
Total synthesis of (+)-brasilenyne via concise construction of an oxonane framework containing a 1,3-cis,cis-diene†
Changjin
Lim
ab,
Jungmin
Ahn
b,
Jaehoon
Sim
ab,
Hwayoung
Yun
 c,
Joonseong
Hur
b,
Hongchan
An
b,
Jaebong
Jang
b,
Seungbeom
Lee
b and
Young-Ger
Suh
*ab
c,
Joonseong
Hur
b,
Hongchan
An
b,
Jaebong
Jang
b,
Seungbeom
Lee
b and
Young-Ger
Suh
*ab
aCollege of Pharmacy, CHA University, 120 Haeryong-ro, Pocheon-si, Gyeonggi-do 11160, Republic of Korea. E-mail: ygsuh@cha.ac.kr; ygsuh@snu.ac.kr; Tel: +82-31-850-9300
bCollege of Pharmacy, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Republic of Korea
cCollege of Pharmacy, Pusan National University, Busan 46241, Republic of Korea
First published on 11th December 2017
Abstract
The enantioselective total synthesis of (+)-brasilenyne has been accomplished. The key features of the synthesis include the convergent preparation of a highly functionalized endocyclization precursor via selective epoxide opening, the construction of an oxonene skeleton through perfect regioselective Pd(0)-catalyzed endocyclization, and the installation of a 1,3-cis,cis-diene unit via a decarboxylative photophenylselenylation and site-selective selenoxide elimination sequence.
Medium-sized cyclic ether metabolites have been continuously isolated from Laurencia red algae and marine organisms that feed on Laurencia species.1 These Laurencia oxacycles have attracted considerable attention of synthetic chemists2 because of their unique structural features including various ring sizes, diverse stereogenic centers, one or more halogen atoms, and terminal enyne or allene units.1 In particular, marine natural products consisting of oxonane, a nine-membered cyclic ether, have been continuously reported, although synthetic studies on the oxonanes are less than the oxocanes, eight-membered cyclic ethers.2
(+)-Brasilenyne (1), one of the Laurencia oxacycles that contains an α,α′-trans-substituted oxonane skeleton (Fig. 1), was isolated from Aplysia brasiliana and reported to show antifeedant activity.3 This secondary metabolite has only been synthesized via an elegant intramolecular silicon-assisted cross-coupling of two vinyl groups by the Denmark4 group partly due to the difficulty in the installation of the 1,3-cis,cis-diene unit in the oxonane framework by other approaches including ring closing metathesis.5 We herein report the total synthesis of 1via concise construction of an oxonane framework containing a 1,3-cis,cis-diene.
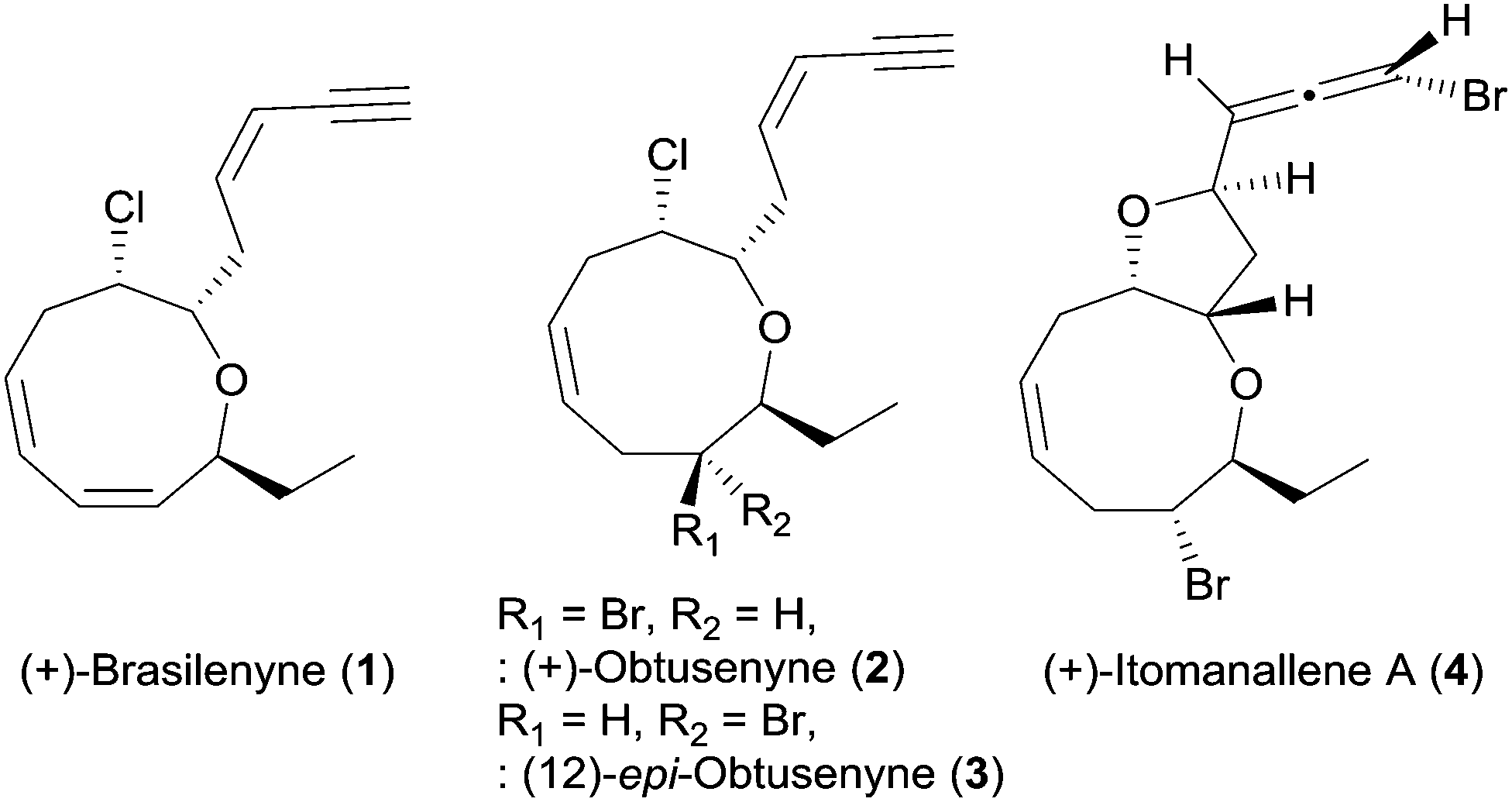 | ||
| Fig. 1 (+)-Brasilenyne (1) and the related oxonane natural product from the Laurencia family. | ||
Our retrosynthetic plan is outlined in Scheme 1. The labile enyne moiety was planned to be introduced into 5 by Julia olefination6 at the final stage. The C7-chloride would be directly introduced via halide displacement of the C7-hydroxy group. The important 1,3-cis,cis-diene would be constructed by a regioselective selenoxide elimination of selenide 6 with the pre-existing ring olefin intact. The oxonane intermediate 6, which possesses appropriate substituents to be converted to 1, can be derived from 7 by desulfonylation and concise conversion of ester to phenylselenide.7 The key α,α′-trans-substituted oxonene 7 was anticipated to be effectively prepared by selective Pd(0)-catalyzed endocyclization, which was previously studied by Hoffmann8 and our group,9 of allylic carbonate 8. The endocyclization precursor 8 was considered to be quite appropriate for the concise construction of the oxonene core 5. As the construction of the 1,3-cis,cis-diene unit in the oxonane system could not be efficiently accomplished from the cyclization precursors reported by Hoffmann8 and our group,9 we optimized the nucleophilic part with benzenesulfonyl acetate.10 The benzenesulfonyl acetate moiety was anticipated to be effectively transformed into phenylselenide via a radical chain reaction. The cyclization precursor 8 could be convergently prepared by the diastereoselective epoxide ring opening of 9 with allylic alcohol 10 and with the assistance of copper triflate.11
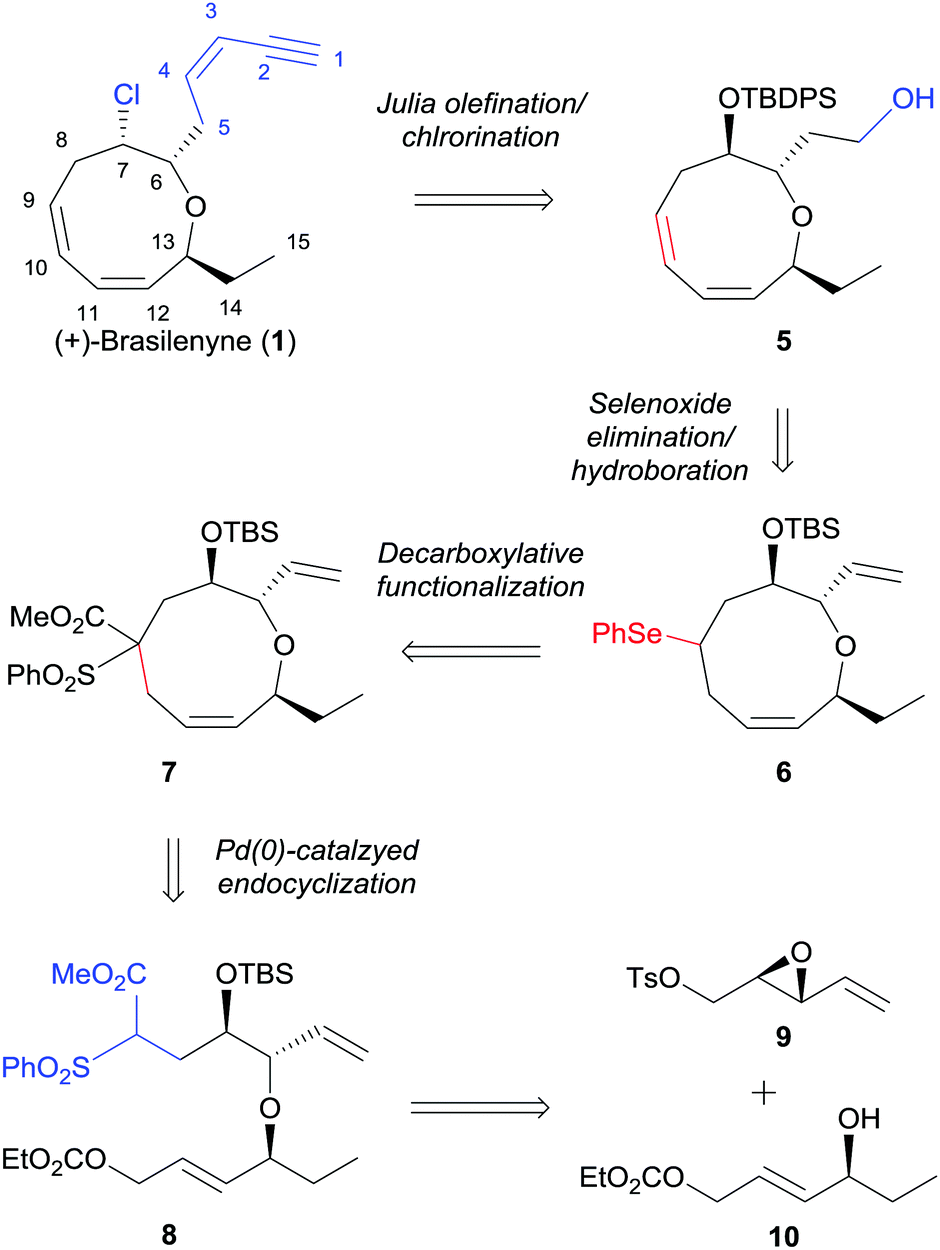 | ||
| Scheme 1 Strategy for the asymmetric total synthesis of (+)-brasilenyne (1). TBDPS = tert-butyldiphenylsilyl, TBS = tert-butyldimethylsilyl, Ph = phenyl, Ts = p-toluenesulfonyl. | ||
The synthesis of 1 was commenced by preparing the cyclization precursor 8 as shown in Scheme 2. DIBAL reduction of the known ester 1112 and ethoxycarbonylation of the resulting alcohol afforded allylic carbonate 10 in 68% yield for two steps. Diastereoselective epoxide opening11 of 9, which is prepared from divinylcarbinol via a three-step sequence,13 in the presence of copper triflate produced ether 12 in 59% yield. Unfortunately, the regioisomeric ether was consistently produced in 29% yield despite our continuous efforts. Iodide displacement of tosylate 12 and TBS protection of alcohol followed by reaction with the benzenesulfonyl acetate anion produced the cyclization precursor 8 in 61% yield for three steps.
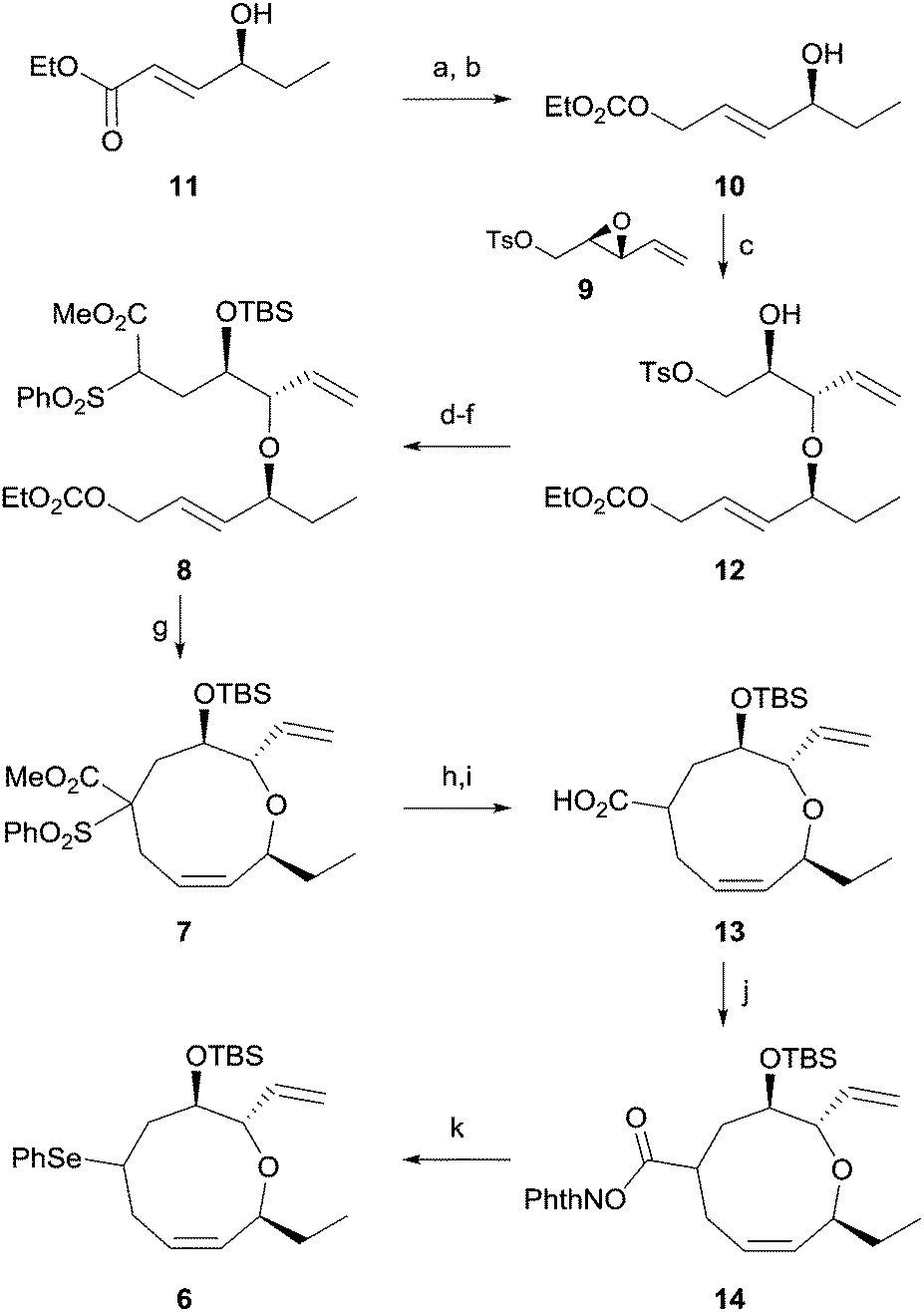 | ||
Scheme 2 Synthesis of the oxonene intermediate. (a) DIBAL-H, THF, −78 °C, 30 min, 81%; (b) ClCO2Et, 2,6-lutidine, MeCN, −20 °C, 2 h, 84%; (c) Cu(OTf)2, CH2Cl2, 0 °C, 1 h, 59%; (d) NaI, acetone, reflux, 12 h; (e) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 1 h, 86% for 2 steps; (f) PhSO2CH2CO2Me, NaH, DMF, 80 °C, 20 h, 71%; (g) Pd(dppe)2, DMSO, 60 °C, 2 h, 75%; (h) SmI2, THF, −78 °C, 30 min; (i) LiOH·H2O, THF/H2O/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1), 8 h, 90% for 2 steps; (j) N-hydroxyl phthalimide, DCC, DMAP, THF, 4 h, 91%; (k) blue LEDs, (PhSe)2, Ru(bpy)3Cl2·6H2O, BNAH, THF/H2O (2:1), 2 h, 79%. dppe = 1,2-bis(diphenylphosphino)ethane, DCC = N,N′-dicyclohexylcarbodiimide, DMAP = 4-(dimethylamino)pyridine, BNAH = 1-benzyl-1,4-dihydronicotinamide, bpy = 2,2′-bipyridyl. :1:1), 8 h, 90% for 2 steps; (j) N-hydroxyl phthalimide, DCC, DMAP, THF, 4 h, 91%; (k) blue LEDs, (PhSe)2, Ru(bpy)3Cl2·6H2O, BNAH, THF/H2O (2:1), 2 h, 79%. dppe = 1,2-bis(diphenylphosphino)ethane, DCC = N,N′-dicyclohexylcarbodiimide, DMAP = 4-(dimethylamino)pyridine, BNAH = 1-benzyl-1,4-dihydronicotinamide, bpy = 2,2′-bipyridyl. | ||
With the cyclization precursor in hand, we initially confirmed the feasibility of the key endocyclization through intensive cyclization studies including different solvents and temperatures. Treatment of allylic carbonate 8 with Pd(dppe)2 in DMSO at 60 °C for 2 h produced the desired nine-membered cyclic ether 7 in 75% yield. The regioisomeric seven-membered cyclic ether was not observed. The excellent regioselectivity of the endocyclization is likely due to the preferred attack of the highly bulky benzenesulfonyl acetate anion at the sterically less hindered terminal carbon of the π-allyl palladium complex.14 In addition, production of the isomeric 6,13-cis cyclic ether via epimerization at C13 through the cleavage of ether linkage,8b,8c of which we were concerned, was not observed.
We next focused on efficient installation of the diene moiety in the oxonene skeleton. Initial attempts at decarboxylation of oxonene 7 followed by direct elimination15 of the benzenesulfonyl group to obtain a diene system were not successful. Thus, removal of the benzenesulfonyl group of 7 with samarium iodide followed by ester hydrolysis afforded acid 13 in 90% yield for two steps. We tried to directly transform the acid to the corresponding halide16 or selenide7avia the Hunsdiecker type reaction, which proved to be ineffective. In addition, efforts to directly eliminate the acid17 were also unsuccessful. Therefore, we finally decided to utilize decarboxylative photo-phenylselenylation. Reaction of acid 13 with N-hydroxyphthalimide in the presence of DCC produced ester 14, which was transformed into phenylseleno oxonene 6 in 72% yield for two steps via treatment with diphenyl diselenide and BNAH in the presence of a ruthenium catalyst.7b To the best of our knowledge, few synthetic applications of decarboxylative phenylselenylation using photoredox catalysis have been reported.18
As shown in Scheme 3, facile regioselective elimination of 6 delightfully proceeded upon hydrogen peroxide treatment to afford the corresponding diene 16 in 67% yield along with a small amount of the regioisomeric triene and the trans-olefinic isomer. However, replacing the TBS protecting group of selenide with a TBDPS protecting group produced the triene 17 in 90% isolated yield with only a small amount of the regioisomeric triene. Formation of the favorable conformation for the excellent regioselective syn-elimination of the selenoxide produced from 15 is likely induced by a steric effect of the bulky TBDPS group.19
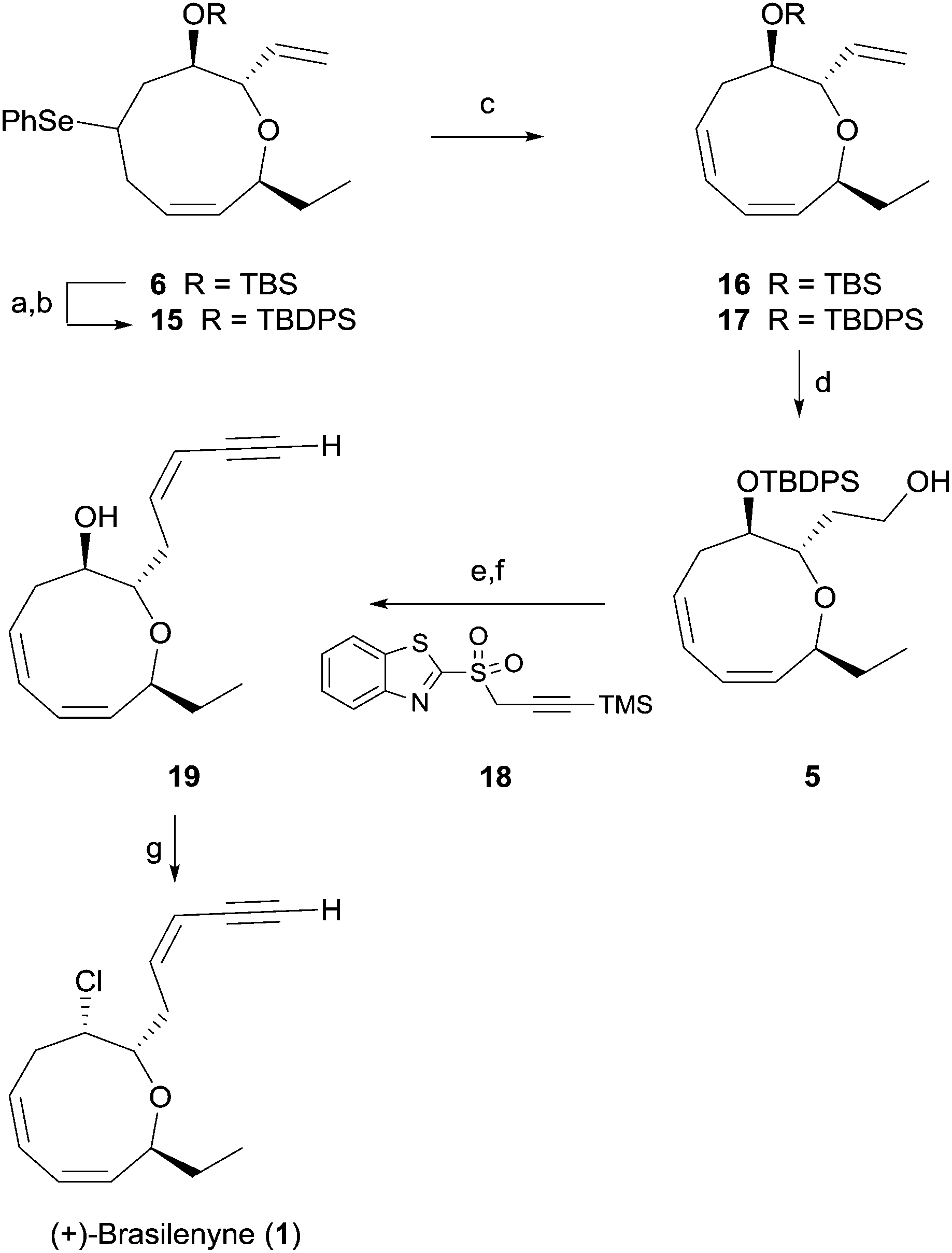 | ||
| Scheme 3 Completion of (+)-brasilenyne (1) synthesis. (a) TBAF, THF, 0 °C, 3 h; (b) TBDPSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 4 h, 83% for 2 steps; (c) H2O2, 2-methyl-2-butene, pyridine, CH2Cl2, 0 °C, 12 h, 67% for 16, 90% for 17; (d) (9-BBN) 2, THF, 8 h then, 2N NaOH, H2O2, 0 °C, 1 h, 72%; (e) DMP, NaHCO3, CH2Cl2, 1 h then, 18, DBU, CH2Cl2, −55 °C, 2 h, 83%; (f) TBAF, THF, 6 h, 99%; (g) CCl4, n-Oct3P, toluene, 60 °C, 12 h, 93%. TBAF = tetrabutylammonium fluoride, BBN = 9-borabicyclo[3,3,1]nonane, DMP = Dess–Martin periodinane, DBU = 1,8-diazabicyclo[5,4,0]undec-7-ene. | ||
Triene 17 was reacted with 9-BBN to give alcohol 5 in 72% yield. For completion of the synthesis, geometrically selective formation of the enyne moiety was extensively examined. The reaction6 of the aldehyde obtained from the Dess–Martin periodinane oxidation of 5 with benzothiazolyl sulfone 18 in the presence of DBU produced the cis-enyne product (Z/E > 20:1) in 83% isolated yield, which is superior in terms of yield and geometric selectivity to the Peterson type olefination used in previous synthesis.4 Global deprotection of the TBDPS and TMS groups with TBAF afforded the hydroxy enyne 19 in 99% yield. Finally, reaction of alcohol 19 with carbon tetrachloride and trioctylphophine4 produced 1 in 93% yield. Synthetic 1 was identical in all respects to natural 1.3
In summary, we accomplished the asymmetric total synthesis of (+)-brasilenyne (1) in 18 steps from the known ester 11 (21 steps from the commercially available starting materials). The key features of our synthesis include stereoselective installation of the stereocenters at the α,α′-positions of the ether linkage and the C7-stereocenter via a selective epoxide ring-opening reaction, concise construction of the oxonane framework by perfectly regioselective endocyclization, effective elaboration of the 1,3-cis,cis-diene unit via a sequence of decarboxylative photophenylselenylation followed by selenoxide elimination, and geometrically selective introduction of the cis-enyne side chain via the one-pot process of oxidation/Julia olefination of alcohol 5. This versatile synthetic approach is expected to be widely utilized for the syntheses of the Laurencia oxonane natural products.
This work was supported by the Global Frontier Project grant (NRF-2015M3A6A4065798) of National Research Foundation funded by the Ministry of Science and ICT of Korea and by the GRRC program of Gyeonggi province (GRRC-CHA2017-B02).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Z. F. Zhou, M. Menna, Y. S. Cai and Y. W. Guo, Chem. Rev., 2015, 115, 1543 CrossRef CAS PubMed; (b) R. B. Pereira, P. B. Andrade and P. Valentao, Mar. Drugs, 2016, 14, 39 CrossRef PubMed; (c) J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2014, 31, 160 RSC and references cited therein.
- For selected reviews, see: (a) T. Martin, J. I. Padron and V. S. Martin, Synlett, 2014, 12 CAS; (b) K. Fujiwara, in Marine Natural Products, ed. H. Kiyota, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, p. 97 Search PubMed; (c) A. S. Kleinke, D. Webb and T. F. Jamison, Tetrahedron, 2012, 68, 6999 CrossRef CAS.
- R. B. Kinnel, R. K. Dieter, J. Meinwald, D. Vanengen, J. Clardy, T. Eisner, M. O. Stallard and W. Fenical, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 3576 CrossRef CAS.
- (a) S. E. Denmark and S. M. Yang, J. Am. Chem. Soc., 2002, 124, 15196 CrossRef CAS PubMed; (b) S. E. Denmark and S. M. Yang, J. Am. Chem. Soc., 2004, 126, 12432 CrossRef CAS PubMed.
- (a) K. Fujiwara, D. Awakura, M. Tsunashima, A. Nakamura, T. Honma and A. Murai, J. Org. Chem., 1999, 64, 2616 CrossRef CAS PubMed; (b) S. Y. F. Mak, N. R. Curtis, A. N. Payne, M. S. Congreve, C. L. Francis, J. W. Burton and A. B. Holmes, Synthesis, 2005, 3199 CAS; (c) S. Y. F. Mak, N. R. Curtis, A. N. Payne, M. S. Congreve, A. J. Wildsmith, C. L. Francis, J. E. Davies, S. I. Pascu, J. W. Burton and A. B. Holmes, Chem. – Eur. J., 2008, 14, 2867 CrossRef CAS PubMed; (d) M. T. Crimmins and K. A. Emmitte, J. Am. Chem. Soc., 2001, 123, 1533 CrossRef CAS PubMed; (e) M. T. Crimmins, K. A. Emmitte and A. L. Choy, Tetrahedron, 2002, 58, 1817 CrossRef CAS; (f) M. T. Crimmins and M. T. Powell, J. Am. Chem. Soc., 2003, 125, 7592 CrossRef CAS PubMed; (g) W. Jeong, M. J. Kim, H. Kim, S. Kim, D. Kim and K. J. Shin, Angew. Chem., Int. Ed., 2010, 49, 752 CrossRef CAS PubMed; (h) M. J. Kim, T. I. Sohn, D. Kim and R. S. Paton, J. Am. Chem. Soc., 2012, 134, 20178 CrossRef CAS PubMed; (i) T. Suzuki, N. Yoshino, T. Uemura, H. Hagiwara and T. Hoshi, Chem. Lett., 2007, 36, 278 CrossRef CAS; (j) T. Uemura, T. Suzuki, N. Onodera, H. Hagiwara and T. Hoshi, Tetrahedron Lett., 2007, 48, 715 CrossRef CAS.
- (a) C. Bonini, L. Chiummiento and V. Videtta, Synlett, 2006, 2079 CrossRef CAS; (b) R. Kumar and B. Zajc, J. Org. Chem., 2012, 77, 8417 CrossRef CAS PubMed; (c) G. Kim, T. I. Sohn, D. Kim and R. S. Paton, Angew. Chem., Int. Ed., 2014, 53, 272 CrossRef CAS PubMed.
- (a) D. H. R. Barton, D. Bridon, Y. Herve, P. Potier, J. Thierry and S. Z. Zard, Tetrahedron, 1986, 42, 4983 CrossRef CAS; (b) K. Okada, K. Okubo, N. Morita and M. Oda, Chem. Lett., 1993, 2021 CrossRef CAS.
- (a) H. M. R. Hoffmann and A. Brandes, Tetrahedron, 1995, 51, 155 CrossRef CAS; (b) A. Brandes and H. M. R. Hoffmann, Tetrahedron, 1995, 51, 145 CrossRef CAS; (c) J. Pohlmann, C. Sabater and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 1998, 37, 633 CrossRef CAS.
- Y. G. Suh, B. A. Koo, E. N. Kim and N. S. Choi, Tetrahedron Lett., 1995, 36, 2089 CrossRef CAS.
- (a) S. Y. Seo, J. K. Jung, S. M. Paek, Y. S. Lee, S. H. Kim, K. O. Lee and Y. G. Suh, Org. Lett., 2004, 6, 429 CrossRef CAS PubMed; (b) S. Lee, S. M. Paek, H. Yun, N. J. Kim and Y. G. Suh, Org. Lett., 2011, 13, 3344 CrossRef CAS PubMed; (c) H. Seo, H. Yun, S. Lee, J. Jang, Y. T. Han, D. D. Kim, J. Lee and Y. G. Suh, Org. Lett., 2013, 15, 531 CrossRef CAS PubMed; (d) T. Kim, Y. T. Han, H. An, K. Kim, J. Lee and Y. G. Suh, J. Org. Chem., 2015, 80, 12193 CrossRef CAS PubMed; (e) J. Sim, I. Yoon, H. Yun, H. An and Y. G. Suh, Org. Biomol. Chem., 2016, 14, 1244 RSC.
- (a) G. Prestat, C. Baylon, M. P. Heck and C. Mioskowski, Tetrahedron Lett., 2000, 41, 3829 CrossRef CAS; (b) G. Prestat, C. Baylon, M. P. Heck, G. A. Grasa, S. P. Nolan and C. Mioskowski, J. Org. Chem., 2004, 69, 5770 CrossRef CAS PubMed; (c) S. G. Wen, W. M. Liu and Y. M. Liang, Synthesis, 2007, 3295 CAS.
- (a) H. Nagatomo, Y. Matsushita, K. Sugamoto and T. Matsui, Tetrahedron: Asymmetry, 2003, 14, 2339 CrossRef CAS; (b) K. S. Petersen and G. H. Posner, Org. Lett., 2008, 10, 4685 CrossRef CAS PubMed.
- Epoxide 9 was prepared by tosylation of the known epoxy alcohol S. Torssell and P. Somfai, Org. Biomol. Chem., 2004, 2, 1643 CAS.
- (a) B. M. Trost, Acc. Chem. Res., 1980, 13, 385 CrossRef CAS; (b) J. A. Marshall, R. C. Andrews and L. Lebioda, J. Org. Chem., 1987, 52, 2378 CrossRef CAS; (c) A. S. Kende, I. Kaldor and R. Aslanian, J. Am. Chem. Soc., 1988, 110, 6265 CrossRef CAS PubMed; (d) A. Furstner and H. Weintritt, J. Am. Chem. Soc., 1998, 120, 2817 CrossRef.
- (a) C. H. Dupenhoat and M. Julia, Tetrahedron, 1986, 42, 4807 CrossRef; (b) C. A. G. Baker-Glenn, A. G. M. Barrett, A. A. Gray, P. A. Procopiou and M. Ruston, Tetrahedron Lett., 2005, 46, 7427 CrossRef CAS.
- (a) S. J. Cristol and W. C. Firth, J. Org. Chem., 1961, 26, 280 CrossRef CAS; (b) J. K. Kochi, J. Am. Chem. Soc., 1965, 87, 2500 CrossRef CAS; (c) P. Camps, A. E. Lukach, X. Pujol and S. Vazquez, Tetrahedron, 2000, 56, 2703 CrossRef CAS; (d) K. Kulbitski, G. Nisnevich and M. Gandelman, Adv. Synth. Catal., 2011, 353, 1438 CrossRef CAS; (e) Z. T. Wang, L. Zhu, F. Yin, Z. Q. Su, Z. D. Li and C. Z. Li, J. Am. Chem. Soc., 2012, 134, 4258 CrossRef CAS PubMed.
- (a) L. J. Goossen and N. Rodriguez, Chem. Commun., 2004, 724 RSC; (b) S. Maetani, T. Fukuyama, N. Suzuki, D. Ishihara and I. Ryu, Organometallics, 2011, 30, 1389 CrossRef CAS; (c) J. M. Anderson and J. K. Kochi, J. Org. Chem., 1970, 35, 986 CrossRef CAS; (d) J. D. Bacha and J. K. Kochi, Tetrahedron, 1968, 24, 2215 CrossRef CAS.
- There is only one application of decarboxylative phenylselenylation using photoredox catalysis, see: M. Jiang, H. J. Yang and H. Fu, Org. Lett., 2016, 18, 1968 CrossRef CAS PubMed.
- (a) K. B. Sharpless, M. W. Young and R. F. Lauer, Tetrahedron Lett., 1973, 1979 CrossRef CAS; (b) H. J. Reich and S. K. Shah, J. Am. Chem. Soc., 1975, 97, 3250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds along with copies of 1H and 13C NMR spectra. See DOI: 10.1039/c7cc08329g |
| This journal is © The Royal Society of Chemistry 2018 |