
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manipulating and quantifying spin states in solution as a function of pressure and temperature†
Ross W.
Hogue
 a,
Christopher P.
Lepper
b,
Geoffrey B.
Jameson
*b and
Sally
Brooker
*a
a,
Christopher P.
Lepper
b,
Geoffrey B.
Jameson
*b and
Sally
Brooker
*a
aDepartment of Chemistry and MacDiarmid Institute for Advanced Materials and Nanotechnology, University of Otago, PO Box 56, Dunedin 9054, New Zealand. E-mail: sbrooker@chemistry.otago.ac.nz
bChemistry – Institute of Fundamental Sciences and MacDiarmid Institute for Advanced Materials and Nanotechnology, Massey University, Private Bag 11 222, Palmerston North 4442, New Zealand. E-mail: G.B.Jameson@massey.ac.nz
First published on 30th November 2017
Abstract
Monitoring the spin states of species in solution is a crucial aspect of understanding magnetic properties as well as spin-labile sensing, supramolecular, catalytic and biochemical processes. Herein, we describe the first quantitative variable-pressure and variable-temperature method of determining spin states in solution, demonstrate that it is accurate, and identify a simultaneous T and P sensor system.
Determining the spin state of a system is fundamental to understanding its magnetic properties and chemical reactivity. The study of molecular magnetic phenomena1 relies upon the measurement of spin states under different conditions. Understanding reactivity2 and catalytic processes3 is greatly aided by being able to determine the spin states of various reactants, intermediates and products. Measurements of spin states in the solution phase are of particular importance, especially for the study of biologically relevant processes, including photosynthesis4 and iron-containing enzyme activity,5 noting that life persists at pressures in excess of 100 MPa (∼1000 atmospheres). However, there is no established method of quantifying spin states in solution as a function of both pressure and temperature.
The application of hydrostatic pressure to spin-labile compounds typically favours the low spin (LS) state due to the smaller molecular volume compared to the high spin (HS) state.6 While this has been observed in pressure-induced solution spin crossover (SCO), by the Gouy method,7 and UV-vis8 and 1H NMR9 spectroscopies, these have all been qualitative measures of spin states. Herein, we establish a method for the accurate quantitative measurement of spin states in solution at variable pressures and temperatures, by extension of the ambient pressure Evans 1H NMR ‘tube-in-tube’ method10 to enable the use of a ‘single-tube’ that can be pressurised to 240 MPa. In addition to quantitative spin-state information, the 1H NMR spectra themselves can also yield useful chemical and 3D structural information about the subject complex.11 Thus, this method adds spin-state capabilities to high-pressure NMR techniques such as those already applied to host–guest,12 catalytic,13 and biomacromolecular systems.14
In order to develop this method of quantifying solution spin states under pressure, three of our recently reported solution-phase (thermally-induced) SCO-active dinuclear iron(II) complexes of 4-substituted-3,5-bis{[(2-pyridylmethyl)-sulfanyl]methyl}-4H-1,2,4-triazole (PSRT) ligands, where R = Ph, MePh or iBu, i.e. [Fe2(PSPhT)2](BF4)4, [Fe2(PSMePhT)2](BF4)4, and [Fe2(PSiBuT)2](BF4)4 (Fig. 1), were investigated by high-pressure 1H NMR spectroscopy. Spectra were recorded in CD3CN solution at 30 MPa intervals between 0.1 MPa (atmospheric pressure) and 240 MPa, and at 5 different temperatures in the range of 278–313 K.
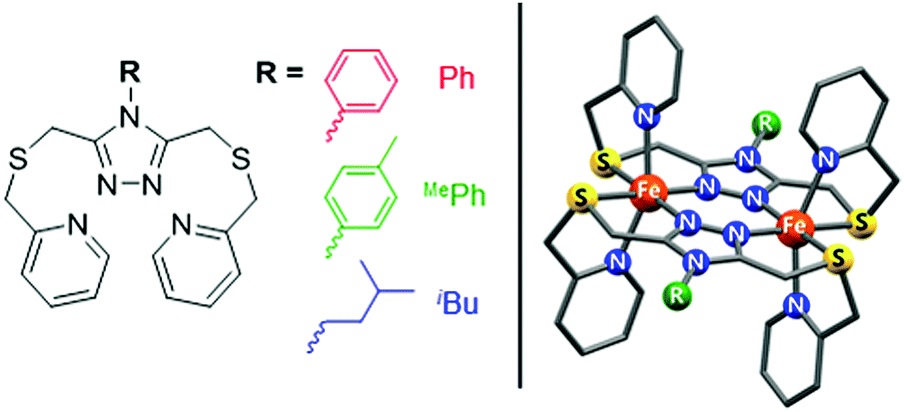 | ||
| Fig. 1 PSRT ligand with the three R groups used in this study (left), and the structure of the [Fe2(PSRT)2]4+ cations (right). | ||
In each spectrum (Fig. 2 and Fig. S1–S3, ESI†), very broad and highly downfield shifted (up to ∼100 ppm) proton signals typical of a paramagnetic substance are observed, indicating population of the HS state. As pressure is increased, these downfield signals shift upfield or towards a “normal” position expected for a diamagnetic material, indicating that a pressure-induced SCO towards the LS state occurs. At 240 MPa, the proton signals are still relatively far downfield, indicating that a significant population of the HS state remains, consistent with the SCO being incomplete within the pressure range studied. Proton signals that were shifted upfield by the paramagnetism (to ∼−5 ppm) concomitantly move downfield towards their diamagnetic values as pressure is increased.
 | ||
| Fig. 2 Stacked 1H NMR spectra of [Fe2(PSPhT)2](BF4)4 in CD3CN solution showing a qualitative SCO at room temperature (293 K) from 0.1 MPa (top) to 240 MPa (bottom). Note that the spectra have been truncated to highlight the downfield shifted signals of some methylene and pyridyl protons (left) and phenyl protons (centre), and the upfield shifted pyridyl proton signal (right). For the full spectra, see Fig. S1, ESI.† | ||
To quantitatively analyse the spin states at each pressure, we turned to the Evans method which under standard conditions (ambient pressure) uses a tube-in-tube experiment. The inner tube contains pure deuterated solvent and the outer tube contains the paramagnetic material in the same solvent. The frequency shift of the solvent signal, Δf, in the outer tube compared to the pure solvent (to which the NMR spectrum is locked), is dependent on the magnetic susceptibility of the paramagnetic material, and hence χMT can be calculated from Δf (Hz), the concentration m (g cm−3), and the spectrometer frequency f (Hz) using the Evans method (eqn (1)) to obtain the mass susceptibility χg (cm3 g−1).10,15
| χg = 3Δf/(4πmf) | (1) |
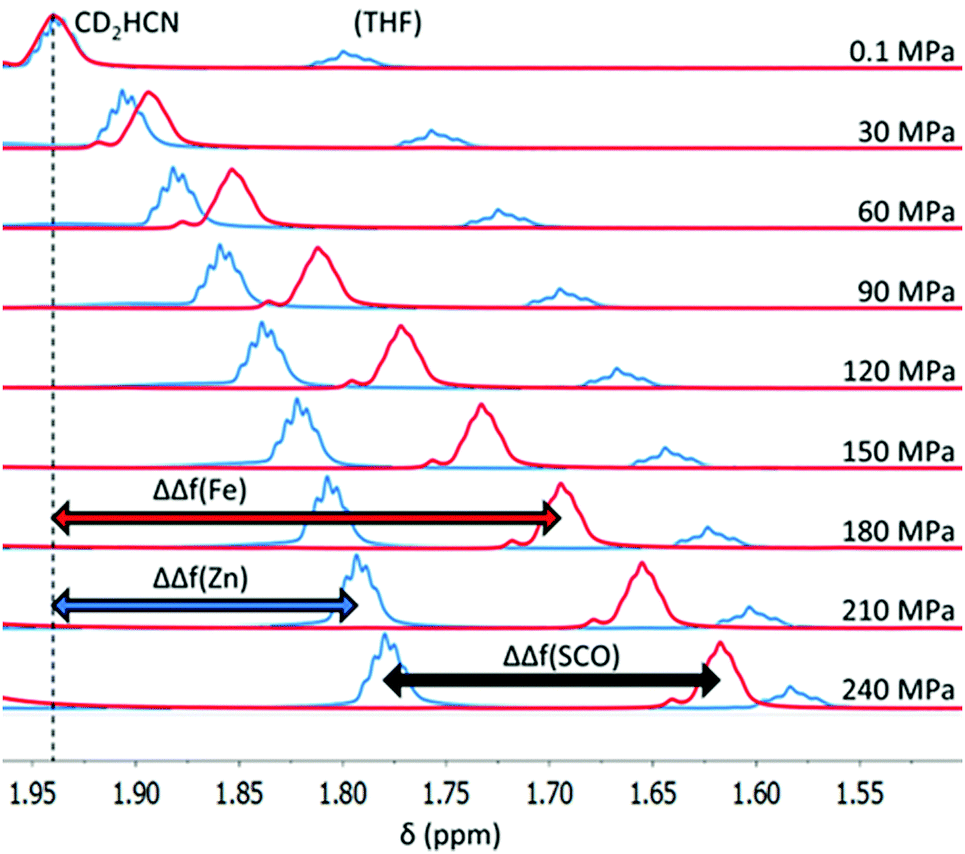 | ||
| Fig. 3 Frequency shift of the CD2HCN signal for [Fe2(PSPhT)2](BF4)4 (red spectra) and the diamagnetic analogue [Zn2(PSiBuT)2](BF4)4 (blue spectra) from 0.1 MPa to 240 MPa at 293 K. The magnitude of the shifts, ΔΔf(Fe) and ΔΔf(Zn), for the Fe(II) and Zn(II) solutions respectively, are indicated, as well as their difference, ΔΔf(SCO), which is due to a decrease in magnetic susceptibility in the Fe(II) solution. Note: to aid visualisation, both [Fe2(PSPhT)2](BF4)4 spectra (red) and [Zn2(PSiBuT)2](BF4)4 spectra (blue) are manually referenced such that the CD2HCN signal is at 1.94 ppm at 0.1 MPa, because ΔΔf(Fe), ΔΔf(Zn), and ΔΔf(SCO) are defined as relative to atmospheric pressure. | ||
For each Fe(II) solution, as the pressure increased the CD2HCN signal shifted upfield (Fig. 3), resulting in increasingly negative values of ΔΔf(Fe) (Fig. 3, red), which is consistent with the SCO towards the LS state. However, with the lock signal off, the CD2HCN peak will gradually drift upfield with time in the absence of any pressure/temperature changes or change in magnetic susceptibility of the sample. This is due to the magnetic field strength of the NMR slowly, and reliably, decreasing with time after tuning. The magnitude of this effect is small in comparison to the signal shift upon pressure changes, but nonetheless time corrections were applied to ΔΔf(Fe) values to minimise error (see the ESI† for details).
A correction must also be made for the CD2HCN signal frequency shift with increasing pressure for a diamagnetic solution, so that the magnitude of the shift in the Fe(II) solution due to the SCO can be ascertained. For this purpose, the Zn(II) analogues of each complex, [Zn2(PSRT)2](BF4)4, were synthesised (see the ESI†). Typically in paramagnetic NMR studies, a free ligand is used as a diamagnetic reference; however, as this is the first high-pressure NMR magnetic susceptibility study, in order to check for any unforeseen chaotrope/kosmotrope effects with varying pressure, the Zn(II) complexes, which are of the same size, shape and charge as the Fe(II) complexes and so were considered to be superior to the free ligands, were also tested. Conveniently, this test demonstrates that the free ligand is in fact a perfectly acceptable diamagnetic reference, as it responds in the same way as the Zn(II) complexes do (Fig. S4, ESI†). 1H NMR spectra of the Zn(II) analogues, at the same concentration in CD3CN as the corresponding Fe(II) complexes, were recorded to determine the frequency shift of CD2HCN relative to atmospheric pressure for the diamagnetic Zn(II) solutions, defined as ΔΔf(Zn) (Fig. 3, blue). This enabled point-by-point corrections, for ΔΔf(Zn) at each pressure and temperature investigated, to be made to the Fe(II) spectra. The resulting ΔΔf(SCO) = ΔΔf(Fe) − ΔΔf(Zn) values are the frequency shifts of CD2HCN relative to atmospheric pressure due only to a change in magnetic susceptibility of the Fe(II) solution (Fig. 3).
From the known χMT at atmospheric pressure,17eqn (1) can be used to calculate the Δf for the particular temperature and concentration of the Fe(II) solution, which we label Δf0, the frequency shift of CD2HCN at atmospheric pressure relative to that of an internal reference standard of pure CD3CN, if it was present in the high pressure cell. ΔΔf(SCO) is then the change in this frequency shift, arising from the change in spin state only, as pressure is applied. Hence, the frequency shift of CD2HCN at each pressure relative to pure CD3CN at that pressure is obtained simply by Δf = Δf0 + ΔΔf(SCO). Finally, χg at each pressure can then be calculated from these Δf values using eqn (1), considering the changes in the density of acetonitrile (and hence concentration of the complex), to give a quantitative description of the spin state at any pressure/temperature.
Applying this method to CD3CN solutions of [Fe2(PSRT)2](BF4)4 reveals the solution SCO induced by both pressure and temperature for each of the three complexes (Fig. 4, Fig. S7–S12, and Tables S9–S11, ESI†). The SCO is gradual and incomplete within the pressure range available. The [Fe2(PSRT)2](BF4)4 series of complexes are potential candidates for solution sensor applications, i.e. for simultaneous sensing of pressure and temperature in solution over, in particular, a wide range of pressure, based on their different magnetic susceptibility responses.18
 | ||
| Fig. 4 χ M T vs. pressure vs. temperature for CD3CN solutions of (left) [Fe2(PSPhT)2](BF4)4, (centre) [Fe2(PSMePhT)2](BF4)4, and (right) [Fe2(PSiBuT)2](BF4)4. The colour coding shows the wide and varied magnetic responses across the three complexes as a function of temperature and pressure. | ||
The proton signals observed in the 1H NMR spectrum (Fig. 2) are the population-weighted average signals of the HS and LS states, and therefore the extent to which the signals are shifted from the fully LS spectrum reflects the magnetic susceptibility of the complex in solution. Assuming the ideal Curie behaviour, the chemical shift, δ, of a proton signal has a linear relationship with χM.11b This allows for the correlation of the processed data, χM, as calculated by our adapted Evans method, with raw δ spectral data for pyridyl protons py-H4 and py-H5 (Fig. 5 and Fig. S14–S19, ESI†). Indeed good linear relationships are observed, indicating a high level of accuracy in the adapted high pressure Evans method of calculating χM or χMT.
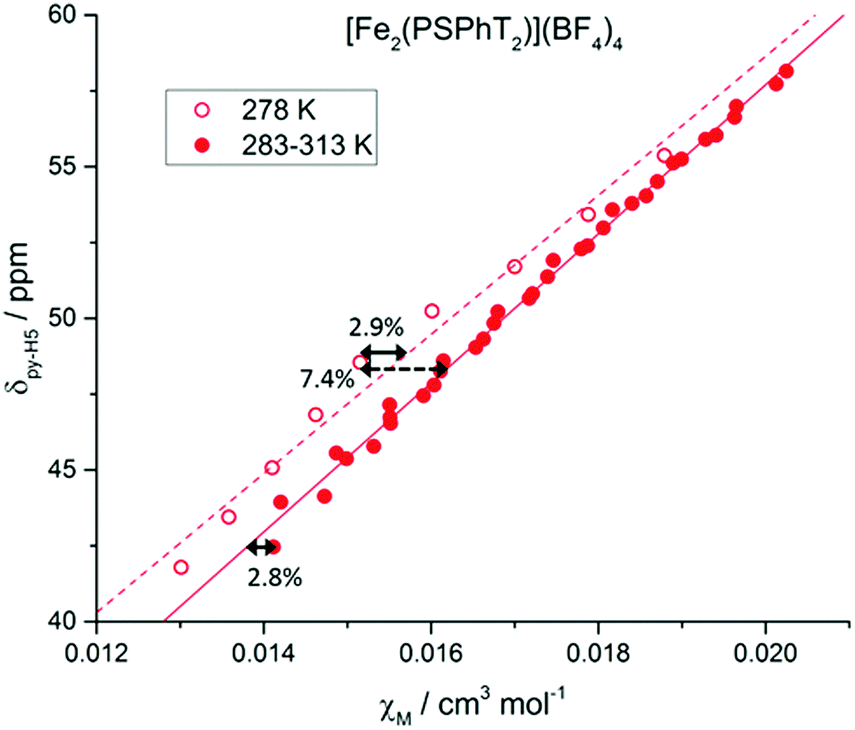 | ||
| Fig. 5 δ(py-H5) vs. χM for [FeII2(PSPhT)2](BF4)4 in CD3CN at variable pressure/temperature. Hollow data points represent the spectra recorded at 278 K in one solution, with the linear fit shown as the dashed line. Solid data points recorded on another solution at 283–313 K, with a solid line for the linear fit. For the χM values calculated for [FeII2(PSPhT)2](BF4)4, the maximum error between data points for the same solution is 2.8–2.9%, and the maximum error between data points for different solutions is 7.4%. See the ESI† for more details. | ||
The most significant error here is in knowing the exact concentration of the solutions used, which is always the case for the standard Evans method and typically results in 5–10% error.19 The error associated with the extra corrections needed (time, Zn analogues) for our adapted method is smaller (3–5%) and contains the error in accurately reading the signal position, which is also present in the standard method. Therefore, no significantly large errors are introduced through our extension of the Evans method from a tube-in-tube method to a single tube method that can be applied to the study of pressure and temperature effects.
It is possible to calculate the fraction HS, γHS, directly from proton chemical shifts11b,c instead of using the Evans method. However, these methods require a fully HS baseline to be reached11b and/or a very wide (200 K) temperature range.11c Although our method requires careful application of corrections, it produces quantitative χMT values (but not γHS) and can operate in narrow ranges of magnetic susceptibility and temperature.
We have described a robust method for determining the spin states quantitatively in high-pressure solutions. In the test case of the [Fe2(PSRT)2](BF4)4 complexes, an SCO was observed, and the spin state is tuned by both pressure and temperature in solution, raising the possibility of solution-based simultaneous pressure/temperature sensing. Unlike a thermally induced SCO, which is limited by the freezing/boiling points of the solvent, the pressure is limited only by instrumentation and so has the potential to allow access to a much wider range of spin states in solution. Although applied here only to simple Fe(II) SCO compounds, this robust method could be extended to study the effects of pressure on spin-labile (bio)catalysts, sensors or host–guest chemical processes.
We thank the University of Otago (for a PhD scholarship to RWH), the Marsden Fund administered by the Royal Society of New Zealand (UOO1318 to SB and 09-MAU-140 to GBJ) and the Allan Wilson Centre for Molecular Evolution and Ecology for supporting this research.
Conflicts of interest
There are no conflicts to declare.References
- (a) E. Coronado, P. Delhaès, D. Gatteschi and J. S. Miller, Molecular Magnetism: From Molecular Assemblies to the Devices, Springer, Netherlands, Dordrecht, 1996 Search PubMed; (b) M. A. Halcrow, Spin-Crossover Materials: Properties and Applications, John Wiley & Sons, Ltd, Chichester, 1st edn, 2013 Search PubMed; (c) O. Kahn, Molecular Magnetism, VCH Publishers Inc., New York, 1993 Search PubMed; (d) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- (a) J. N. Harvey, R. Poli and K. M. Smith, Coord. Chem. Rev., 2003, 238–239, 347–361 CrossRef CAS; (b) R. Poli, Chem. Rev., 1996, 96, 2135–2204 CrossRef CAS PubMed.
- (a) Y.-Y. Jiang, J.-L. Jiang and Y. Fu, Organometallics, 2016, 35, 3388–3396 CrossRef CAS; (b) D. Schröder, S. Shaik and H. Schwarz, Acc. Chem. Res., 2000, 33, 139–145 CrossRef; (c) P. L. Holland, Acc. Chem. Res., 2015, 48, 1696–1702 CrossRef CAS PubMed; (d) R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485–1493 CrossRef CAS PubMed; (e) S. B. Muñoz Iii, S. L. Daifuku, W. W. Brennessel and M. L. Neidig, J. Am. Chem. Soc., 2016, 138, 7492–7495 CrossRef PubMed.
- (a) J. C. De Paula and G. W. Brudvig, J. Am. Chem. Soc., 1985, 107, 2643–2648 CrossRef CAS; (b) R. Chatterjee, G. Han, J. Kern, S. Gul, F. D. Fuller, A. Garachtchenko, I. D. Young, T.-C. Weng, D. Nordlund, R. Alonso-Mori, U. Bergmann, D. Sokaras, M. Hatakeyama, V. K. Yachandra and J. Yano, Chem. Sci., 2016, 7, 5236–5248 RSC; (c) V. Krewald, M. Retegan, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Inorg. Chem., 2016, 55, 488–501 CrossRef CAS PubMed; (d) H. Kim, J. Park, I. Park, K. Jin, S. E. Jerng, S. H. Kim, K. T. Nam and K. Kang, Nat. Commun., 2015, 6, 8253 CrossRef CAS PubMed; (e) V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- (a) A. R. McDonald and L. Que Jr, Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS; (b) Y. Sugiura, J. Kuwahara, T. Nagasawa and H. Yamada, J. Am. Chem. Soc., 1987, 109, 5848–5850 CrossRef CAS; (c) A. J. Boone, C. H. Chang, S. N. Greene, T. Herz and N. G. J. Richards, Coord. Chem. Rev., 2003, 238–239, 291–314 CrossRef CAS; (d) D. Sahoo, M. G. Quesne, S. P. de Visser and S. P. Rath, Angew. Chem., Int. Ed., 2015, 54, 4796–4800 CrossRef CAS PubMed; (e) R.-J. Cheng, P.-Y. Chen, P.-R. Gau, C.-C. Chen and S.-M. Peng, J. Am. Chem. Soc., 1997, 119, 2563–2569 CrossRef CAS.
- (a) P. Gütlich, V. Ksenofontov and A. B. Gaspar, Coord. Chem. Rev., 2005, 249, 1811–1829 CrossRef; (b) H. G. Drickamer and C. W. Frank, Electronic Transitions and the High Pressure Chemistry and Physics of Solids, Springer, Netherlands, Dordrecht, 1973, pp. 126–151 Search PubMed.
- (a) A. H. Ewald, R. L. Martin, E. Sinn and A. H. White, Inorg. Chem., 1969, 8, 1837–1846 CrossRef CAS; (b) A. H. Ewald, R. L. Martin, I. G. Ross and A. H. White, Proc. R. Soc. London, Ser. A, 1964, 280, 235–257 CrossRef CAS.
- W. S. Hammack, A. J. Conti, D. N. Hendrickson and H. G. Drickamer, J. Am. Chem. Soc., 1989, 111, 1738–1741 CrossRef CAS.
- O. Troeppner, R. Lippert, T. E. Shubina, A. Zahl, N. Jux and I. Ivanović-Burmazović, Angew. Chem., Int. Ed., 2014, 53, 11452–11457 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- (a) I. Bertini, C. Luchinat, G. Parigi and E. Ravera, NMR of Paramagnetic Molecules, Elsevier, Boston, 2nd edn, 2017, pp. 1–24 Search PubMed; (b) B. Weber and F. A. Walker, Inorg. Chem., 2007, 46, 6794–6803 CrossRef CAS PubMed; (c) H. Petzold, P. Djomgoue, G. Horner, J. M. Speck, T. Ruffer and D. Schaarschmidt, Dalton Trans., 2016, 45, 13798–13809 RSC; (d) C. Piguet, E. Rivara-Minten, G. Bernardinelli, J.-C. G. Bunzli and G. Hopfgartner, J. Chem. Soc., Dalton Trans., 1997, 421–434 RSC; (e) K. L. Bren, Spin States in Biochemistry and Inorganic Chemistry, John Wiley & Sons, Ltd, 2015, pp. 409–434 Search PubMed; (f) Z. Ni and M. P. Shores, J. Am. Chem. Soc., 2009, 131, 32–33 CrossRef CAS PubMed.
- A. V. Davis, D. Fiedler, G. Seeber, A. Zahl, R. van Eldik and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 1324–1333 CrossRef CAS PubMed.
- (a) L. Damoense, M. Datt, M. Green and C. Steenkamp, Coord. Chem. Rev., 2004, 248, 2393–2407 CrossRef CAS; (b) C. L. Dwyer, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree, Elsevier, Oxford, 2007, pp. 483–507 Search PubMed.
- R. Fourme, E. Girard and K. Akasaka, Curr. Opin. Struct. Biol., 2012, 22, 636–642 CrossRef CAS PubMed.
- C. Piguet, J. Chem. Educ., 1997, 74, 815–816 CrossRef CAS.
- J. Jonas, in High Pressure Molecular Science, ed. R. Winter and J. Jonas, Springer, Netherlands, Dordrecht, 1999, pp. 231–259 Search PubMed.
- R. W. Hogue, H. L. C. Feltham, R. G. Miller and S. Brooker, Inorg. Chem., 2016, 55, 4152–4165 CrossRef CAS PubMed.
- (a) J. Linares, E. Codjovi and Y. Garcia, Sensors, 2012, 12, 4479–4492 CrossRef CAS PubMed; (b) C.-M. Jureschi, J. Linares, A. Boulmaali, P. Dahoo, A. Rotaru and Y. Garcia, Sensors, 2016, 16, 187 CrossRef PubMed; (c) K. Y. Maryunina, X. Zhang, S. Nishihara, K. Inoue, V. A. Morozov, G. V. Romanenko and V. I. Ovcharenko, J. Mater. Chem. C, 2015, 3, 7788–7791 RSC.
- L. A. Yatsunyk and F. A. Walker, Inorg. Chem., 2004, 43, 757–777 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, high pressure instrumentation, detailed NMR data collection and processing, and X-ray crystallography. CCDC 1554635. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc08104a |
| This journal is © The Royal Society of Chemistry 2018 |