reduction to 2. | ||
The aromaticity of [1-H2]2+o is consistent with the nucleus-independent chemical shifts (NICS) calculated for two cavities at the centres of the 16-membered rings.15 These values are in the range expected for diatropic porphyrinoids, yet for [1-H2]2+o the NICS index in cavity II increases by 5.6 ppm compared to 1 (Table S3, ESI†).
The chemical reduction of 1 in a dichloromethane solution with zinc powder in the presence of gaseous HCl afforded azulene-1,3-diyl-strapped A,D-dithia[28]hexaphyrin 2 (Scheme 2). The spectroscopic features of 2 clearly confirm the generation of a highly antiaromatic 28π-electron macrocycle as demonstrated by the downfield chemical shifts of the inner NHs (18.26 ppm) and azulene H37 hydrogen (16.37 ppm) (Fig. S1a, ESI†).
The insertion of palladium(II) into 1 (Scheme 3) afforded the monopalladium(II) complex 1-Pd+. The 1H NMR spectrum of 1-Pd+ has somehow been broadened, yet clearly shows the preserved aromaticity of the macrocycle, with the missing H37 signal confirming metal–carbon bond formation (Fig. 4). The geometry of the coordinated subunit in DFT-optimized 1-Pd+o (Fig. S14, ESI†) resembles that of palladium(II) 23-thiaazuliporphyrin.10 The thiophene ring is bent out of the N32–C37–N36 carbaporphyrin core plane, while the two pyrrole rings and azulene are only slightly tipped away from that plane.
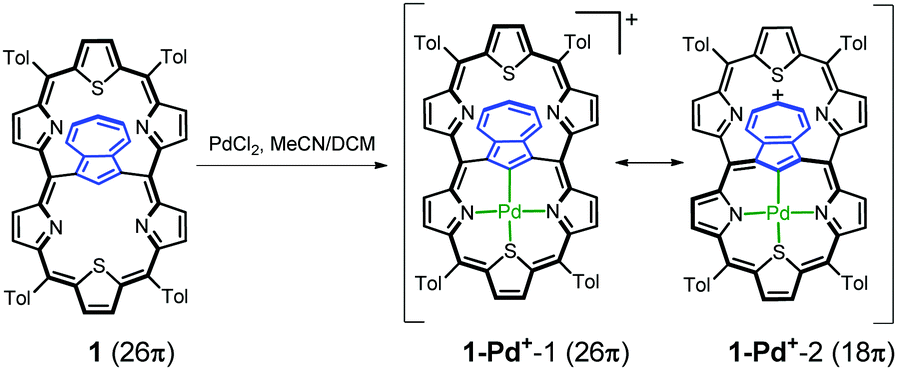 | ||
| Scheme 3 Insertion of palladium(II) into 1. | ||
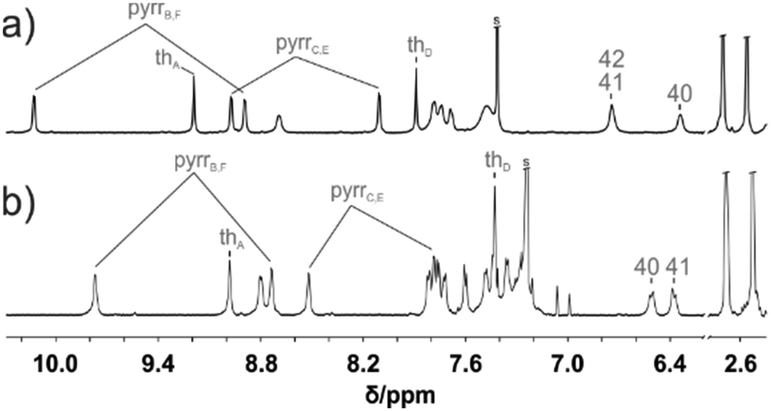 | ||
| Fig. 4 1H NMR spectra (600 MHz, CDCl3, 250 K) of (a) 1-Pd+ and (b) 3-Pd. | ||
The NICS(0) value in the centre of unoccupied cavity II equals −7.2 ppm (Table S3, ESI†). Thus, it is significantly higher than that in 1, yet still within the range expected for aromatic macrocycles. The electrostatic potential map (Fig. S13, ESI†) shows the enrichment of the 7-membered ring in a positive charge consistent with the porphyrin-like aromaticity of the coordinated subunit of 1-Pd+. A relationship between the accumulation of positive charges on the 7-membered ring and the preference of an 18π- over a 26π-electron delocalization pathway seems to be noticeable.
In azuliporphyrinoid chemistry this accumulation makes the tropylium ring vulnerable to nucleophilic attacks. Under oxidative conditions this resulted in ring contraction, leading, in the case of azuliporphyrins, to fully conjugated benzocarbaporphyrins.10,11,16–19 A stable cationic species in the form of 1-Pd+ was a suitable reagent for testing the possibility of contraction in the case of an expanded porphyrinoid.
The insertion of palladium(II) into 1 in DMF, instead of DCM/MeCN, allowed for the identification of a palladium(II) complex with an indene-1,3-diyl bridge (3-Pd) and inseparable traces of two derivatives formylated on the indene ring, 3-Pda and 3-Pdb (Scheme 4). The transformation of 1-Pd+, whose formation should be the initial step, to 3-Pd requires the contraction of a 7-membered ring into a 6-membered one, converting the azulene bridge into an indene one.
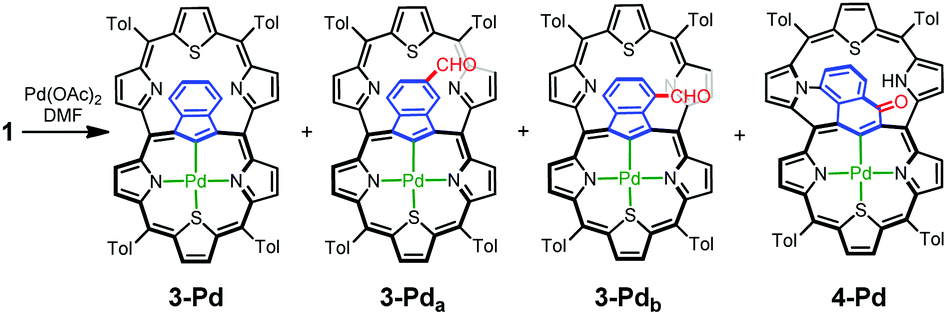 | ||
| Scheme 4 Azulene transformations leading to palladium(II) meso-linked thiacarbaporphyrins 3-Pd and 4-Pd. | ||
The 1H NMR spectrum of 3-Pd (Fig. 4b) possesses a characteristic symmetric AA′BB′ pattern of fused benzene centred at around 6.4 ppm and a deshielded thiatripyrrinic moiety (pyrrole doublets centred at 9.77 and 8.73 ppm and a thiophene singlet at 8.98 ppm), which remains contrasted with the slight aromaticity of the uncoordinated thiatripyrrinic moiety (pyrrole doublets centred at 8.52 and 7.78 ppm and a thiophene singlet at 7.43 ppm).
Another isolated product showed an unprecedented rearrangement, i.e. the formation of a fused derivative of the palladium(II) complex with an oxynaphthalene-1,3-diyl bridge (4-Pd) – the constitutional isomer of 3-Pda/b (Scheme 4). Here, the oxynaphthalene unit is additionally linked to the pyrrolic nitrogen N33.
Formally, analysing the molecular frame, this conversion resembles the isomerization of azulene to naphthalene.20 Mechanistic considerations (Scheme 5) suggest that this peculiar transformation to 4-Pd is presumably initiated by hydroxylation, for the first time, at the quaternary carbon atom of azulene instead of the tertiary ones invoked in the formation of the 3-Pd series. This is reflected by the oxygen atom position in the final structure. The initial steps do not require the presence of an external oxidant as an aromatic macrocycle allows for an intramolecular redox process in which the hexaphyrin frame is reduced, while the bridging azulene undergoes formal oxidation accompanied by the isomerization.21
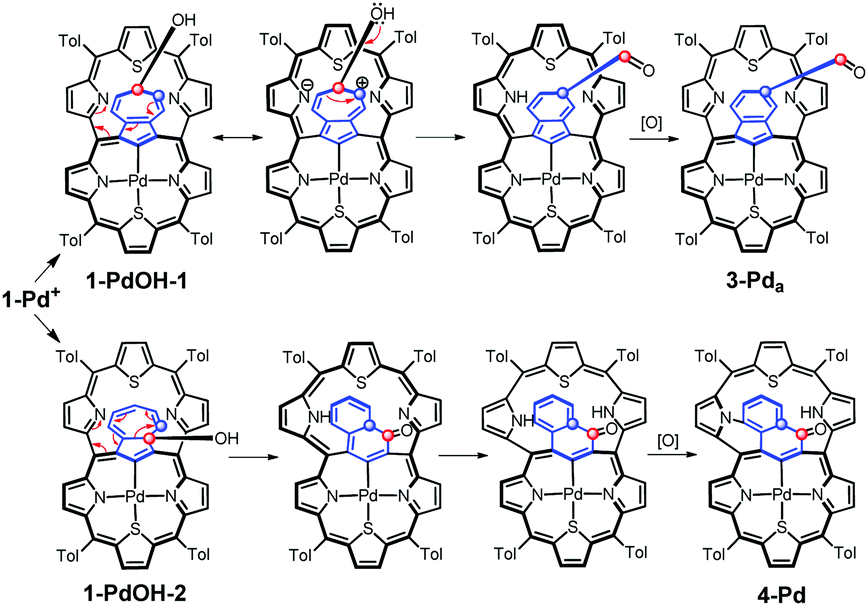 | ||
| Scheme 5 Formation of 3-Pda and 4-Pd through an intramolecular redox process. Formally, the dithiahexaphyrin is reduced while the azulene bridge is oxidized. | ||
Eventually, external oxidation affords 3-Pda/b or 4-Pd. According to DFT data, 4-Pdo is also 28.6 kcal mol−1 more stable than isomeric 3-Pda,o.
4-Pd adopts a noticeably folded conformation imposed by the incorporation of a fused oxynaphthalene moiety, as determined by the X-ray diffraction study (Fig. 5).
 | ||
| Fig. 5 The molecular structure of 4-Pd: (a) perspective view and (b) side view. | ||
The 3-Pd subunit encompassing palladium(II) contributes to the 1H NMR spectrum (Fig. 4) with a set of resonances closely resembling the one reported for aromatic palladium(II) 23-thiabenzocarbaporphyrin.10 In comparison, β-H signals of an unoccupied cavity are significantly upfield shifted.
Similarly, the palladium(II) containing subunit of 4-Pd (Fig. S2, ESI†) reflects the aromatic features of the hypothetical 24-thiaoxynaphthiporphyrin consistently with the known diatropic properties of oxynaphthiporphyrin and its silver(III) complex.22
The NICS(0) values calculated in the centres of the unoccupied cavities of 3-Pdo and 4-Pdo (Table S3, ESI†) suggest a negligible ring current effect in these molecular fragments. In classical terms, following azulene rearrangement the hexaphyrinic 26π-electron delocalization pathway of 1 was switched to the porphyrinic 18π-electron one of 3 or 4, as readily visualized through the comparison of the appropriate resonance structures in Schemes 3 and 4.
Finally, the reaction of palladium(II) with 2 afforded pink dipalladium(II) azulene-1,3-diyl-strapped A,D-dithia[28]hexaphyrin 2-Pd2. The preliminary X-ray studies confirmed the asymmetric coordination to two PdCl2 units, each to one S and N donor from both cavities located on opposite sides of the dithiahexaphyrin plane and served as the starting point for DFT optimization (Fig. 6).
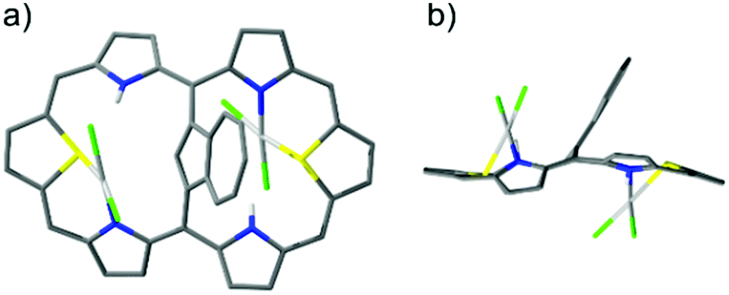 | ||
| Fig. 6 DFT-optimized model of 2-Pd2: (a) perspective view and (b) side view. | ||
The 1H NMR spectrum (Fig. S3, ESI†) shows that 2-Pd2 is asymmetric in solution and preserves the antiaromaticity of 2, which is yet diminished due to increased framework folding.
The present work reveals an intriguing Janus-like facet of porphyrinoid aromaticity visualized by internally-bridged expanded porphyrins, explored as (annuleno)annulene variants to display dual electronic circuits.4–9,23–25
Here, the dual electronic circuit transformations of azulene-1,3-diyl-strapped A,D-dithiahexaphyrin was achieved by diprotonation or coordination. The tropylium ring contraction in 1-Pd+ yielded palladium(II) indene-1,3-diyl-strapped A,D-dithiahexaphyrin 3-Pd or the fused derivative of palladium(II) oxynaphthalene-1,3-diyl-strapped A,D-dithiahexaphyrin 4-Pd, totally switching the 26π-electron so characteristic for regular hexaphyrins to the 18π-electron one of 23-thiabenzocarbaporphyrin or 24-thiaoxynaphthiporphyrin (Scheme 6).
 | ||
| Scheme 6 Rearrangement from azulene-1,3-diyl-strapped A,D-dithia[28]hexaphyrin to meso-linked thiacarbaporphyrins. | ||
Financial support from the National Science Center (Grant 2012/04/A/ST5/00593) and support provided to M. J. B. by the Foundation for Polish Science are kindly acknowledged. DFT calculations were carried out using resources provided by the Wroclaw Centre for Networking and Supercomputing, Grant 329.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584 CrossRef CAS PubMed.
- B. Szyszko, M. J. Białek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839 CrossRef CAS PubMed.
- M. Menand, M. Sollogoub, B. Boitrel and S. Le Gac, Angew. Chem., Int. Ed., 2016, 55, 297 CrossRef CAS PubMed.
- M. Stępień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288 CrossRef PubMed.
- J. M. Lim, K. Ganesan, Y. M. Sung, A. Srinivasan, T. K. Chandrashekar and D. Kim, Chem. Commun., 2014, 50, 4358 RSC.
- Y. M. Sung, J. Oh, W. Kim, H. Mori, A. Osuka and D. Kim, J. Am. Chem. Soc., 2015, 137, 11856 CrossRef CAS PubMed.
- G. Karthik, M. Sneha, V. P. Raja, J. M. Lim, D. Kim, A. Srinivasan and T. K. Chandrashekar, Chem. – Eur. J., 2013, 19, 1886 CrossRef CAS PubMed.
- H. Mori, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2013, 52, 12997 CrossRef CAS PubMed.
- M. Suzuki and A. Osuka, J. Am. Chem. Soc., 2007, 129, 464 CrossRef CAS PubMed.
- M. J. Białek and L. Latos-Grażyński, Inorg. Chem., 2016, 55, 1758 CrossRef PubMed.
- T. D. Lash, Chem. Rev., 2017, 117, 2313 CrossRef CAS PubMed.
- B. Szyszko and L. Latos-Grażyński, Chem. Soc. Rev., 2015, 44, 3588 RSC.
- S. Venkatraman, V. G. Anand, H. PrabhuRaja, H. Rath, J. Sankar, T. K. Chandrashekar, W. Teng and K. Ruhlandt-Senge, Chem. Commun., 2002, 1660 RSC.
- M. Stępień, B. Szyszko and L. Latos-Grażyński, J. Am. Chem. Soc., 2010, 132, 3140 CrossRef PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- K. Berlin, C. Steinbeck and E. Breitmaier, Synthesis, 1996, 336 CrossRef CAS.
- T. D. Lash, J. Chem. Soc., Chem. Commun., 1998, 1683 RSC.
- T. D. Lash, D. A. Colby, S. R. Graham and S. T. Chaney, J. Org. Chem., 2004, 69, 8851 CrossRef CAS PubMed.
- T. D. Lash, J. A. El-Beck and G. M. Ferrence, Org. Biomol. Chem., 2014, 12, 316 CAS.
- L. T. Scott and M. A. Kirms, J. Am. Chem. Soc., 1981, 103, 5875 CrossRef CAS.
- M. Pawlicki, D. Bykowski, L. Szterenberg and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2012, 51, 2500 CrossRef CAS PubMed.
- T. D. Lash, J. M. Rasmussen, K. M. Bergman and D. A. Colby, Org. Lett., 2004, 6, 549 CrossRef CAS PubMed.
- T. Soya, H. Mori, Y. Hong, Y. H. Koo, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2017, 56, 3232 CrossRef CAS PubMed.
- V. G. Anand, S. Saito, S. Shimizu and A. Osuka, Angew. Chem., Int. Ed., 2005, 44, 7244 CrossRef CAS PubMed.
- T. Kim, A. Ghosh, Z. Zhang, X.-S. Ke, R. Ali, V. M. Lynch, J. Jung, W. Kim, S. Lee, S. Fukuzumi, J. S. Park, J. L. Sessler, T. K. Chandrashekar and D. Kim, Nat. Chem., 2017, 9, 1243 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, additional discussion, spectra, crystallographic data, and DFT studies and coordinates. CCDC 1577493 and 1577494. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc08754c |
| This journal is © The Royal Society of Chemistry 2018 |