
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidative organocatalytic chemoselective N-acylation of heterocycles with aromatic and conjugated aldehydes†
Linda
Ta
 and
Henrik
Sundén
*
and
Henrik
Sundén
*
Chemistry and Chemical Engineering, Chalmers University of Technology, Kemivägen 10, 412 96 Göteborg, Sweden. E-mail: sundenh@chalmers.se
First published on 21st December 2017
Abstract
Selective acylation of indoles is cumbersome often involving the need for sensitive and reactive acyl chloride derivatives or coupling reagents. Here we report a mild, functional group tolerant and highly chemoselective oxidative carbene catalyzed N-acylation of indoles with aldehydes. The acylation has a broad substrate scope and is compatible with substituents on both the aldehyde and the indole reaction partner. Furthermore, aza-heterocycles such as pyrrole and indazole can also be used as nucleophiles in this reaction providing the corresponding amide congeners in good yield.
The power to selectively functionalize heterocycles with multiple reactive sites is a challenging and important task. By selective functionalization, an atom efficient and protecting group free strategy can be achieved for the preparation of complex molecules.1 In this respect, chemoselective N- or C-functionalization of indoles has been a longstanding challenge in organic synthesis.2 Selective N-acylation of indoles is of particular importance as N-acylindoles, are found in numerous biologically active compounds, such as, indomethacin,3 oxamethacin, acemetacin, L-768,2424 and have also been used as imaging agents for beta amyloid plaques (1, Scheme 1a).5 Synthetically, N-acylindoles can serve as protected carboxylate derivatives,6 and can be set up to undergo several forms of annulation reactions with the C2-carbon.7
Generally, acylation of indoles occurs at the C3-position2 and can for instance be selectively performed in the presence of a carboxylic acid derivative and a Lewis acid,8 or with the Vilsmeier–Haack reaction.9 Selective N-acylation on the other hand, is normally conducted in the presence of a reactive electrophile such as a chloride containing acylating reagent and a stoichiometric base or a carboxylic acid that needs to be activated with a coupling reagent.10 The use of inorganic bases and/or sensitive reagents can potentially have a negative impact on the functional group compatibility, thus restricting further development of these protocols. To overcome these concerns, Sarpong and co-workers showed that N-acylation of indoles could be selectively performed under mild conditions in the presence of a stoichiometric carbonylimidazole derivative.11 Moreover, Scheidt and co-workers have shown that acylated indoles can be formed through a tetrapropylammonium perruthenate-catalyzed dihydrogenative coupling of alcohols.12
In the field of organocatalysis, N-heterocyclic carbenes (NHCs) have received widespread attention as potent catalysts for a number of diverse reaction paths13 and are particularly useful in converting aldehydes into acyl donors. The key intermediate in these reactions is the acyl azolium intermediate (Scheme 1b)14 that can be generated either through an internal oxidation of the Breslow intermediate or in the presence of an external oxidant15 such as the Kharasch oxidant (2).16 The Kharasch oxidant has been used in, for instance, acylation reactions involving alcohols,17 azides,18 macrolactonizations,19 and amidations20 (Scheme 1b). With our recent interest in oxidative N-heterocyclic carbene catalysis21 we wanted to investigate if the acyl azolium would generate selectivity in the acylation of densely polyfunctionalized molecules such as indoles and other heterocycles. Thus, enabling the use of available aldehydes as mild acylating reagents. Previous studies have shown that N-functionalization of indoles is indeed possible through an intramolecular reaction cascade22 or through an imination reaction with isocyanides.23 Here we report the NHC-catalyzed oxidative, chemoselective N-acylation of indoles with aldehydes.
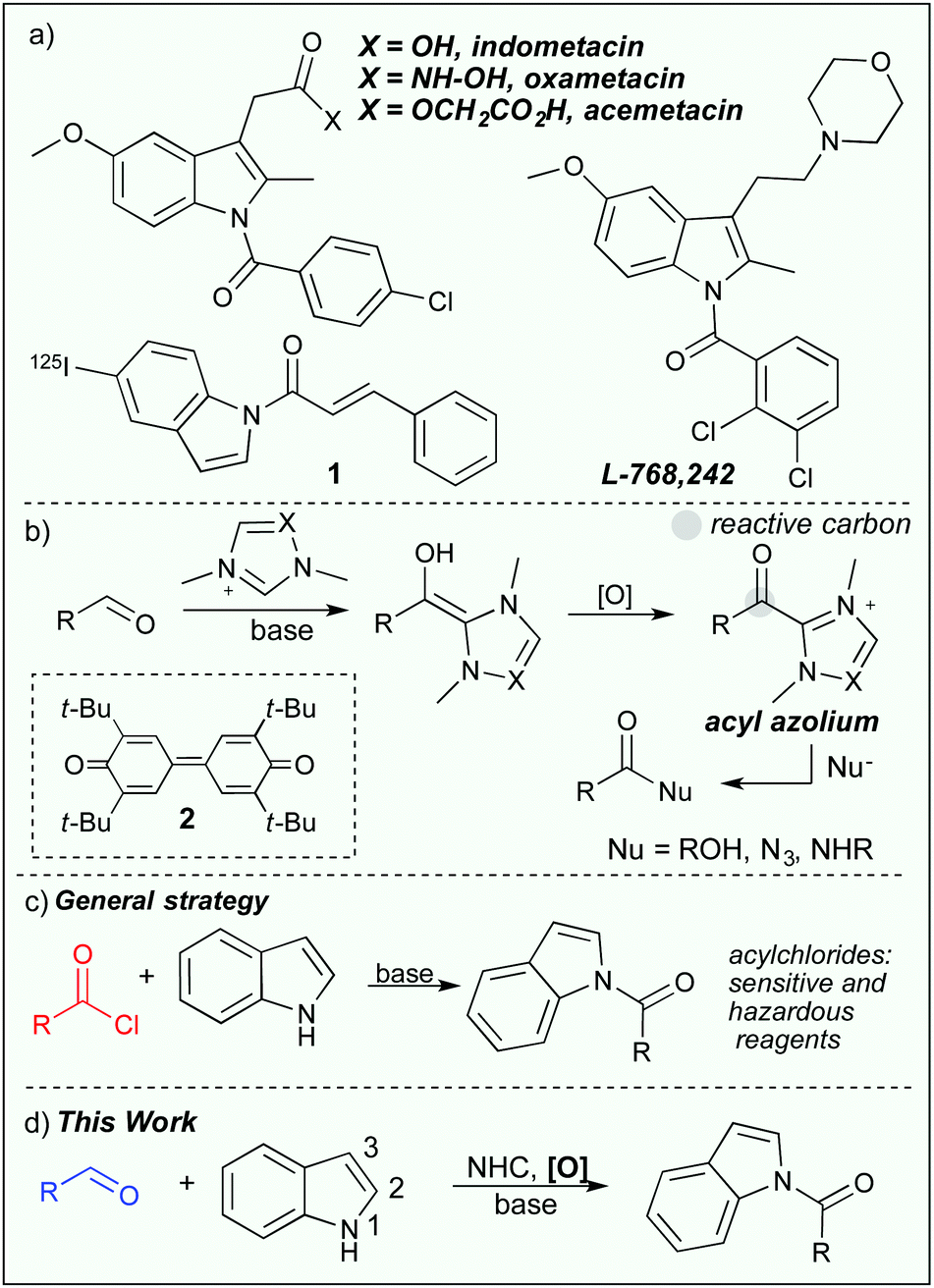 | ||
| Scheme 1 (a) Bioactive acylindoles. (b) Oxidative carbene-catalyzed acylation of alcohols and amines. (c) Carbene catalyzed oxidative acylation of indoles with aldehydes. (d) Oxidative NHC catalyzed N-acylation of indols. | ||
Our study commenced with a survey of reaction conditions where both imidazolium and triazolium salts were found to be suitable precatalysts for this reaction with 7 being the superior one in the series (Table 1, entries 1–3). Among the bases tested DBU (entry 1) was the best alternative as compared to Cs2CO3 and trimethylamine (entries 4 and 5). Dichloromethane was shown to be the best solvent for the reaction compared to acetonitrile, toluene and THF (entries 6–8). The most effective reaction conditions were found with the combination of an increased base-loading and molecular sieves (MS 4 Å) (entry 9). To conclude that the NHC has an active role as a catalyst in the reaction, a series of reactions were performed systematically excluding the NHC, base, and oxidant (entries 10 and 11). In all these cases, no product formation could be detected indicating that oxidative carbene catalysis is indeed the reaction pathway.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Precat. | Base | Additive | Yieldb |
| a 4 (1 eq.), cinnamaldehyde (2 eq.), base (0.5 eq.), precatalyst (10 mol%), solvent (1 ml) and 2 (1 eq.). b Determined by 1H NMR on the crude reaction mixture with durene as an internal standard. c 1.5 ml. d 0.8 ml. e 1 eq. f Isolated yield. g Without 2. | |||||
| 1 | DCM | 7 | DBU | — | 54 |
| 2 | DCM | 8 | DBU | — | 31 |
| 3 | DCM | 9 | DBU | — | 25 |
| 4 | DCMc | 7 | Cs2CO3 | — | 4 |
| 5 | DCMc | 7 | Et3N | — | 0 |
| 6 | MeCNd | 7 | DBUe | — | 46 |
| 7 | Toluened | 7 | DBUe | — | 36 |
| 8 | THFd | 7 | DBUe | — | 17 |
| 9 | DCMd | 7 | DBUe | MS 4 Å | 81/71f |
| 10 | DCMd | — | DBU | — | 0 |
| 11 | DCMd | — | — | — | 0 |
| 12g | DCMd | 7 | DBU | MS 4 Å | 0 |
|
|||||

With our optimal reaction conditions in hand, the reaction scope was investigated (Table 2). Different α,β-unsaturated aldehydes were generally well tolerated by the reaction delivering the α,β-unsaturated acylindoles in up to 96% yield. α,β-unsaturated acylindoles are of particular importance as they can be further functionalized in a number of metal-catalyzed reactions.7b–d Both electron donating and withdrawing substituents were compatible under our oxidative reaction conditions. For example, p-dimethylamino, o- and p-methoxy cinnamaldehyde delivers the corresponding acylindole in 70–96% yield, respectively (compounds 10, 11 and 12). Halogen substituted cinnamaldehydes are less efficient in promoting the reaction and require longer reaction times as compared to the electron rich aldehydes (compounds 13 and 14). Benzaldehydes with substituents on all positions also work as an acylating agent in this reaction delivering the benzoylated indoles in 63–97% yield (compound 16–19).24 Furthermore, aliphatic conjugated aldehydes are also compatible with our reaction conditions and 20 is isolated in 73% yield.25
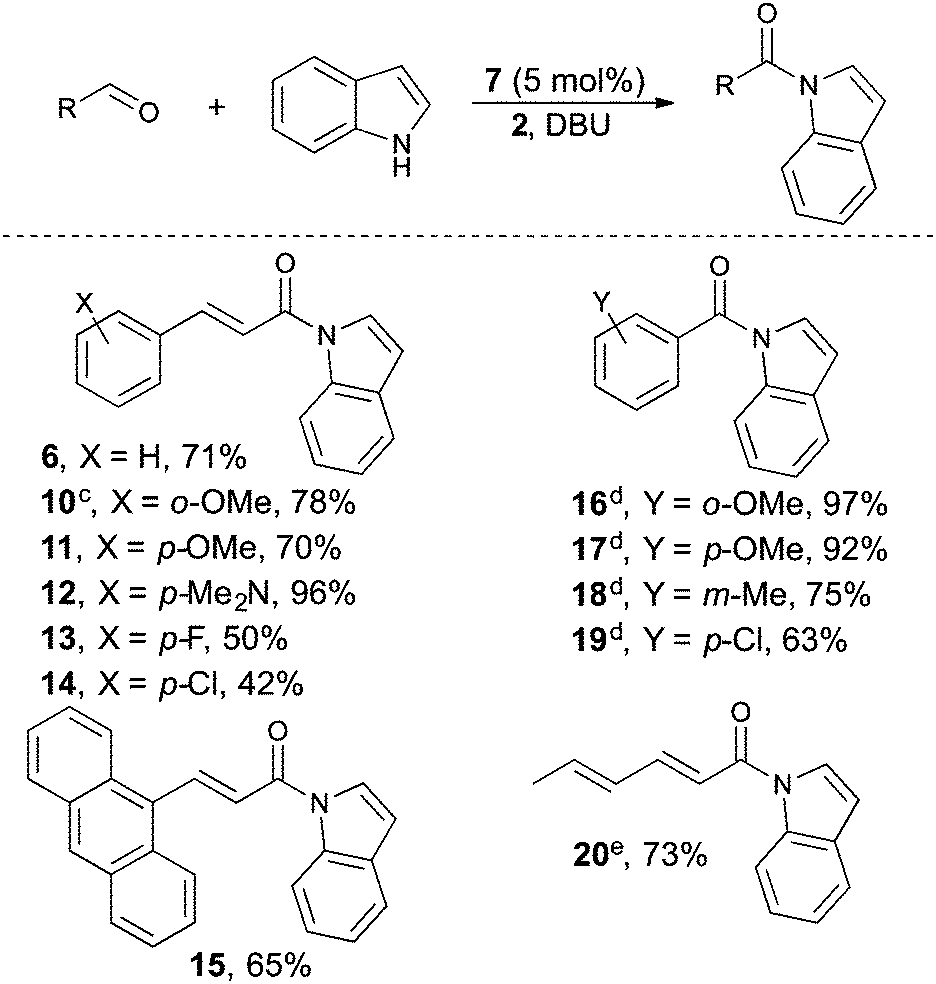
Next the indole reaction partner was investigated (Table 3). Generally, the reactions work well for indoles incorporating a high degree of different functional groups. For instance, halogens are tolerated in positions 4, 5 and 6 and the acylated indole derivatives can be isolated in good to excellent yields (53–90% compounds 21–31). Electron donating groups such as methyl and methoxy give good reactivity and compounds 22, 24, 27, 23 and 31 can be isolated in 56–90% yield.26 Moreover, tryptamine analogues incorporating functional groups such as CN and a tertiary amine are also tolerated by the reaction and compound 29 and 30 can be isolated in 56% and 75% yield, respectively. In all cases the reactions exhibited a high degree of chemoselectivity toward N-acetylation, and the C3-acylated compound was never detected. In control experiments with the N-site protected using 1-methylindole no conversion of starting materials could be observed. Other nitrogen heterocycles such as pyrrole and indazole, afforded the products in good to excellent yields (32 and 33). For instance, 6-aminoindazole can be selectively acylated in the presence of an unprotected primary amine to give 33 in 69% yield.

To improve the E-factor of the reaction, aerial oxygen was investigated as the terminal oxidant (Scheme 2).
 | ||
| Scheme 2 NHC catalyzed oxidative aerial acylation of 4. FePc = iron phthalocyanine. | ||
In brief, the oxidation was accomplished with a system of electron transfer mediators (FePc and 2) and oxygen and efficiently replaces the stoichiometric use of 2 providing 12 in 90% yield.21d,f–h
The acyl indoles are good synthetic building blocks and can readily undergo further transformations (Scheme 3). For example, amide 17 can be converted to yield the corresponding benzophenone 34 by a Pd-catalyzed Suzuki–Miyaura reaction, in 53% yield. This is a rare example of a Pd-catalyzed C–N bond activation of an indole-based amide and combined with our oxidative amidation offers a rapid entry to a metal catalyzed coupling of aldehydes.27
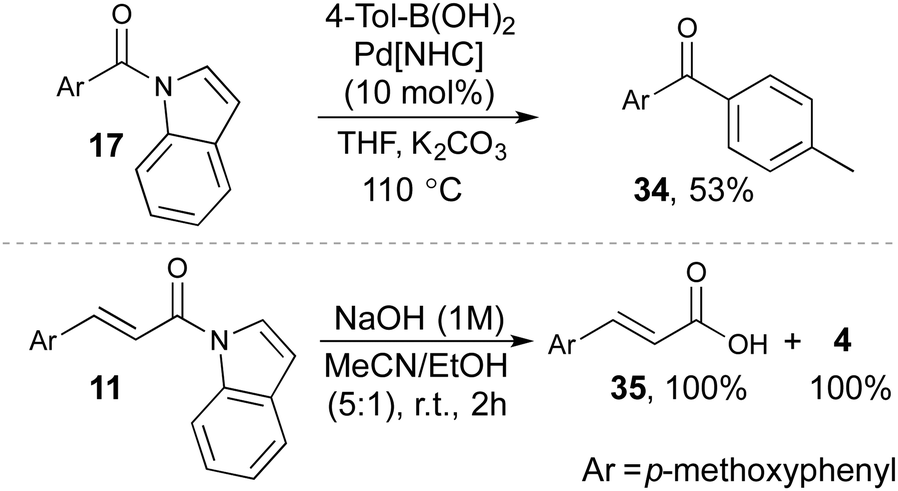 | ||
| Scheme 3 Synthetic transformations involving acylindoles. For details; see, ESI.† Pd[NHC] = [1,3-Bis(2,6-diisopropylphenyl)imidazol-2-ylidene]chloro[3-phenylallyl]palladium(II). | ||
Furthermore, the C–N bond in compound 11 smoothly undergoes hydrolysis in the presence of NaOH to yield carboxylic acid 35 and indole in quantitative yields. This transformation underpins the importance of the acylindole as a prominent protecting group for both carboxylic acids and indoles.
The proposed catalytic cycle starts with base promoted formation of carbene I. Carbene I adds to the aldehyde forming Breslow intermediate II that is oxidized by 2 to form the acyl azolium (IV). In the next step the deprotonated indole adds to the carbonyl carbon of intermediate IV forming the acylated indole V and regenerating catalyst I (Scheme 4).
 | ||
| Scheme 4 Proposed catalytic cycle. | ||
In conclusion, a chemoselective synthesis of synthetically valuable N-acylated indoles has been developed using oxidative NHC-catalysis at ambient conditions. This reaction offers acylindoles in good to excellent yields with a wide substrate scope. Electron donating and electron withdrawing substituents on both aldehyde and on the indole, are tolerated. Our oxidative NHC-catalyzed reaction also works with other heteroaromatic compounds such as pyrrole and indazole. The oxidative acylation of N-heterocycles with aldehydes is a promising alternative to the harsh reaction conditions that normally accompany these forms of transformations.
Funding from the Swedish Research Council (VR and Formas) is gratefully acknowledged. Furthermore, we thank Magnus Bergvalls stiftelse for supporting the running costs of this project.
Conflicts of interest
There are no conflicts to declareNotes and references
- (a) R. W. Hoffmann, Synthesis, 2006, 3531–3541 CrossRef CAS; (b) P. S. Baran, T. J. Maimone and J. M. Richter, Nature, 2007, 446, 404–408 CrossRef CAS PubMed; (c) I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193–205 CrossRef CAS PubMed.
- R. J. Sundberg, The chemistry of indoles, ed. R. J. Sundberg, Academic Press, New York and London, vol. 18, pp. 401–430 Search PubMed.
- T. Y. Shen, T. B. Windholz, A. Rosegay, B. E. Witzel, A. N. Wilson, J. D. Willett, W. J. Holtz, R. L. Ellis, A. R. Matzuk, S. Lucas, C. H. Stammer, F. W. Holly, L. H. Sarett, E. A. Risley, G. W. Nuss and C. A. Winter, J. Am. Chem. Soc., 1963, 85, 488–489 CrossRef CAS.
- M. Gallant, C. Dufresne, Y. Gareau, D. Guay, Y. Leblanc, P. Prasit, C. Rochette, N. Sawyer, D. M. Slipetz, N. Tremblay, K. M. Metters and M. Labelle, Bioorg. Med. Chem. Lett., 1996, 6, 2263–2268 CrossRef CAS.
- Y. Yang, X.-H. Duan, J.-Y. Deng, B. Jin, H.-M. Jia and B.-L. Liu, Bioorg. Med. Chem. Lett., 2011, 21, 5594–5597 CrossRef CAS PubMed.
- (a) M. J. V. de Oliveira Baptista, A. G. M. Barrett, D. H. R. Barton, M. Girijavallabhan, R. C. Jennings, J. Kelly, V. J. Papadimitriou, J. V. Turner and N. A. Usher, J. Chem. Soc., Perkin Trans. 1, 1977, 1477–1500, 10.1039/P19770001477,; (b) P. Linda, A. Stener, A. Cipiciani and G. Savelli, J. Heterocycl. Chem., 1983, 20, 247–248 CrossRef CAS; (c) A. G. M. Barrett and D. Dhanak, Tetrahedron Lett., 1987, 28, 3327–3330 CrossRef CAS; (d) E. Arai, H. Tokuyama, M. S. Linsell and T. Fukuyama, Tetrahedron Lett., 1998, 39, 71–74 CrossRef CAS; (e) J. Isaacson, M. Loo and Y. Kobayashi, Org. Lett., 2008, 10, 1461–1463 CrossRef CAS PubMed.
- (a) T. A. Dwight, N. R. Rue, D. Charyk, R. Josselyn and B. DeBoef, Org. Lett., 2007, 9, 3137–3139 CrossRef CAS PubMed; (b) D. V. Patil, M. A. Cavitt and S. France, Org. Lett., 2011, 13, 5820–5823 CrossRef CAS PubMed; (c) S. R. Kandukuri, J. A. Schiffner and M. Oestreich, Angew. Chem., Int. Ed., 2012, 51, 1265–1269 CrossRef CAS PubMed; (d) J. Liu, S. Zhao, W. Song, R. Li, X. Guo, K. Zhuo and Y. Yue, Adv. Synth. Catal., 2017, 359, 609–615 CrossRef CAS; (e) J. Xu, X. Yu and Q. Song, Org. Lett., 2017, 19, 980–983 CrossRef CAS PubMed.
- (a) T. Okauchi, M. Itonaga, T. Minami, T. Owa, K. Kitoh and H. Yoshino, Org. Lett., 2000, 2, 1485–1487 CrossRef CAS PubMed; (b) O. Ottoni, A. d. V. F. Neder, A. K. B. Dias, R. P. A. Cruz and L. B. Aquino, Org. Lett., 2001, 3, 1005–1007 CAS.
- G. F. Smith, J. Chem. Soc., 1954, 3842–3846 RSC.
- For selected examples of N-acyalation of idoles, see: (a) W. J. Welstead, H. F. Stauffer and L. F. Sancilio, J. Med. Chem., 1974, 17, 544–547 CrossRef CAS PubMed; (b) V. O. Illi, Synthesis, 1979, 387–388 CrossRef CAS; (c) Y. Kikugawa, Synthesis, 1981, 460–461 CrossRef CAS; (d) O. Ottoni, R. Cruz and R. Alves, Tetrahedron, 1998, 54, 13915–13928 CrossRef CAS; (e) D. S. Dhanoa, S. W. Bagley, R. S. L. Chang, V. J. Lotti, T. B. Chen, S. D. Kivlighn, G. J. Zingaro, P. K. S. Siegl, A. A. Patchett and W. J. Greenlee, J. Med. Chem., 1993, 36, 4230–4238 CrossRef CAS PubMed; (f) J. B. Bremner, S. Samosorn and J. I. Ambrus, Synthesis, 2004, 2653–2658 CrossRef CAS; (g) A. Umehara, H. Ueda and H. Tokuyama, J. Org. Chem., 2016, 81, 11444–11453 CrossRef CAS PubMed.
- S. T. Heller, E. E. Schultz and R. Sarpong, Angew. Chem., Int. Ed., 2012, 51, 8304 CrossRef CAS PubMed.
- B. E. Maki and K. A. Scheidt, Org. Lett., 2009, 11, 1651–1654 CrossRef CAS PubMed.
- (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (b) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS PubMed; (c) Stereoselective Organocatalysis: Bond Formation Methodologies and Activation Modes, ed. R. R. Torres, John Wiley & Sons, Inc., 2013 Search PubMed; (d) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- (a) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696–707 CrossRef CAS PubMed; (b) C. Zhang, J. F. Hooper and D. W. Lupton, ACS Catal., 2017, 7, 2583–2596 CrossRef CAS.
- (a) C. E. I. Knappke, A. Imami and A. J. von Wangelin, ChemCatChem, 2012, 4, 937–941 CrossRef CAS; (b) S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem. – Eur. J., 2013, 19, 4664–4678 CrossRef CAS PubMed.
- M. S. Kharasch and B. S. Joshi, J. Org. Chem., 1957, 22, 1439–1443 CrossRef CAS.
- (a) S. De Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef CAS PubMed; (b) S. De Sarkar, A. Biswas, C. H. Song and A. Studer, Synthesis, 2011, 1974–1983 CAS; (c) R. C. Samanta, S. De Sarkar, R. Froehlich, S. Grimme and A. Studer, Chem. Sci., 2013, 4, 2177–2184 RSC; (d) M. T. Berry, D. Castrejon and J. E. Hein, Org. Lett., 2014, 16, 3676–3679 CrossRef CAS PubMed.
- S. De Sarkar and A. Studer, Org. Lett., 2010, 12, 1992–1995 CrossRef CAS PubMed.
- K. Lee, H. Kim and J. Hong, Angew. Chem., Int. Ed., 2012, 51, 5735–5738 CrossRef CAS PubMed.
- (a) C. A. Gondo and J. W. Bode, Synlett, 2013, 1205–1210 CAS; (b) A. Porey, S. Santra and J. Guin, Asian J. Org. Chem., 2016, 5, 870–873 CrossRef CAS; (c) C. Zheng, X. Liu and C. Ma, J. Org. Chem., 2017, 82, 6940–6945 CrossRef CAS PubMed; (d) S. Premaletha, A. Ghosh, S. Joseph, S. R. Yetra and A. T. Biju, Chem. Commun., 2017, 53, 1478–1481 RSC.
- (a) L. Ta, A. Axelsson, J. Bijl, M. Haukka and H. Sundén, Chem. – Eur. J., 2014, 20, 13889–13893 CrossRef CAS PubMed; (b) A. Axelsson, L. Ta and H. Sundén, Catalysts, 2015, 5, 2052 CrossRef CAS; (c) H. Sundén, L. Ta and A. Axelsson, J. Vis. Exp., 2015, 105, e53213, DOI:10.3791/53213; (d) A. Axelsson, E. Hammarvid, L. Ta and H. Sunden, Chem. Commun., 2016, 52, 11571–11574 RSC; (e) A. Axelsson, L. Ta and H. Sundén, Eur. J. Org. Chem., 2016, 3339–3343 CrossRef CAS; (f) L. Ta, A. Axelsson and H. Sundén, Green. Chem., 2016, 18, 686–690 RSC; (g) A. Axelsson, A. Antoine-Michard and H. Sunden, Green. Chem., 2017, 19, 2477–2481 RSC; (h) A. Axelsson, L. Ta and H. Sundén, Synlett, 2017, 873–878 CAS.
- Y.-J. Yang, Y. Ji, L. Qi, G. Wang and X.-P. Hui, Org. Lett., 2017, 19, 3271–3274 CrossRef CAS PubMed.
- J. Kim and S. H. Hong, Org. Lett., 2017, 19, 3259–3262 CrossRef CAS PubMed.
- Nitro-substituted aldehydes reacts slowly and the reaction shuts down after 10–20% conversion.
- Aliphatic aldehydes are unreactive under these reaction conditions.
- The 2- and 7-position on the indole are troublesome and both 2-methylindole, 7-methyl indole and 7-cloroindole fail to promote the reaction. This is probably a steric effect that arises when the indole is attacking the acyl azolium (see, catalytic cycle).
- Szostack and co-workers have shown that N-acylpyrroles and pyrazoles are prominent reactions partners in the synthesis of biaryl ketones under similar reaction conditions: G. Meng, R. Szostak and M. Szostak, Org. Lett., 2017, 19, 3596–3599 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and 1H NMR, 13C NMR and 19F NMR data. See DOI: 10.1039/c7cc08672e |
| This journal is © The Royal Society of Chemistry 2018 |