
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Growth and dissolution of NaO2 in an ether-based electrolyte as the discharge product in the Na–O2 cell†
Iain M.
Aldous
 and
Laurence J.
Hardwick
*
and
Laurence J.
Hardwick
*
Stephenson Institute for Renewable Energy, Department of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK. E-mail: hardwick@liverpool.ac.uk
First published on 16th March 2018
Abstract
The deposition and dissolution of sodium superoxide (NaO2) was investigated by atomic force microscopy. Rectangular prisms consisting of 8 smaller sub-structures grew from NaO2 platelets, when discharged in 0.5 M NaClO4, diethylene glycol dimethyl ether on highly ordered pyrolytic graphite. During oxidation the 8 sub-structures are conserved. Ring-like structures of Na2CO3 of 200 nm diameter remain at the end of oxidation.
Sodium–oxygen (Na–O2) batteries are under investigation due to the advantageous theoretical specific energy of 1106 Wh kg−1 for the reaction (eqn (1)):1
| Na+ + e− + O2 → NaO2 E° = 2.27 V vs. Na+/Na | (1) |
The reduction of dissolved O2 in the presence of Na+ results in the precipitation of large micrometre-sized cubes (1–50 μm)2,3 of NaO2 on the surface of the electrode. Understanding the growth and dissolution mechanism of NaO2 cubes formed by the one-electron reduction of O2 is critical towards the realisation of Na–O2 as a practical technology. NaO2 cubes are often of uniform size; however, their morphology changes based on the rate of formation. At high rates (>600 μA cm−2)2 icosahedral features are formed on the electrode surface.2 At lower rates (<400 μA cm−2)2 they precipitate out as cubes.4 The explanation of these observations is that at higher rates the amount of soluble NaO2 formed rapidly saturates the electrolyte near the electrode interface and precipitates out as smaller cubes at many nucleation points.2 Whereas the slower formation and saturation of the electrolyte at lower rates results in larger cubes at a decreased amount of nucleation points. Subsequently, the reaction is halted by the formation of compact insulating NaO2 films.5,6
Recent reports on the growth mechanism of NaO2 suggest that the soluble NaO2 formed, aided by HO27 catalysed intermediates, agglomerate within the solution to form cubic structures.7 The sudden death of the cell chemistry is attributed to the formation of a passivation film between the cubic structures at the end of the discharge process, where the thickness is discharge rate dependent.8
Although many factors based on NaO2 growth and morphology have been reported including rate,2,8,9 impurity concentration,7,10 and solvent dependence,11–18 a unified model of the fundamental control parameters of the discharge process remains unclear, with different conclusions operating under similar conditions being made. Although there has been an extensive review carried out by Bender et al.1 that ascertains the differences and similarities, Bi et al.19 analysed the cell setup concluding that glass cell setups afford different discharge products (mainly Na2O2·2H2O) in comparison to stainless steel cell setups (NaO2 only).
Atomic force microscopy (AFM) is a powerful method for the investigation of NaO2 growth and dissolution reaction, as the size and morphology of the deposits can be directly measured with nanometer precision. This experimental approach has been already successfully undertaken in a lithium–oxygen (Li–O2) battery system by Wen et al.20 on highly ordered pyrolytic graphite (HOPG) and by Liu et al.21 on Au. On HOPG, agglomerates of LiO2 nanoparticles are collected and grow at step edges forming Li2O2 nanoplates.20 On Au, due to the lack of step edges and a more uniform surface reactivity, Liu et al.21 concluded that a soluble LiO2 species was initially formed that subsequently precipitates out on the surface as Li2O2.22 S. E. Herrera et al.23 studied the formation of Li2O2 in DMSO on HOPG using ex situ AFM, and herein an analogous AFM study in the Na–O2 system of the growth and dissolution of NaO2 precipitates on HOPG is presented.
A CV of O2 saturated 0.5 M NaClO4, diethylene glycol dimethyl ether (DEGDME) on a HOPG surface is shown in Fig. 1a. All stated potentials are vs. Na+/Na. The reductive current increase begins at the thermodynamic potential of NaO2 formation (2.33 V) where it peaks at 1.5 V.
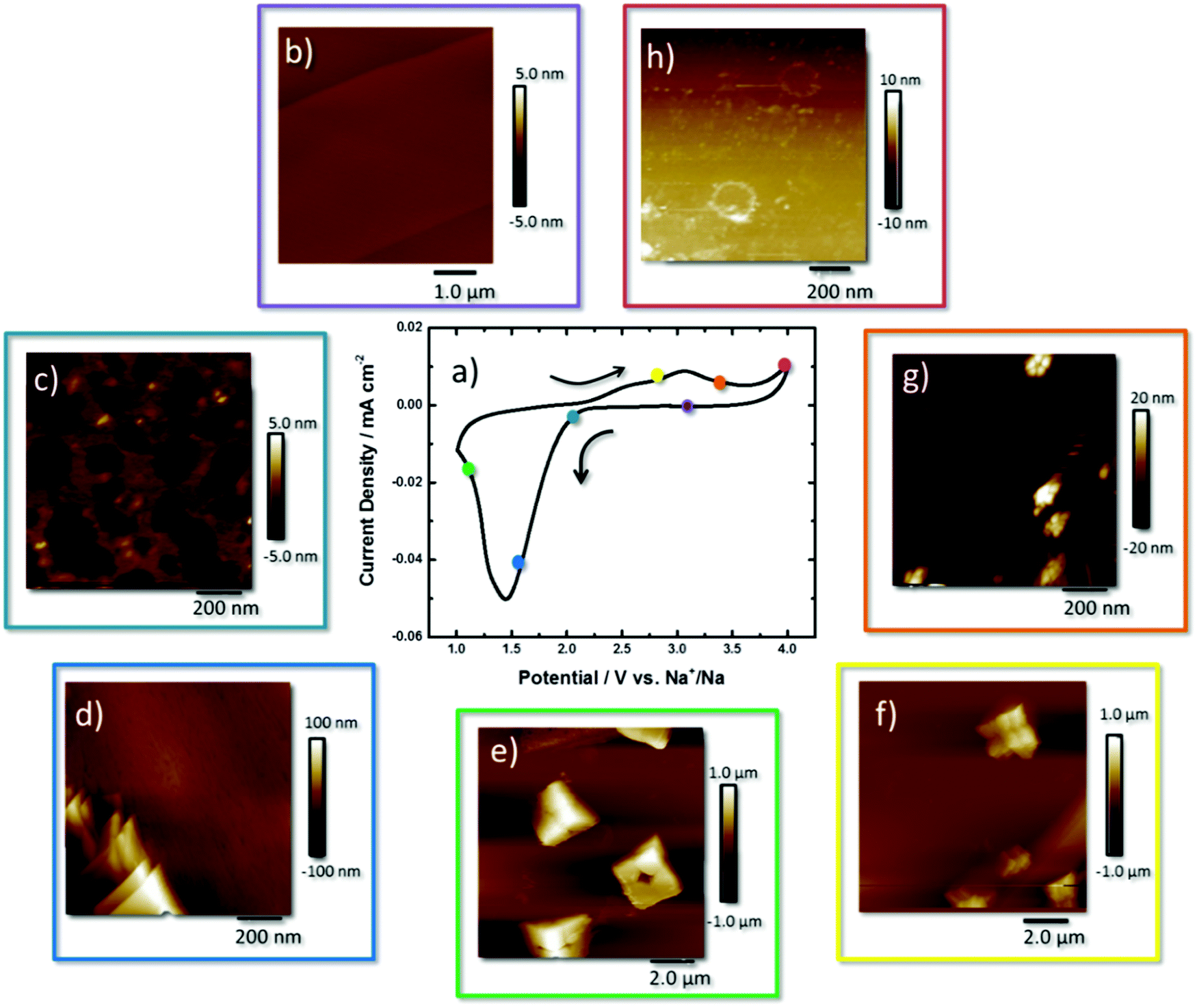 | ||
| Fig. 1 (a) Cyclic voltammogram of HOPG in oxygen saturated, 0.5 M NaClO4, DEGDME, AFM images taken at various potentials around the CV (b) 2.8, (c) 2.10, (d) 1.6, (e) 1.1, (f) 2.8, (g) 3.3, and (h) 4.0 V vs. Na+/Na. | ||
During the reverse scan a broad peak beginning at 2.35 V, reaching a maximum at 3.06 V, signals the electrooxidation of NaO2 back to O2 and Na+. The current increases at 4.0 V, highlighting further oxidation of the remaining reaction products.12,16,24
The sequential description of the AFM images collected around the CV curve begins at the OCP (2.8 V) where the small striations of the freshly cleaved HOPG surface are observed that follow the step edges of the stacked graphene sheets, and the height profiles show that the step edges are 0.8 nm in height (Fig. 1b and Fig. S1, ESI†). At 2.2 V (Fig. 1c) an inhomogeneous 5 nm film of NaO2 was observed on the surface with 20 × 20 nm sized deposits of 5 nm thickness. Calculation of the mass of the discharge product, via the charge passed, estimates an idealised film of 4.7 nm formed on the surface (Table S1, ESI†). Height profiles are shown in the ESI† for all measurements (Fig. S2–S7, ESI†).
The formation of 50–150 nm-thick plate-like structures of NaO2 was observed at 1.9 V (Fig. 1d and Fig. S3, ESI†), and at 1.5 V the formation of cube-like NaO2 crystals at the interface is observed (Fig. 1e and Fig. S4, ESI†). The NaO2 crystals have dimensions of 2 × 2 × 1 μm, with axial lines visible on the surface along with a centralised recess that is half the deposits’ height. These deposits are smaller than those observed in the literature which are generally 2–10 μm in size.25 The Raman spectra (Fig. S8, ESI†) of these deposits show a peak at 1165 cm−1, consistent with a superoxide species. The axial lines reveal a structure whereby the discrete crystal consists of stacked particles that have agglomerated on the surface and grown together. Some crystals appear prismatic or tetragonal in shape coming out of the surface, but still contain the same axial lines defining different cube-like features together, but emerging out of the plane. The depth of the hole within the centre of the crystal is ∼0.5 μm in height as observed from the line profile (Fig. S4, ESI†).
Taking an extended view of the surface, the NaO2 crystals formed at the HOPG surface are all fairly uniform in size and are both scattered across the basal plane and agglomerated along the step edge (Fig. S9, ESI†). Visible along the x- and y-axes of these features are faint lines that reveal cubic building blocks of the overall structure (Fig. 1e). The scale of the hole here suggests that the depth of this hole is in fact the height of the cube (1.0 μm). Upon closer inspection of the height profile, it is actually 0.5 μm (Fig. S4, ESI†). For other deposits, the electrode surface is visible in the middle of the cube (Fig. S10, ESI†). The secondary image taken on top of NaO2 shows imperfect facets of the cubic structure. This is interesting as the majority of studies show uniform NaO2 cubes that appear as perfect plate-like structures.26
Upon reversing the potential back to 2.6 V, the NaO2 are diminished in size with the axial outlines of smaller cubes making up the larger crystals still apparent (Fig. 1f and Fig. S7, ESI†). This observation concurs with the previous state where these NaO2 crystals formed are built essentially from 8 smaller building blocks. By moving the overpotential to 3.25 V (Fig. 1g and Fig. S8, ESI†), beyond the peak current density of the redox process, very little NaO2 is left on the HOPG surface. Only tiny agglomerates of the nanoparticles (ca. 20 nm in height) are observed. At 4.0 V little remains on the surface of the HOPG electrode (Fig. 1h and Fig. S9, ESI†). Nonetheless, in comparison with HOPG at the OCP (Fig. 1b), the image shows some residual material on the electrode surface, some of which form rings of ca. 200 nm diameter and 1.5–7 nm height, suggesting incomplete oxidation of the discharge products. This is possibly the imprints of NaO2 deposits on the surface of the electrode (the line profile shown in Fig. S9, ESI†). The plates could be a coating of side products obtained from other reactions as indicated by Ledano et al.26 and Black et al.27 Only the carbon G band (1583 cm−1) of HOPG is present in the Raman spectrum (Fig. S11a, ESI†). However in the FTIR spectrum, a band for Na2CO3 at 1429 cm−1 (Fig. S11b, ESI†) is detected.
Observations from the AFM study are summarised within Scheme S1 (ESI†) where initially a thin film (2–3 nm) is formed on the surface with deposits of nanoparticles of 5 nm. Platelets an order of magnitude (100 nm) higher than the film appear on the surface. On the surface as the overpotential is increased, agglomeration of cubic structures that grow into rectangular prisms of NaO2 that consist of 8 discrete building blocks is observed. With the start of the oxidation process the edges of each crystal loses definition, leading to the presence of nano-agglomerates left at the interface. The resulting “ghost” shells of the crystal can be seen on the interface identified as Na2CO3.
Further AFM measurements (Fig. S12, ESI†) showed that NaO2 crystallisation chemistry was found to be salt dependent and reduction of O2 in 0.5 M NaTFSI, DEGDME in the electrolyte resulted in the formation of a holed plate-like deposit with larger dimensions of 16 × 16 × 0.5 μm. This identifies that further work needs to be undertaken to fully understand the role of the anion, as well as the ion coordination and solvent types in the electrolytes upon NaO2 precipitation.28
The observation of imperfections on the NaO2 rectangular prism including both holes and axial lines indicates the high mobility of NaO2 particles at the interface to agglomerate and grow in discrete, potential dependent morphologies. This has implications for practical cells where controlling the type of NaO2 precipitate will be necessary for predictable cell behaviour. The high mobility of NaO2 indicates that agglomerates will migrate along the surface until an energy favourable site, such as a defect, is available to deposit. At the initial stage of reduction, the NaO2 is dissolved into the electrolyte. Once the double layer is saturated with NaO2, the deposition and formation of NaO2 crystals can occur.
Evidence of coarsening or Ostwald ripening is found in the initial film and final stages of the reduction of O2 (Fig. 1c) and the oxidation of NaO2 (Fig. 1g). This is provided by the spherical nature of the particles mapped within the AFM images. These are thermodynamically more stable due to surface energy minimisation of the particles through particle dissolution and ion precipitation.29 At this stage of formation, the precipitation of primary nanoparticles is similar to classical nanoparticle growth. Therefore at these stages of the discharge, a high concentration of monomers i.e. O2− and Na+ should prevent particle coarsening and changes in morphology.30,31 These effects would be heightened at an electrode interface due to the formation of the double layer structure. The anion has a considerable influence on the structure of the double layer in each system, and hence, the electrolyte anion affects these stages of NaO2 growth, as demonstrated by the variation in deposit size between NaClO4 (Fig. 1) and NaTFSI (Fig. S12, ESI†). However, classical crystallisation does not characterise the complete mechanism here due to the observation of Rubik cube-like structures.
Precursor layered plates and then Rubik cube-like structures are unsurprising given the observations of oxide formation in this solvent in the lithium analogue of this system.20 From evidence mentioned here the steps between spherical nanoparticles of NaO2 and rectangular prismatic structures suggest that a reoriented aggregation (RO) or mesocrystal mechanism is taking place. RO mechanisms simply encompass the aggregation of building block nanoparticles which then reorient to form single-faced structures again minimising the surface energy. Within the film the initially formed NaO2 nanoparticles are observed (Fig. 1c). These favourable aggregation sites will then form the precipitation sites at the interface. Here collisions of aligned nanocrystals in suspension or the rotation of collided misaligned nanoparticles form the basis of growth at the interface.31,32 Once an agglomerate of a certain size is reached, in some instances coarse ripening has been shown to smooth the edges of the final crystal structures. However, this model is not the best fit. Mesocrystal formation is a better model in this case because of the initial stages of growth proceeding into subsequent smoothing of the final precipitate. In mesocrystal formation the primary nanoparticles are iso-oriented into a crystal via oriented aggregation (OA).33 Organic medium has been shown to temporarily stabilise these particles which form a mesocrystal via mesoscale assembly.33 Fusion can then occur to form an oriented crystal and then finally to a single crystal. These previous observations help to explain the observations of the Rubik cube-like structures within this data series. Within this the final stage of fusion to form a single crystal is not observed (in contrast to many literature reports33,34), most likely due to the much shorter timescale of the voltammetry experiment and the high current rate generation of superoxide.
The process of fusion of mesocrystals, which is thermodynamically favoured under the displacement of the solvent molecules, can often include part of the organic molecule as a coating,33 and this is proposed as a pathway for the formation of a Na2CO3 coating upon the formation of crystals, and a crust of Na2CO3 remaining on the surface of the electrode after oxidation of NaO2.26
The dissolution mechanism should firstly involve the oxidation of the surface layer in contact with the surface and the edges of the rectangular prism agglomerates. The stages characterised in this study would suggest that similar stages of dissolution are present upon dissolution as upon formation of these mesocrystals.
The scan rate used (100 mV s−1) resulted in a high rate of O2 reduction leading to initial film formation by supersaturation of the double layer providing many smaller nucleation points.35 In the medium ionic strength electrolyte (NaClO4/DEGDME), the amount of time a particle of NaO2 can reorient itself before being fixed is lowered.8 This observation is different from that reported by Ledrano et al.6 through in situ electrochemical quartz mass balance measurements on Au electrodes. They concluded that there is only solution-based NaO2 in this instance and that no film is formed until the end of the discharge. Recently Lutz et al.36 have highlighted the different behaviour of this process due to the substrate, especially the difference in the observed morphology between carbon (cubic NaO2) and Au (NaO2 flakes) surfaces. Undoubtedly the electrode surface, solvent, salt and H2O level have major influences on the growth and morphology of NaO2 crystals, and further investigations are required to understand these influences.
In conclusion, ex situ AFM images on HOPG demonstrate the rich and intricate dissolution and recrystallisation chemistry during O2 reduction in an aprotic ether-based solvent in the presence of Na+. An initial formation of a 5 nm NaO2 film followed by subsequent deposition of stacked NaO2 platelets is observed. These plates then grow into crystalline rectangular prisms (2 × 2 × 1 μm) consisting of 8 smaller sub-structures. These were found to preferentially agglomerate at the step edges of HOPG, and also upon the basal plane itself. The observation of building block rectangular prisms that have stacked on top of each other, with striations on the deposit resembling those found on the Rubik's Cube puzzle, was unexpected, and this method of NaO2 precipitation results in the observed 0.5 μm deep holes present in the central face of the NaO2. Upon oxidation the dissolution of the rectangular prismatic structure conserves an unevenly shaped collection of 8 sub-structures that degrades further to groups of 40 nm-sized deposits. Rings of 200 nm diameter remain on the surface after oxidation, and these rings are thought to be the unoxidised side reaction product (Na2CO3) that forms on the surface of NaO2.
Support from the Engineering and Physical Sciences Research Council grant EP/J020265/1 and the Energy SuperStore, Early Career Researcher Award is acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. L. Bender, D. Schröder, R. Pinedo, P. Adelhelm and J. Janek, Angew. Chem., Int. Ed., 2016, 55, 4640–4649 CrossRef CAS PubMed.
- D. Schröder, C. L. Bender, M. Osenberg, A. Hilger, I. Manke and J. Janek, Sci. Rep., 2016, 6, 24288 CrossRef PubMed.
- S. Y. Sayed, K. P. C. Yao, D. G. Kwabi, T. P. Batcho, C. V. Amanchukwu, S. Feng, C. V. Thompson and Y. Shao-Horn, Chem. Commun., 2017, 53, 460 RSC.
- N. Ortiz-Vitoriano, T. P. Batcho, D. G. Kwabi, B. Han, N. Pour, K. P. C. Yao, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 2636–2643 CrossRef CAS PubMed.
- K. B. Knudsen, J. E. Nichols, T. Vegge, A. C. Luntz, B. D. McCloskey and J. Hjelm, J. Phys. Chem. C, 2016, 120, 10799–10805 CAS.
- I. Landa-Medrano, J. T. Frith, I. Ruiz de Larramendi, I. Lozano, N. Ortiz-Vitoriano, N. Garcia-Araez and T. Rojo, J. Power Sources, 2017, 345, 237–246 CrossRef CAS.
- C. Xia, R. Black, R. Fernandes, B. Adams and L. F. Nazar, Nat. Chem., 2015, 7, 496–501 CrossRef CAS PubMed.
- J. E. Nichols and B. D. McCloskey, J. Phys. Chem. C, 2017, 121, 85–96 CAS.
- N. Zhao, C. Li and X. Guo, Phys. Chem. Chem. Phys., 2014, 16, 15646 RSC.
- J. Kim, H. Park, B. Lee, W. M. Seong, H. D. Lim, Y. Bae, H. Kim, W. K. Kim, K. H. Ryu and K. Kang, Nat. Commun., 2016, 7, 10670–10679 CrossRef CAS PubMed.
- L. Lutz, W. Yin, A. Grimaud, D. Alves Dalla Corte, M. Tang, L. Johnson, E. Azaceta, V. Sarou-Kanian, A. J. Naylor, S. Hamad, J. A. Anta, E. Salager, R. Tena-Zaera, P. G. Bruce and J. M. Tarascon, J. Phys. Chem. C, 2016, 120, 20068–20076 CAS.
- I. M. Aldous and L. J. Hardwick, Angew. Chem., Int. Ed., 2016, 55, 8254–8257 CrossRef CAS PubMed.
- B. Wang, N. Zhao, Y. Wang, W. Zhang, W. Lu, X. Guo and J. Liu, Phys. Chem. Chem. Phys., 2017, 19, 2940–2949 RSC.
- J. Kim, H.-D. Lim, H. Gwon and K. Kang, Phys. Chem. Chem. Phys., 2013, 15, 3623 RSC.
- I. I. Abate, L. E. Thompson, H. C. Kim and N. B. Aetukuri, J. Phys. Chem. Lett., 2016, 7, 2164–2169 CrossRef CAS PubMed.
- B. Wang, N. Zhao, Y. Wang, W. Zhang, W. Lu, X. Guo and J. Liu, Phys. Chem. Chem. Phys., 2016, 19, 2940–2949 RSC.
- B. Sun, K. Kretschmer, X. Xie, P. Munroe, Z. Peng and G. Wang, Adv. Mater., 2017, 1606816 CrossRef PubMed.
- N. Zhao and X. Guo, J. Phys. Chem. C, 2015, 119, 25319–25326 CAS.
- X. Bi, R. Wang, L. Ma, D. Zhang, K. Amine and J. Lu, Small Methods, 2017, 1, 1700102 CrossRef.
- R. Wen, M. Hong and H. R. Byon, J. Am. Chem. Soc., 2013, 135, 10870–10876 CrossRef CAS PubMed.
- C. Liu and S. Ye, J. Phys. Chem. C, 2016, 120, 25246–25255 CAS.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J.-M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- S. E. Herrera, A. Y. Tesio, R. Clarenc and E. J. Calvo, Phys. Chem. Chem. Phys., 2014, 16, 9925–9929 RSC.
- J. Kim, H.-D. Lim, H. Gwon and K. Kang, Phys. Chem. Chem. Phys., 2013, 15, 3623–3629 RSC.
- N. Ortiz-Vitoriano, T. P. Batcho, D. G. Kwabi, B. Han, N. Pour, K. P. C. Yao, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 2636–2643 CrossRef CAS PubMed.
- I. Landa-Medrano, A. Sorrentino, L. Stievano, I. Ruiz de Larramendi, E. Pereiro, L. Lezama, T. Rojo and D. Tonti, Nano Energy, 2017, 37, 224–231 CrossRef CAS.
- R. Black, A. Shyamsunder, P. Adeli, D. Kundu, G. K. Murphy and L. F. Nazar, ChemSusChem, 2016, 9, 1795–1803 CrossRef CAS PubMed.
- M. He, K. C. Lau, X. Ren, N. Xiao, W. D. McCulloch, L. A. Curtiss and Y. Wu, Angew. Chem., Int. Ed., 2016, 55, 15310–15314 CrossRef CAS PubMed.
- I. M. Lifshitz and V. V. Slyozov, J. Phys. Chem. Solids, 1961, 19, 35–50 CrossRef.
- X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS PubMed.
- E. J. H. Lee, C. Ribeiro, E. Longo and E. R. Leite, J. Phys. Chem. B, 2005, 109, 20842–20846 CrossRef CAS PubMed.
- C. Ribeiro, E. J. H. Lee, T. R. Giraldi, E. Longo, J. A. Varela and E. R. Leite, J. Phys. Chem. B, 2004, 108, 15612–15617 CrossRef CAS.
- M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC.
- J. Fang, B. Ding, X. Song and Y. Han, Appl. Phys. Lett., 2008, 92, 17120–17123 Search PubMed.
- L. C. Soare, P. Bowen, J. Lemaitre and H. Hofmann, J. Phys. Chem. B, 2006, 110, 17763–17771 CrossRef CAS PubMed.
- L. Lutz, D. A. D. Corte, Y. Chen, D. Batuk, L. R. Johnson, A. Abakumov, L. Yate, E. Azaceta, P. G. Bruce, J.-M. Tarascon and A. Grimaud, Adv. Energy Mater., 2017, 1701581 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, line profiles from AFM images and Raman spectra of NaO2 deposits and FTIR characterisation of residual films after oxidation. See DOI: 10.1039/c7cc08201k |
| This journal is © The Royal Society of Chemistry 2018 |