
Practical synthesis of pharmaceutically relevant molecules enriched in sp3 character†
Peter S.
Campbell
a,
Craig
Jamieson
 *a,
Iain
Simpson
b and
Allan J. B.
Watson
a
*a,
Iain
Simpson
b and
Allan J. B.
Watson
a
aDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: craig.jamieson@strath.ac.uk
bMedicinal Chemistry, Oncology IMED Biotech Unit, AstraZeneca, Darwin Building, Unit 310 Cambridge Science Park, Cambridge, CB10 4EW, UK
First published on 27th November 2017
Abstract
The expedient synthesis of compounds enriched in sp3 character is key goal in modern drug discovery. Herein, we report how a single pot Suzuki–Miyaura-hydrogenation can be used to furnish lead and fragment-like products in good to excellent yields. The approach has been successfully applied in formats amenable to parallel synthesis, in an asymmetric sense, and in the preparation of molecules with annotated biological activity.
An increasingly important consideration in modern medicinal chemistry is control of physicochemical properties with the aspi-ration of improving overall quality of candidate drugs.1 In relation to this high-level objective, there has been significant focus on an increased degree of saturation (i.e. fewer aromatic rings) in compound design as this has been linked to increased probability of improved physical properties, such as high solubility.2 Greater sp3 character bestows three-dimensionality, which allows molecules to sample a wider area of chemical space and potentially achieve superior selectivity.2,3 In addition, a higher degree of saturation is believed to impart an increased likelihood of clinical success.3 The overarching design principles discussed above become apparent when considering molecules emerging from medicinal chemistry laboratories which have fewer aromatic rings and are frequently typified by C-aryl linked saturated heterocycles represented generally by 1 (Fig. 1). These efforts have furnished a range of compounds of this general chemotype, and are associated with broad biological activity, as illustrated by the exemplar compounds (2–4) shown in Fig. 1(a).4
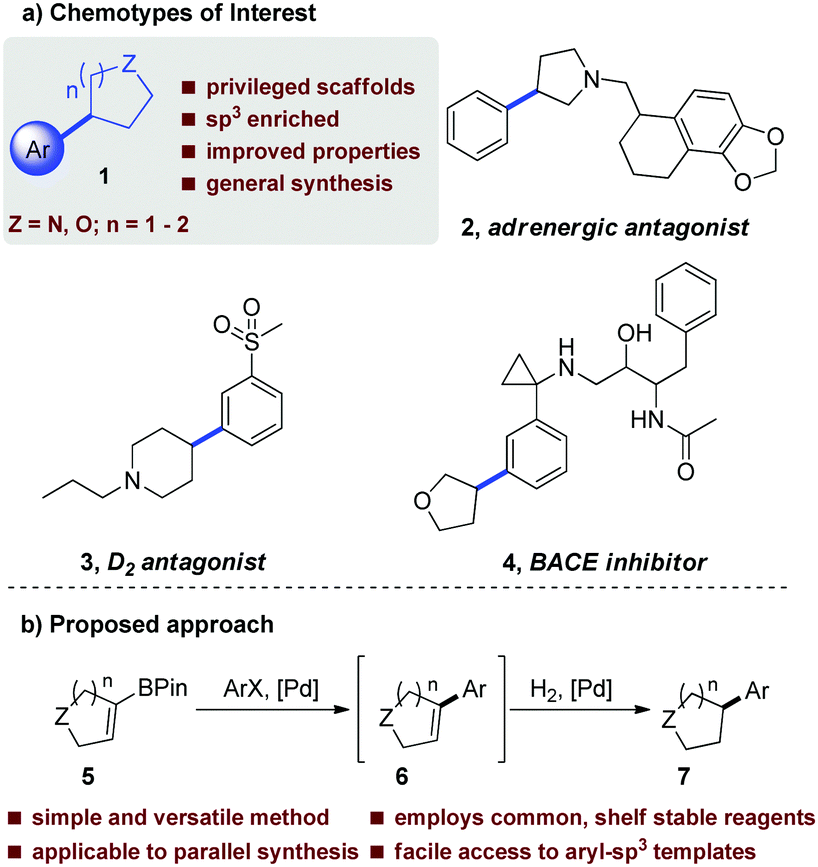 | ||
| Fig. 1 (a) Biologically active molecules exemplifying the motif of interest with key sp2–sp3 bond indicated. (b) Proposed current study. | ||
Accordingly, efficient methods for the synthesis of molecules from this emerging chemotype are currently being actively pursued, and typically involve a metal-catalysed cross-coupling reaction of an organometallic reagent (boron, zinc, tin or magnesium)5–8 with a suitable aryl halide derivative to form the sp2–sp3 bond indicated in Fig. 1. Despite these efforts, some of the currently available methods suffer from a number of drawbacks. For example, alkylboron reagents are prone to protodeboronation and β-hydride elimination.9 Additionally, secondary alkylboron reagents have a propensity to isomerize during cross coupling, often resulting in inseparable mixtures of products.9 Taken as a whole, these issues often preclude the application of many of the current methods to synthesize target molecules in a high throughput fashion.
Since our aim was to furnish biologically relevant frameworks with emphasis on 3D character in a manner which was amenable to a medicinal chemistry setting, an important objective at the outset of our study was that the methodology was sufficiently robust to be applicable diverse range of templates. Currently, there is no general consensus on the optimal catalyst/ligand combinations that should be utilized in these transformations, with cross-coupling protocols often requiring a significant degree of optimisation. Another practical consideration in a medicinal chemistry setting is the ability to synthesize target compounds in a high-throughput manner, meaning that ideally the chemistry selected should be sufficiently robust to be amenable to array format, which may preclude application of more sensitive reagent and catalyst systems.
With these issues in mind, we aimed to develop a robust, one-pot Suzuki–Miyaura (SM)-hydrogenation protocol10 to enable facile access to molecules of the general chemotype shown in Fig. 1(b), employing common laboratory reagents, and avoiding the use of alkylboron species. From a practical perspective, it was envisaged that following Suzuki–Miyaura cross coupling, introduction of a suitable hydrogen source could then enable reduction of the newly formed olefin 6in situ, furnishing the desired sp2–sp3 linked system, 7. Our initial efforts were focused on optimizing the SM component of the reaction, followed by investigation into the hydrogenation step; however, it was found that the SM conditions employed had a significant impact on the extent of hydrogenation. The most important factor was the choice of phosphine ligand and the ratio of the two palladium catalysts. Upon initial screening, excellent SM yields were achieved with minimal optimization. Attention then turned to the incorporation of an in situ hydrogenation protocol using a Pd/C co-catalyst system as use of the SM catalyst system alone was not competent in promoting hydrogenation. Our investigation commenced with evacuation of the vessel and subsequent addition of a hydrogen source following the initial cross-coupling. This was accomplished using hydrogen gas, with a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the two catalysts. Through variation of the palladium catalyst to the monodentate PdXPhosG2, and slightly increasing the Pd/C loading, the full one-pot process could be achieved with under transfer hydrogenation (Table 1, entry 6), thus rendering the transformation amenable to array chemistry (vide infra).
:1 ratio of the two catalysts. Through variation of the palladium catalyst to the monodentate PdXPhosG2, and slightly increasing the Pd/C loading, the full one-pot process could be achieved with under transfer hydrogenation (Table 1, entry 6), thus rendering the transformation amenable to array chemistry (vide infra).
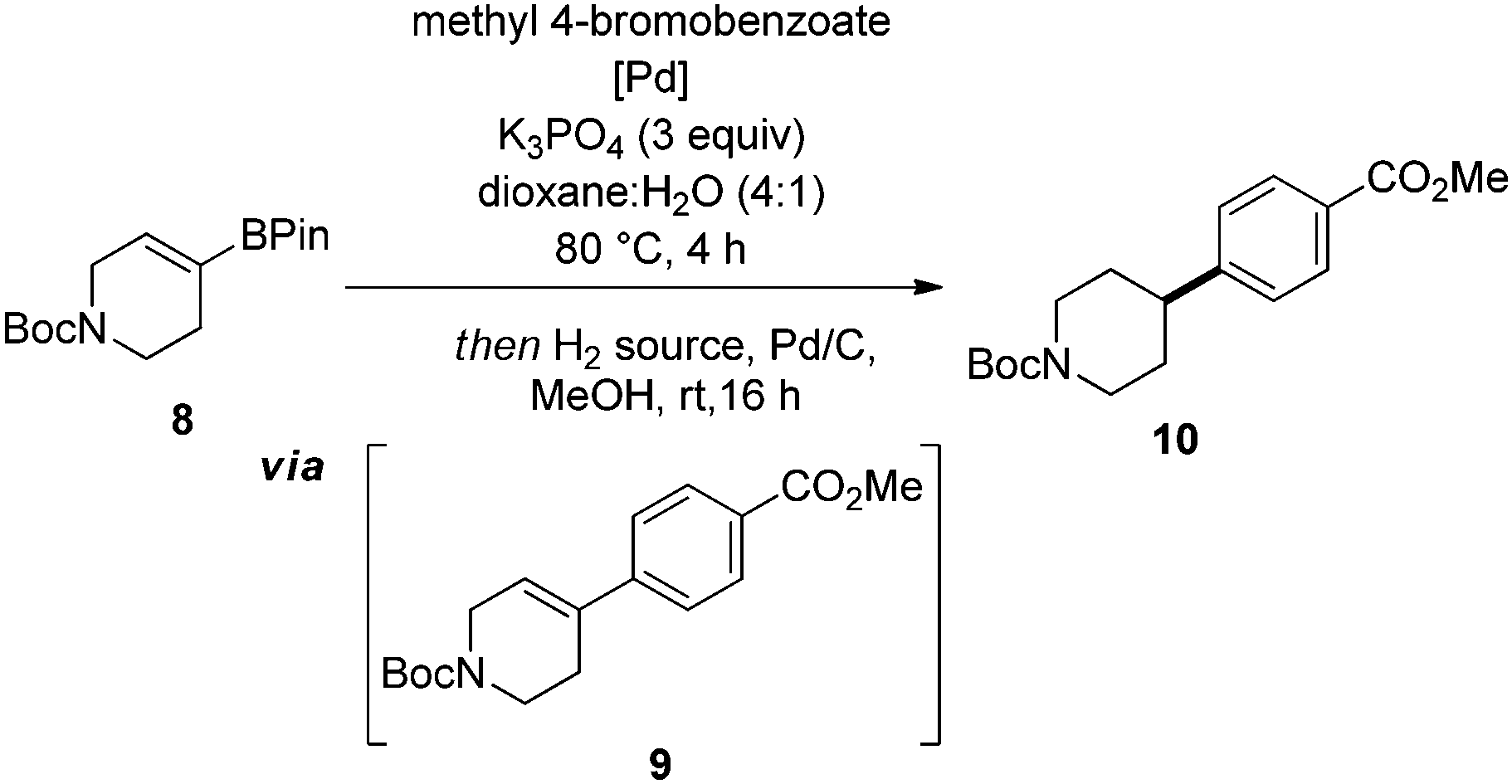
Having identified optimal conditions, we next sought to explore the general applicability of this nascent procedure. In the first instance, the scope was examined with respect to the aryl halide coupling partner, using the vinyl-piperidine derived system (8) as the boronate ester species (Scheme 1). Methyl 4-bromobenzoate coupled in excellent yield. Electron-neutral, electron-rich, and electron-poor aryl halides were all well tolerated, with ortho, meta, and para substitution patterns examined. The reaction manifold was also compatible with heterocyclic substrates which are highly attractive templates from a medicinal chemistry perspective as they are highly lead-like in terms of their physicochemical properties.1,2 In another aspect of this part of the study, we examined whether reducible groups (e.g., nitrobenzene derivatives) or protecting groups labile to hydrogenolysis could be concomitantly transformed using the newly developed procedure, although this may represent a potential limitation of the method if these are required to be retained during subsequent synthetic manipulation.
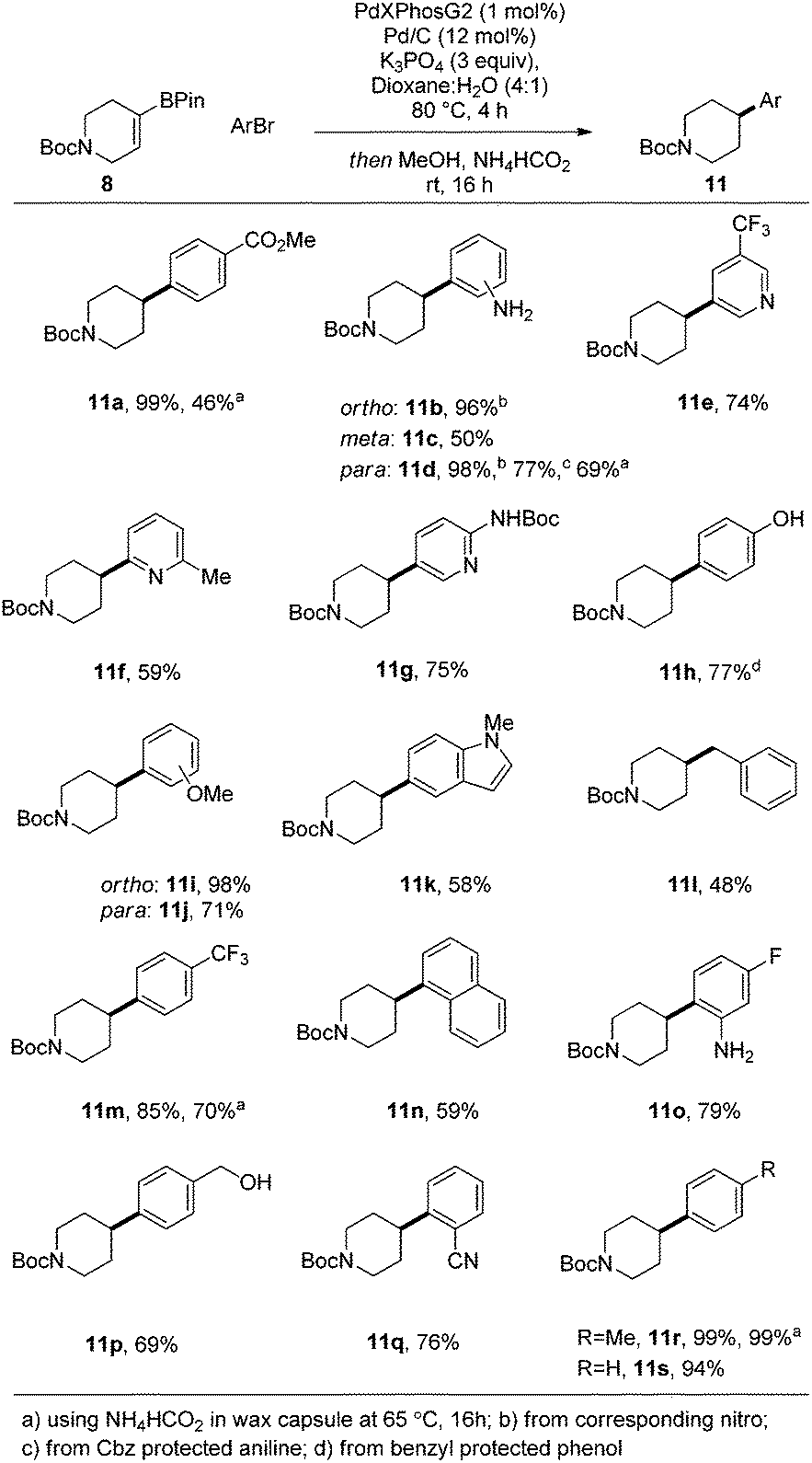 | ||
| Scheme 1 Lead-like molecules exemplifying the motif of interest with key sp2–sp3 bond indicated. | ||
The results of this work indicated that nitrobenzene derivatives could be reduced to the corresponding aniline systems (11b, 11d), while CBz protected amines and benzyl ethers could be removed under the reaction conditions, furnishing compounds 11d and 11h, respectively, providing additional functionality for further derivatization. We then focused our attention on developing a protocol which allowed all reactants to be present from the reaction outset in order to elevate the practicality of the process. Under the standard conditions, addition of ammonium formate at the beginning of the reaction posed two problems: protodehalogenation of the aryl halide, and reduction (followed by protodeboronation) of the vinyl boronate ester, both of which inhibited the SM reaction. To circumvent these issues, we took inspiration from the work of Buchwald et al.11 by employing a wax capsule to physically separate the incompatible reagents. By encapsulating ammonium formate in a degradable vessel, the rate at which the capsule melted, and thus the release of ammonium formate into the system, could be controlled by careful tuning of the reaction temperature. Accordingly, the adapted method was successfully applied to a series of substrates (11a, 11d, 11m and 11r), as indicated in Scheme 1.
Following on from this initial successful demonstration of the utility of the process, we sought to apply the conditions developed to a broader range of substrates (Scheme 2). Variation of both coupling partners enabled highly efficient access to a raft of different substrates, many of which are significantly enriched in sp3 character.
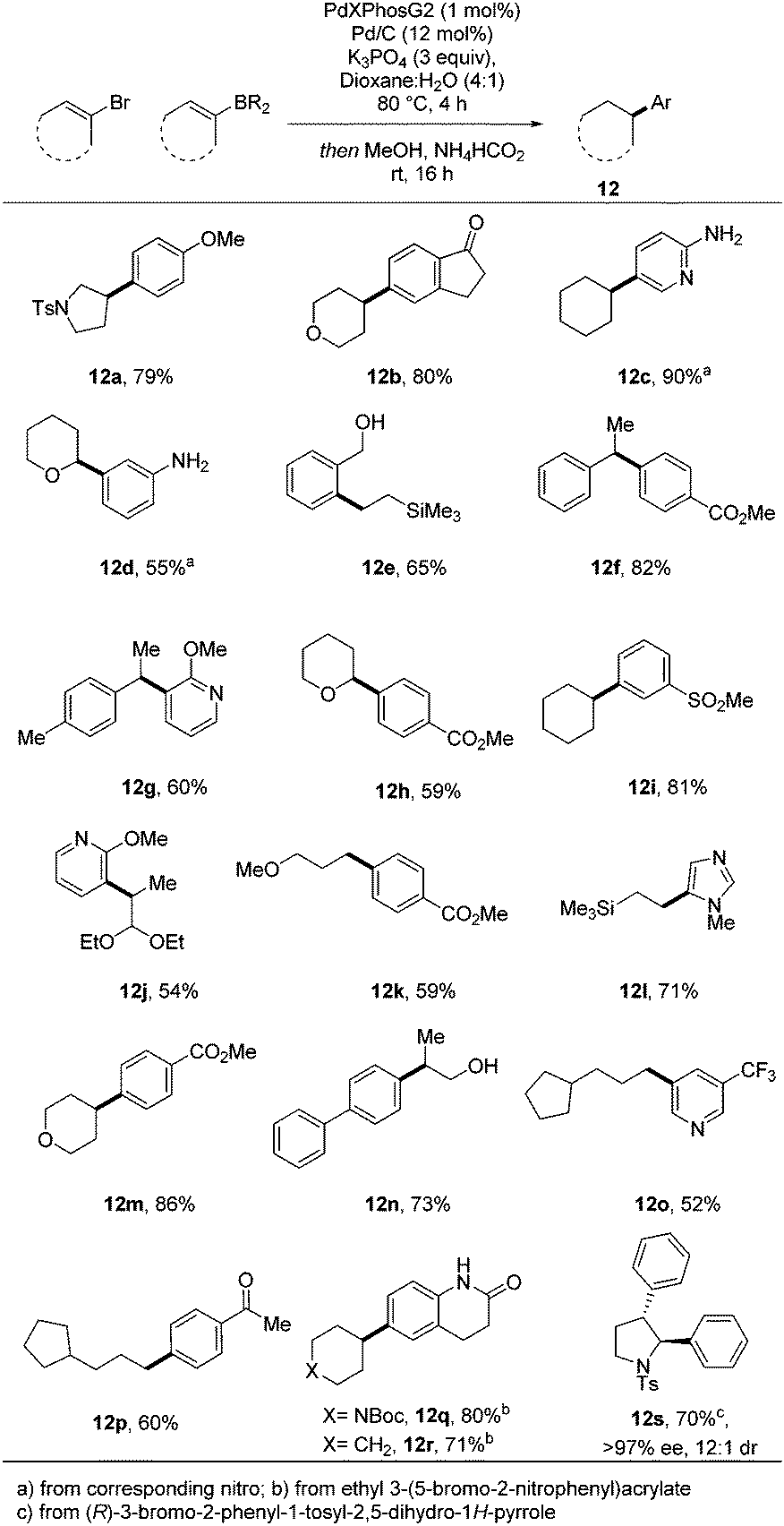 | ||
| Scheme 2 Variation of both coupling partners with key sp2–sp3 bond indicated. | ||
Additional diversity could be achieved when using more functionalized aryl bromide precursors. For example, when ethyl 3-(5-bromo-2-nitrophenyl)acrylate was used, an efficient in situ lactamisation took place, with three reductions and two bond forming steps occurring in one pot, yielding 12q and 12r in excellent overall yield. We have also demonstrated how application of a chiral vinyl bromide substrate can lead to isolation of a fully reduced, cross-coupled product in high enantio- and diastereomeric purity (12s) and good yield. The thermodynamically more stable trans-product is presumably obtained through a hydritic rather than concerted reduction of the alkene.12 To illustrate the practicality of this approach in a medicinal chemistry setting, the chemistry was applied to an array synthesis format. This allows the preparation of multiple sp3-enriched analogues in parallel from commercial monomers in a highly efficient manner. We carried out a 4 × 6 array with a range of sp2-functionalised organoboron esters and aryl bromides which enabled the successful preparation of 23 compounds, 22 of which were obtained in greater than 50% conversion as determined by 1H NMR (Fig. 2).
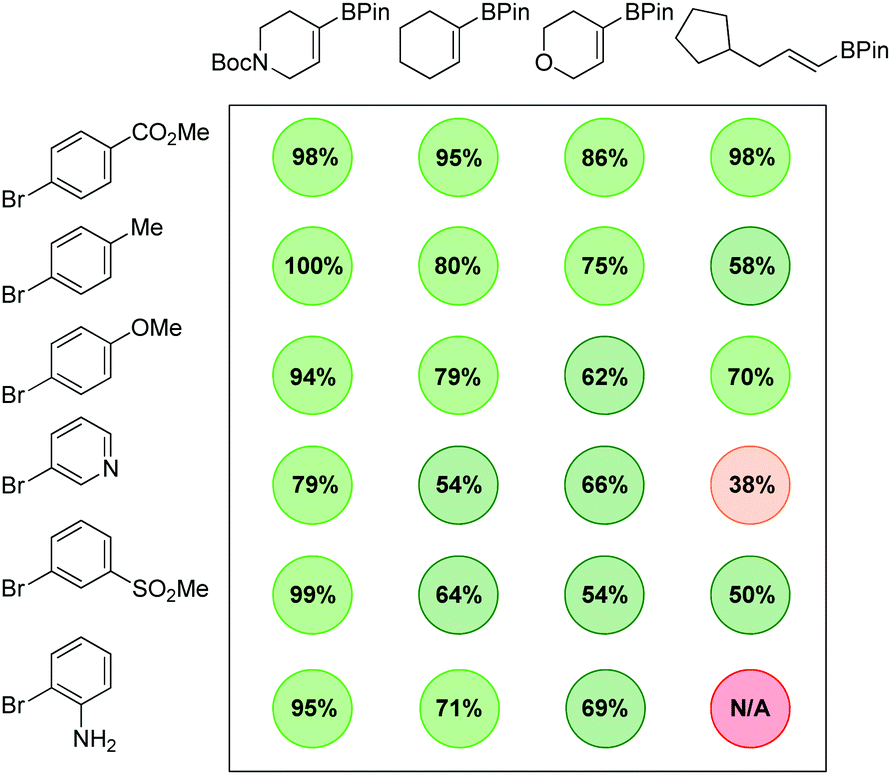 | ||
| Fig. 2 24-compound array synthesis. | ||
To further underline the utility of the developed process we also examined the synthesis of two bioactive compounds. Firstly, pridopidine (3, Scheme 3),4c a dopaminergic antagonist, was efficiently accessed in 3 steps, which considerably improves upon the previously reported synthesis of 7 steps.4c Pleasingly, the SM-hydrogenation step could be also be accomplished in comparable yield through use of the encapsulation approach described previously. Our second target compound was a 2-amino-3-(5-methyl-3-hydroxyisoxazol-4-yl)-propanoic acid (AMPA) receptor modulator, 15 (Scheme 3).13 Vinyl bromide 14 and biphenyl boronic acid (13) were coupled to provide the protected sulfonamide in 71% yield, with subsequent acidolysis of the of 2,4-dimethoxybenzyl (DMB) group providing the desired compound (15) in high yield (Scheme 3).
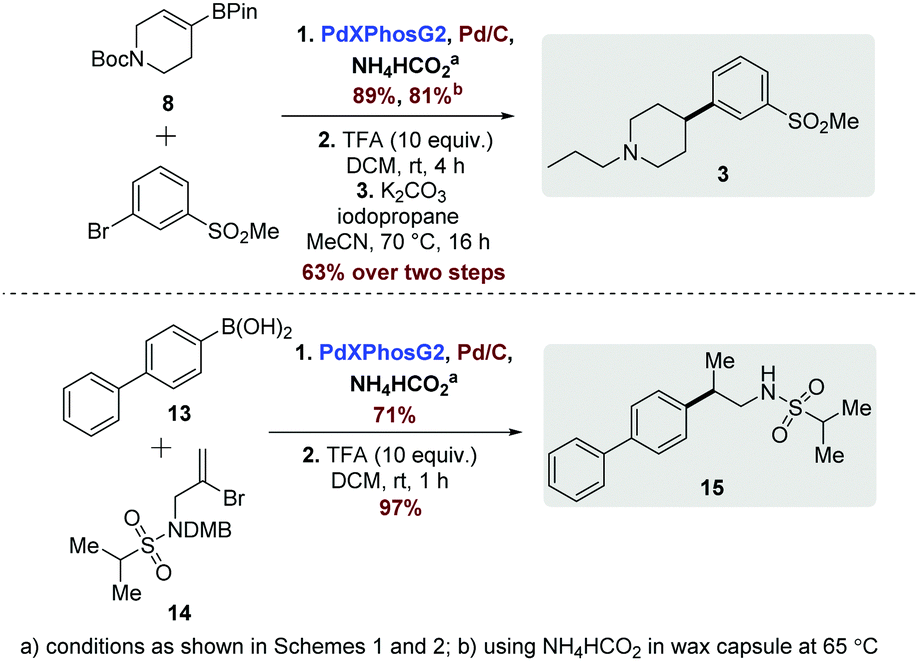 | ||
| Scheme 3 Application to synthesis of biologically active compounds. | ||
In conclusion, a general and versatile method for formal sp2–sp3 coupling has been developed which is applicable to a palette of substrates representing frameworks of pharmaceutical relevance. Computational analysis14 of set shows it has a high mean fraction sp3 (fsp3) of 0.52, which based on Lovering's data3 is well above the high watermark for candidate drugs (mean fsp3 = 0.38 for phase I clinical candidates), with the caveat that some of the molecules contain protecting groups which may influence the overall average fsp3. Successful application of the method in an array format further supports its utility in the context of pharmaceutical research and development. Work is currently focused on further extension of the process towards asymmetry, and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. H. Kerns and L. Di, Drug Like Properties: Concepts, Structure, Design and Methods, Academic Press, Burlington MA, 2008 Search PubMed.
- T. J. Ritchie, S. J. F. Macdonald, R. J. Young and S. D. Pickett, Drug Discovery Today, 2011, 16, 164 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- (a) R. E. Zelle, A. A. Hancock, S. A. Buckner, F. Z. Basha, K. Tietje, J. F. DeBernardis and M. D. Meyer, Bioorg. Med. Chem. Lett., 1994, 4, 1319 CrossRef CAS; (b) A. P. Truong, G. Tóth, G. D. Probst, J. M. Sealy, S. Bowers, D. W. G. Wone, D. Dressen, R. K. Hom, A. W. Konradi, H. L. Sham, J. Wu, B. T. Peterson, L. Ruslim, M. P. Bova, D. Kholodenko, R. N. Motter, F. Bard, P. Santiago, H. Ni, D. Chian, F. Soriano, T. Cole, E. F. Brigham, K. Wong, W. Zmolek, E. Goldbach, B. Samant, L. Chen, H. Zhang, D. F. Nakamura, K. P. Quinn, T. A. Yednock and J. M. Sauer, Bioorg. Med. Chem. Lett., 2010, 20, 6231 CrossRef CAS PubMed; (c) F. Pettersson, H. Pontén, N. Waters, S. Waters and C. Sonesson, J. Med. Chem., 2010, 53, 2510 CrossRef CAS PubMed.
- For examples of cross-coupling with alkylboron reagents see: (a) B. Potter, A. A. Szymaniak, E. K. Edelstein and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 17918 CrossRef CAS PubMed; (b) C. Sun, B. Potter and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 6534 CrossRef CAS PubMed; (c) G. A. Molander and S. R. Wisniewski, J. Am. Chem. Soc., 2012, 134, 16856 CrossRef CAS PubMed; (d) D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195 CrossRef CAS PubMed; (e) S. D. Dreher, S.-E. Lim, D. L. Sandrock and G. A. Molander, J. Org. Chem., 2009, 74, 3621 CrossRef PubMed; (f) L. Li, S. Zhao, A. Joshi-Pangu, M. Diane and M. R. Biscoe, J. Am. Chem. Soc., 2014, 136, 14027 CrossRef CAS PubMed.
- For examples of cross-coupling with alkylzinc reagents see: (a) C. Han and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 7532 CrossRef CAS PubMed; (b) Y. Yang, K. Niedermann, C. Han and S. L. Buchwald, Org. Lett., 2014, 16, 4638 CrossRef CAS PubMed; (c) A. Joshi-Pangu, M. Ganesh and M. R. Biscoe, Org. Lett., 2011, 13, 1218 CrossRef CAS PubMed; (d) K. Moriya and P. Knochel, Org. Lett., 2014, 16, 924 CrossRef CAS PubMed; (e) S. Çalimsiz and M. G. Organ, Chem. Commun., 2011, 47, 5181 RSC.
- For examples of cross-coupling with alkyltin reagents see: (a) L. Li, C.-Y. Wang, R. Huang and M. R. Biscoe, Nat. Chem., 2013, 5, 607–612 CrossRef CAS PubMed; (b) M. Goli, A. He and J. R. Falck, Org. Lett., 2011, 13, 344 CrossRef CAS PubMed; (c) A. Herve, A. L. Rodriquez and E. Fouquet, J. Org. Chem., 2005, 70, 1953 CrossRef CAS PubMed.
- For examples of cross-coupling with alkylmagnesium reagents see: (a) A. Joshi-Pangu, C.-Y. Wang and M. R. Biscoe, J. Am. Chem. Soc., 2011, 133, 8478–8481 CrossRef CAS PubMed; (b) M. E. Limmert, A. H. Roy and J. F. Hartwig, J. Org. Chem., 2005, 70, 9364 CrossRef CAS PubMed; (c) J. A. Miller, Tetrahedron Lett., 2002, 43, 7111 CrossRef CAS.
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed.
- For a two-step protocol involving isolation of the alkene intermediate see: K. D. Hesp, D. P. Fernando, W. Jiao and A. T. Londregan, Org. Lett., 2014, 16, 413 CrossRef CAS PubMed.
- A. C. Sather, H. G. Lee, J. R. Colombe, A. Zhang and S. L. Buchwald, Nature, 2015, 524, 208 CrossRef CAS PubMed.
- J. Lopez-Serrano, S. B. Duckett and A. Lledos, J. Am. Chem. Soc., 2006, 128, 9596 CrossRef CAS PubMed.
- P. L. Ornstein, D. M. Zimmerman, M. B. Arnold, T. J. Bleisch, B. Cantrel, R. Simon, H. Zarrinmayeh, S. R. Baker, M. Gates, J. P. Tizzano and D. Bleakman, J. Med. Chem., 2000, 43, 4354 CrossRef CAS PubMed.
- Determined using LLAMA: I. Colomer, C. J. Empson, P. Craven, Z. Owen, R. G. Doveston, I. Churcher, S. P. Marsden and A. Nelson, Chem. Commun., 2016, 52, 7209 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and spectral data for all compounds. See DOI: 10.1039/c7cc08670a |
| This journal is © The Royal Society of Chemistry 2018 |