
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithium diamidodihydridoaluminates: bimetallic cooperativity in catalytic hydroboration and metallation applications†‡
Victoria A.
Pollard
,
Samantha A.
Orr
,
Ross
McLellan
,
Alan R.
Kennedy
 ,
Eva
Hevia
and
Robert E.
Mulvey
*
,
Eva
Hevia
and
Robert E.
Mulvey
*
WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: r.e.mulvey@strath.ac.uk
First published on 16th January 2018
Abstract
Cooperativity between the Li and Al centres is implicated in catalytic hydroboration reactions of aldehydes and ketones with pinacolborane via heteroleptic lithium diamidodihydridoaluminates. In addition to implementing hydroalumination, these versatile heteroleptic ates can also perform as amido bases as illustrated with an acidic triazole.
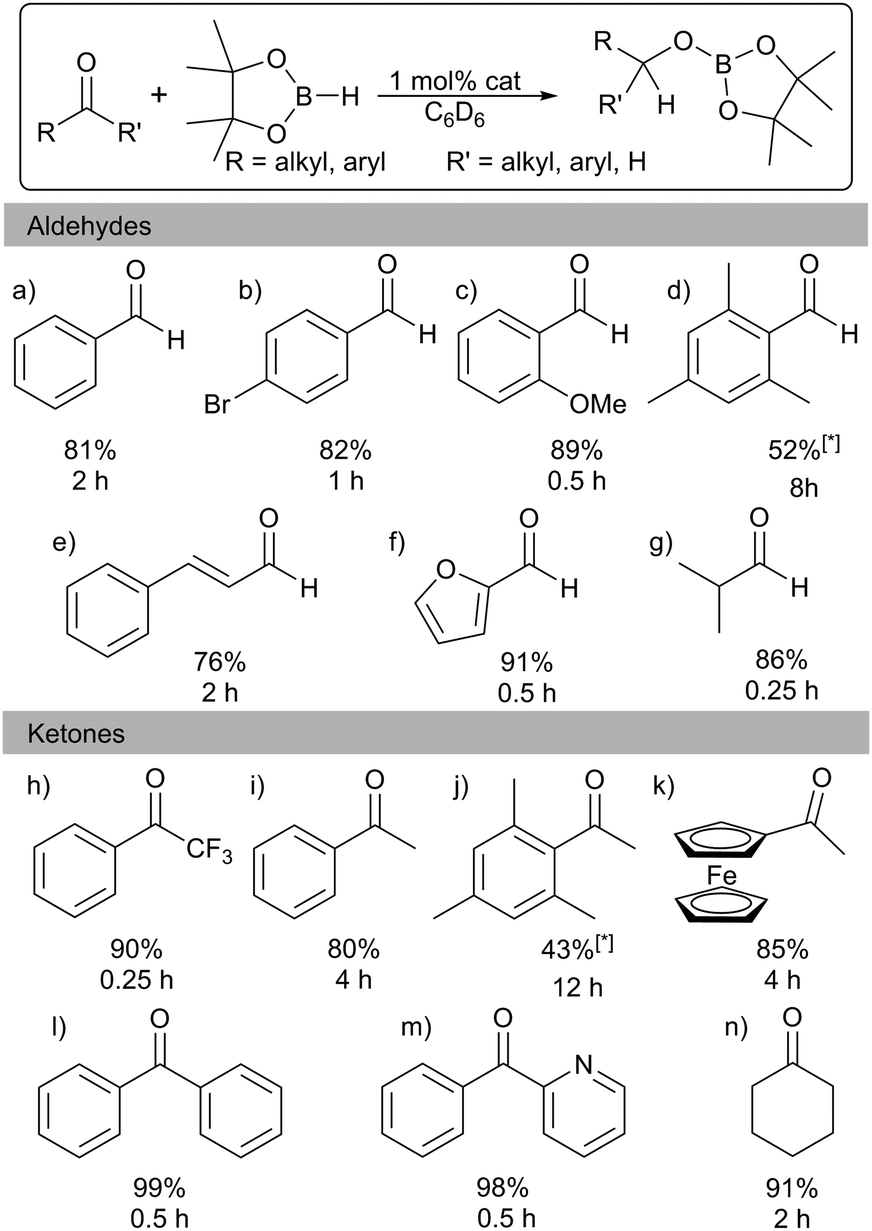
The application of main group complexes in homogeneous catalysis is currently gaining momentum, driven by a quest to supplement expensive and low abundant precious transition metals by more earth abundant sustainable alternatives. Aluminium is the most abundant metal in the earth's crust and has low toxicity. Its compounds have therefore been the focus of an increasing number of studies and find application in a range of chemical processes including as catalysts for dehydrocoupling, hydrosilylation and hydroboration,1–3 as well as small molecule activation by frustrated Lewis pairs (FLP)4–6 and trans-metal-trapping.7
Catalytic hydroboration of aldehydes and ketones has been reported for several Al-based catalysts, such as I–IV (Fig. 1).8–11 Roesky reported one of the first Al hydride complexes stabilised by a β-diketiminate ligand, I (Fig. 1) that is an active catalyst for hydroboration of terminal alkynes and organic carbonyls.1 Notably, Cowley and Thomas utilised DIBAL-H, IV (Fig. 1), and Et3Al·DABCO for alkyne hydroboration.11
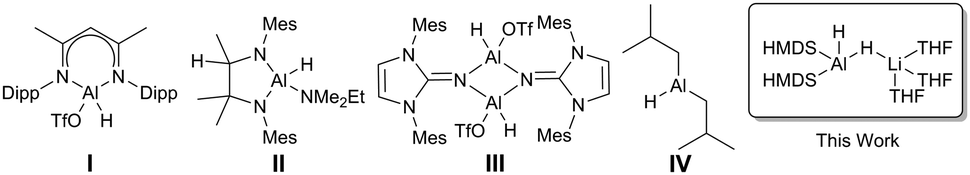 | ||
| Fig. 1 Selected reported aluminium hydride catalysts for hydroboration reactions. | ||
It is significant that to date most Al-based catalysts for such hydroboration reactions involve neutral complexes. Given our interest in bimetallic systems that can function synergistically under stoichiometric regimes,12 and recent advances suggesting that borates are crucial reaction intermediates in hydroboration,13,14 we propose that aluminates would exhibit even greater reactivity. In this contribution we introduce anionic complexes to this important emerging area through a series of heteroleptic lithium diamidodihydridoaluminate complexes with the bulky 1,1,1,3,3,3-hexamethyldisilazide ligand [HMDS, (Me3Si)2N−]. We show that (i) the performance of the anionic [(HMDS)2Al(H)2]− moiety in hydroboration reactions of aldehydes and ketones can be influenced by the nature of Lewis donor solvation of the Li centre; (ii) an intermediate from a stoichiometric reaction with benzaldehyde retains both metal centres following hydride transfer; and (iii) while HMDS acts as a stabilising spectator in the hydroboration reactions, it can operate as a base with a suitably acidic aromatic substrate, establishing the dual functionality of these ates.
Reaction of LiAlH4 with two equivalents of HMDS(H) in THF yielded the heteroleptic diamidodihydride (HMDS)2AlH(μ-H)Li(THF)3, 1, as colourless crystals in 61% yield. Fig. 2a shows the molecular, contacted ion pair (CIP) structure of 1. A Cambridge Structural Database search identified a similar compound, (HMDS)2Al(μ-H)2Li(OEt2)2, 2, prepared by Stalke.15 Complex 1 contains one terminal hydride and one bridging hydride ligand between the Al and Li centres, whereas both hydrides bridge in 2. Towards obtaining a solvent-separated ion pair (SSIP) variant, donor ligand exchange was performed on 2 with 12-crown-4, PMDETA (N,N,N′,N′′,N′′-pentamethyldiethylenetriamine), TMEDA (N,N,N′,N′-tetramethylethylenediamine) and Me6-TREN (Scheme 1). The products (HMDS)2Al(μ-H)2Li(L) (L = 12-crown-4, 3), (HMDS)2AlH(μ-H)Li(L) (L = PMDETA, 4) and [(HMDS)2AlH2][Li(L)] [L = (TMEDA)2, 5 or Me6-TREN, 6] are obtained. Fig. 2 depicts the crystallographically-determined structures of 1, 3 and 5, with those of 4 and 6 in the ESI.‡ In 3 both hydrides bridge and 12-crown-4 binds to Li in a κ4 manner. 4 has only one bridging hydride, with the second bonded solely to Al. TMEDA and Me6-TREN, generated SSIPs 5 and 6 respectively. In 5 and 6 the hydrides reside on Al centres. Li in 5 is in a distorted tetrahedral environment, while in 6 the Me6-TREN chelates Li leaving one face of it open for contacts between Li and H atoms on a HMDS on the neighbouring Al centre [Li⋯H–CH2 distance 2.41(1) Å and 2.99(1) Å]. Note Okuda studied ligand effects on the rate of hydroboration catalysis with [(L)Li][HBPh3], finding Me6-TREN to be superior to other ligands tested.16 A sodium analogue of 1 made using NaAlH4 generated 7, (HMDS)2AlH(μ-H)Na(THF)4 (see ESI‡). For complexes 1 and 4–6 terminal Al–H bond lengths lie in the range 1.54(2)–1.61(5) Å, while the Al–μH range [1.57(2)–1.61(2) Å] is similar. Li–H bond lengths for 1 and 4 are 1.83(2) Å and 1.81(2) Å, respectively. These compare to those in 2 (Al–H 1.62(2)/1.55(2) Å, Li–H 1.89(2)/1.97(2) Å). In comparison, 3 has longer Li–H bond lengths of, 2.13(2) Å and 2.11(2) Å.
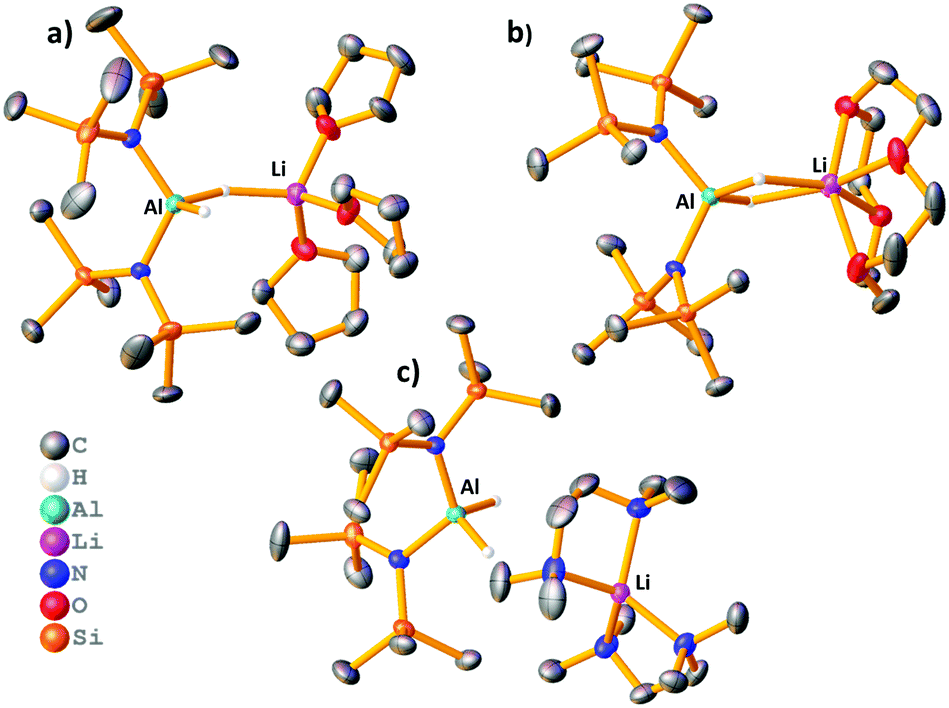 | ||
| Fig. 2 Molecular structures of (a) 1, (b) 3 and (c) 5. Hydrogen atoms except hydrides, and disordered THF in 1 and TMEDA in 5, and co-crystallised toluene in 3 have been omitted. Thermal ellipsoids are drawn at 40% probability. | ||
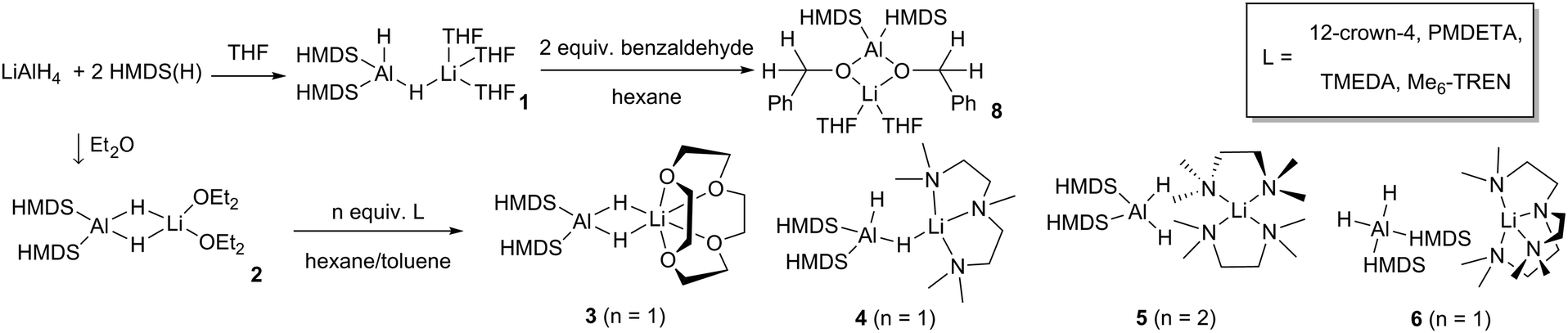 | ||
| Scheme 1 Synthesis of heteroleptic lithium diamidodihydridoaluminates complexes 1–6, and of bis-benzyloxide complex, (HMDS)2Al(μ-OCH2Ph)2Li(THF)2, 8. | ||
In all cases, it was not possible to observe the hydride signals in the 1H NMR spectrum. This is unsurprising since both 27Al and 7Li are quadrupolar nuclei. However, a 1H{27Al} NMR experiment revealed a new, broad singlet at approximately 3.8 ppm in each case. Integrating to two, this signal lies in the expected chemical shift range for an Al–H signal. In crystalline 1 and 4 the hydrides are inequivalent, and so the broad singlet in 1H{27Al} NMR indicates that the exchange of the inequivalent hydrides is fast on the NMR time scale. However, a variable temperature 1H{27Al} NMR experiment with 1 (−40 °C to 70 °C) did not give a sharp singlet, or the resolution of two signals. In comparison, the 1H NMR chemical shifts for the hydride signals in LiAlH4·Me6-TREN appear as a broad singlet at 3.86 ppm.17 A high temperature 27Al NMR experiment on 3 shows that as the temperature is increased the signal becomes sharper, and at 70 °C a triplet is resolved (1JAl–H, 173.6 Hz) (Fig. 3). This is consistent with reported 1JAl–H couplings (e.g., for [AlH2(NEt2)2]−, 175 Hz).18 Despite the SSIP structures in solid 5 and 6, DOSY NMR studies for 1–6 indicate that all species are CIP in C6D6 solution (see ESI‡). Thus, the level of solvation on the Li centre appears to play a key role in the resulting catalytic performance (see below).
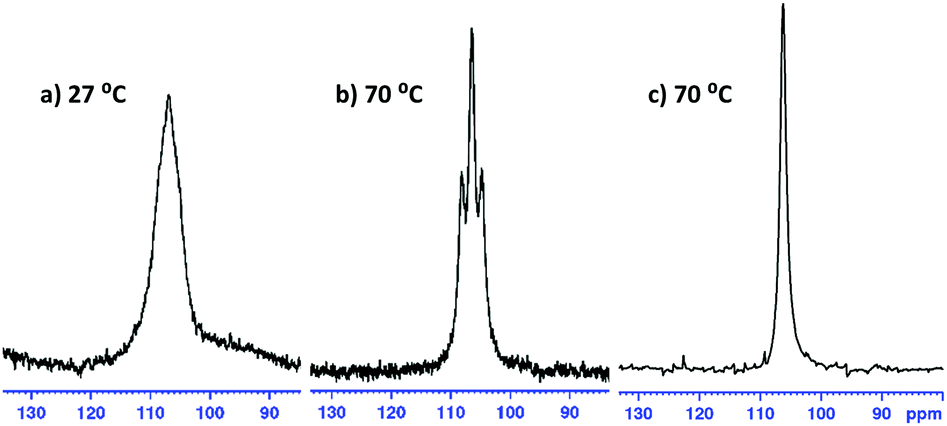 | ||
| Fig. 3 (a and b) 27Al NMR spectra of 3; a triplet resolves at 70 °C, 1JAl–H = 173.6 Hz. (c) 27Al{1H} NMR experiment shows the triplet has collapsed to a singlet. | ||
Several examples exist of Al hydrides that can act as catalysts for hydroboration of carbonyls.1 Encouraged by these studies we screened 1 for catalytic activity. Benzaldehyde, pinacolborane (HBpin), and a catalytic loading of 1 (1 mol%) in C6D6 were placed in a J. Young's NMR tube and the 1H and 11B NMR spectra were monitored over time. After 2 h, 81% of the hydroborated product was obtained. (Table 1; entry a). The substrate scope includes a range of functional groups (Table 1; a–g). Notably, halides are compatible with this system; 4-bromobenzaldehyde was cleanly converted to the borate ester in 82% yield in 2 h. The α,β-unsaturated cinnamaldehyde was selectively hydroborated at the carbonyl group in 76% yield. The bulky mesitaldehyde required heating to 70 °C for 8 h to obtain 52% yield, with no further conversion observed over 16 h. In comparison to 1, Nembenna used 0.5–1 mol% of II (Fig. 1) to achieve good to excellent yields at room temperature in 0.33 h with a range of aldehydes.9
| Catalysis performed in C6D6, at room temperature, 1 mol% pre-catalyst. 1H NMR yields relative to internal standard hexamethylcyclotrisiloxane. [*] heated to 70 °C. |
|---|
|
Hydroboration of ketones was examined next, using 1 mol% of 1 in C6D6 at room temperature. Reaction times were generally found to be longer than with aldehydes. Again, a range of different functionalities were tolerated (Table 1; h–n). Heteroaromatics are compatible: 2-benzoylpyridine is hydroborated cleanly at the carbonyl. As with mesitaldehyde, bulky 2,4,6-trimethyl acetophenone required heating at 70 °C. Deprotonation of the acidic (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)Me group could possibly be interfering in this case, though NMR data did not show any significant amounts of the corresponding Bpin enolate. Alkyl ketones are also tolerated with cyclohexanone requiring 2 h to reach 91% yield. These results are in keeping with those with other Al catalysts: for example, Nembenna used 2 mol% of II (Fig. 1) to hydroborate a range of ketones in high yields in 3–4 h,9 while Roesky used 2 mol% of I for hydroboration of acetophenone (51% in 6 h).81 achieves 80% yield in 4 h with acetophenone.
O)Me group could possibly be interfering in this case, though NMR data did not show any significant amounts of the corresponding Bpin enolate. Alkyl ketones are also tolerated with cyclohexanone requiring 2 h to reach 91% yield. These results are in keeping with those with other Al catalysts: for example, Nembenna used 2 mol% of II (Fig. 1) to hydroborate a range of ketones in high yields in 3–4 h,9 while Roesky used 2 mol% of I for hydroboration of acetophenone (51% in 6 h).81 achieves 80% yield in 4 h with acetophenone.
The stoichiometric reaction of 1 with two equivalents of benzaldehyde yielded crystals of hydrometallated product 8, (HMDS)2Al(μ-OCH2Ph)2Li(THF)2 (Scheme 1 and Fig. 4). Roesky used DFT calculations to propose that the rate determining step of carbonyl hydroboration by Al catalyst, I, is hydroalumination to give a benzyloxide product.88, as the hydrometallated product, is in keeping with this postulate, though here the substrate initially binds to Li not Al. Significantly, applying 8 in a catalytic quantity (1 mol%) for hydroboration of benzaldehyde with HBpin generates the desired product in 80% yield after 2 h. This result is akin to that using catalytic 1, proving that 8 is catalytically competent. The catalysis with Na analogue 7 was found to be less efficient than 1, suggesting a possible alkali metal effect. LiAlH4 was also tested for catalytic activity using solid LiAlH4 in 10 mol% (for ease of weighing owing to its lower molecular weight), and for comparison the catalysis with 1 was repeated using 10 mol% (0.05 mmol). It was found the catalysis with 10 mol% of 1 reached 81% within 0.25 h before the reaction plateaued out, whereas using catalytic LiAlH4 gave 94% within 0.25 h. While this result is slightly better, unlike LiAlH4, 1 hydrolyses slowly, is easily employed in known lower catalyst loadings, and has a well-defined molecular structure that provides insights into the tandem hydroalumination process (e.g. via8). Surprisingly, LiAlH4 has only been used catalytically in a few instances including the hydrosilylation of olefins and dehydrocoupling of amine boranes.19–23
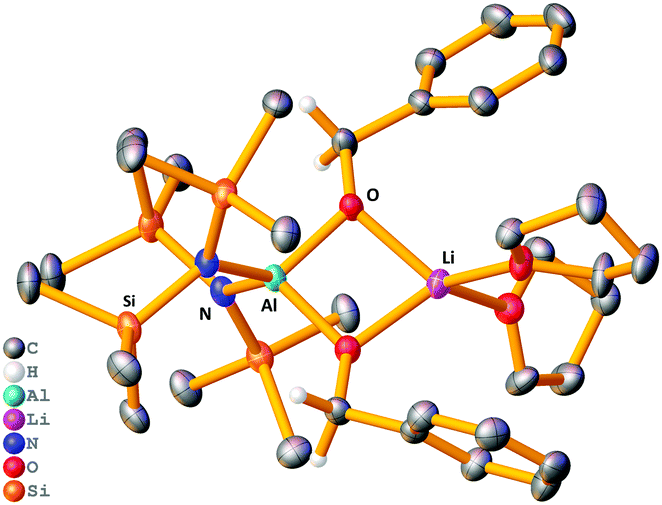 | ||
| Fig. 4 Molecular structure of 8, disordered THF ligands and hydrogen atoms (except benzyloxide CH2) omitted for clarity. Thermal ellipsoids drawn at 40% probability. | ||
Next, the Lewis donor effect on the catalysis was probed using acetophenone as substrate. Table 2 shows a perceptible trend in catalysis efficiency. For example, 1, bearing three labile THF ligands outperforms 4, containing the non-labile chelating PMDETA ligand. This fact may be due to the ease of displacement of THF compared with one arm of the PMDETA ligand, which would inhibit access of substrate to the active metal centre(s) in a catalytic regime. Expanding upon this idea we repeated hydroboration of acetophenone with 1 and 4 in bulk THF-d8, rationalising that catalysis would proceed in similar rates, since excess THF replaces the PMDETA ligand from 4. In each case the NMR yield after 4 h is ca. 95%, giving credence to the dramatic solvent effects displayed in these systems in C6D6. These data imply the catalysts operate through bimetallic cooperation with the hydride transfer emanating from Al, but the initial substrate coordination occurring at Li, the rate of which depends on the relative lability of the donor ligands. This idea is supported by the structure of 8 where the reduced substrate bridges both Al and Li centres.
| Pre-catalyst | X-ray structure | DOSY structure | % yield at 4 h |
|---|---|---|---|
| Reactions performed in C6D6, room temperature, 1 mol% of pre-catalyst, all yields relative to internal standard hexamethylcyclotrisiloxane.a 10 mol% pre-catalyst.b Yield obtained within 0.25 h.c Catalysis performed in d8-THF, 1 mol% pre-catalyst, room temperature (see ESI)d Yield obtained within 2 h. | |||
| 1 | CIP | CIP | 80 |
| 2 | CIP | CIP | 60 |
| 3 | CIP | CIP | 50 |
| 4 | CIP | CIP | 29 |
| 5 | SSIP | CIP | 55 |
| 6 | SSIP | CIP | 15 |
| 7 | CIP | CIP | 57 |
| 1 | CIP | CIP | 81b |
| LiAlH4a | — | — | 94b |
| 1 | CIP | CIP | 95d |
| 4 | CIP | CIP | 99d |
Hydroboration relies on 1 donating a hydride to the carbonyl substrate, however we envisaged other reactivity modes may be available to it. Roesky demonstrated hydroboration of terminal alkynes with HBpin to yield boronate esters with deprotonation a pivotal step.3 In that case hydride was the base, but we pondered whether 1 could also exhibit amido basicity. Preliminary investigations with 1-methyl-1,2,4-triazole gave (HMDS)AlH(μH)[C3H4N3]Li(THF) 9 (yield, 55%) in which the triazole has been aluminated at C5 (Fig. 5). Here one HMDS ligand acts as an internal base.
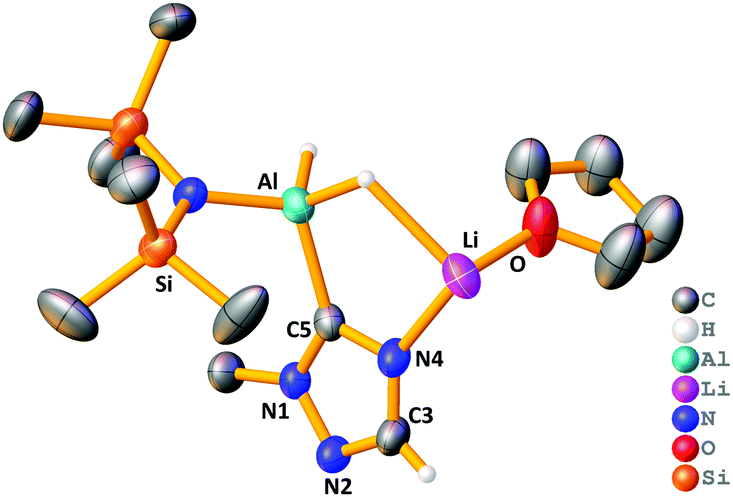 | ||
| Fig. 5 Asymmetric unit of polymeric structure of 9 with triazole ring atoms labelled. Hydrogen atoms (except hydride and C3–H) and disordered THF molecules have been omitted for clarity. Thermal ellipsoids are drawn at 40% probability. | ||
Conforming to the cooperativity between the two metals, the Li also remains within 9, coordinated to N4 on the triazole ring. Crystalline 9 exists as a one-dimensional polymeric chain propagating via Li–N2 bonds between asymmetric units (see ESI‡). Metallation of triazoles typically requires careful control of reaction conditions, with the metallated intermediates fragmenting at ambient temperature,24–26 so it is significant this reaction occurred at room temperature.
In conclusion, this first application of anionic aluminates for hydroboration of aldehydes and ketones establishes that coordination of lithium plays a part though hydride transfer occurs from aluminium, signifying a bimetallic process.
The authors thank the EPSRC (grant, EP/N011384/1 and DTP award to V. A. P.) for funding, and Craig Irving and Alexander Clunie for assistance with analytical facilities. The data set underlying this research can be located at http://www.10.15129/2a1e37f8-5fc5-4df2-aaa6-92751990f568.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- W. Li, X. Ma, M. G. Walawalkar, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2017, 350, 14–29 CrossRef CAS.
- G. I. Nikonov, ACS Catal., 2017, 7, 7257–7266 CrossRef CAS.
- Z. Yang, M. Zhong, X. Ma, K. Nijesh, S. De, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 2548–2551 CrossRef CAS PubMed.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396–8397 CrossRef CAS PubMed.
- C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925–3928 CrossRef CAS PubMed.
- G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2011, 50, 8396–8399 CrossRef PubMed.
- D. R. Armstrong, E. Crosbie, E. Hevia, R. E. Mulvey, D. L. Ramsay and S. D. Robertson, Chem. Sci., 2014, 5, 3031–3045 RSC.
- Z. Yang, M. Zhong, X. Ma, S. De, C. Anusha, P. Parameswaran and H. W. Roesky, Angew. Chem., Int. Ed., 2015, 54, 10225–10229 CrossRef CAS PubMed.
- V. K. Jakhar, M. K. Barman and S. Nembenna, Org. Lett., 2016, 18, 4710–4713 CrossRef CAS PubMed.
- D. Franz, L. Sirtl, A. Pöthig and S. Inoue, Z. Anorg. Allg. Chem., 2016, 642, 1245–1250 CrossRef CAS.
- A. Bismuto, S. P. Thomas and M. J. Cowley, Angew. Chem., Int. Ed., 2016, 55, 15356–15359 CrossRef CAS PubMed.
- R. E. Mulvey and S. D. Robertson, Top. Organomet. Chem., 2014, 47, 129–158 CrossRef CAS.
- Y. Wu, C. Shan, J. Ying, J. Su, J. Zhu, L. L. Liu and Y. Zhao, Green Chem., 2017, 19, 4169–4175 RSC.
- N. W. J. Ang, C. S. Buettner, S. Docherty, A. Bismuto, J. R. Carney, J. H. Docherty, M. J. Cowley and S. P. Thomas, Synthesis DOI:10.1055/s-0036-1591719.
- A. Heine and D. Stalke, Angew. Chem., Int. Ed. Engl., 1992, 31, 854–855 CrossRef.
- H. Osseili, D. Mukherjee, T. P. Spaniol and J. Okuda, Chem. – Eur. J., 2017, 23, 14292–14298 CrossRef CAS PubMed.
- A. R. Kennedy, R. McLellan, G. J. McNeil, R. E. Mulvey and S. D. Robertson, Polyhedron, 2016, 103, 94–99 CrossRef CAS.
- J. W. Akitt, Prog. Nucl. Magn. Reson. Spectrosc., 1989, 21, 1–149 CrossRef.
- S. W. Youn and Y. H. Kim, Synlett, 2000, 880–882 CAS.
- B. Zeynizadeh and L. Sadighnia, Synth. Commun., 2011, 41, 637–644 CrossRef CAS.
- R. J. Less, H. R. Simmonds, S. B. J. Dane and D. S. Wright, Dalton Trans., 2013, 42, 6337–6343 RSC.
- K. Mineo and M. Itoh, Chem. Lett., 1996, 1013–1014 Search PubMed.
- M. Itoh, M. Kobayashi and J. Ishikawa, Organometallics, 1997, 16, 3068–3070 CrossRef CAS.
- S. Ghose and T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 1991, 775–779 RSC.
- R. Raap, Can. J. Chem., 1971, 49, 1792–1798 CrossRef CAS.
- L. Davin, R. McLellan, A. Hernan-Gomez, W. Clegg, A. R. Kennedy, M. Mertens, I. A. Stepek and E. Hevia, Chem. Commun., 2017, 53, 3653–3656 RSC.
Footnotes |
| † Dedicated to Professor Phil Power on the happy occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental, spectroscopic and crystallographic details. CCDC 1579613–1579620. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc08214b |
| This journal is © The Royal Society of Chemistry 2018 |