
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-Mediated molecular differentiation for fluorimetric quantification of chemotherapeutic drugs in human plasma†
Luis A.
Serrano
 a,
Ye
Yang
a,
Elisa
Salvati‡
b,
Francesco
Stellacci
c,
Silke
Krol
de and
Stefan
Guldin
*a
a,
Ye
Yang
a,
Elisa
Salvati‡
b,
Francesco
Stellacci
c,
Silke
Krol
de and
Stefan
Guldin
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. E-mail: s.guldin@ucl.ac.uk
bIFOM, The FIRC Institute for Molecular Oncology Foundation, IFOM-IEO Campus, Via Adamello, 20139 Milan, Italy
cInstitute of Materials, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland
dGiovanni Paolo II/I.R.C.C.S. Cancer Institute, Viale Orazio Flacco, 70124 Bari, Italy
eNanoMed lab, Fondazione IRCCS Institute of Neurology “Carlo Besta”, via Amadeo 42, 20133 Milan, Italy
First published on 23rd January 2018
Abstract
At present, drug dosage is based on standardised approaches that disregard pharmakokinetic differences between patients and lead to non-optimal efficacy and unnecessary side effects. In this work, we demonstrate the potential of pH-mediated fluorescence spectroscopy for therapeutic drug monitoring in complex media. We apply this principle to the simultaneous quantification of the chemotherapeutic prodrug Irinotecan and its active metabolite SN-38 from human plasma across the clinically relevant concentration range, i.e. from micromolar to nanomolar at molar ratios of up to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
Therapeutic drug monitoring (TDM), i.e. the preferably close to real time measurement of medication concentration in the blood, allows to define and maintain a precise therapeutic window for drug administration, which is of particular importance for highly toxic or expensive medication used in chemotherapy and personalised medicine.1 So far, drug dosage is typically calculated on the basis of the body surface of the patient without patient-specific analytical feedback.2 The significant pharmacokinetic variability between patients results in either ineffective treatment or adverse toxic effects and thus failure to reach the therapeutic goal for a large percentage of drugs being administered.3,4
The gold standard for TDM is high-performance liquid chromatography (HPLC) combined with a suitable detection method, such as mass spectrometry, spectrophotometry or spectrofluorimetry, for which many protocols are available.5,6 Nevertheless, HPLC exhibits some inherent drawbacks, in particular the need to operate in a specialised analytical laboratory with off-site transport and long waiting times as well as elaborate sample preparation and analysis leading to overall high costs.7 Another problem, which is often underestimated, is the variability in pre-analytic sample handling and the related inconsistencies between the measured drug concentration and the real drug concentration in blood.8 Consequently, a simplified drug monitoring that is closer to the point of care would offer a better definition of the therapeutic window as well as continuous patient feedback for optimised efficacy.
Fluorescence spectroscopy is a valuable technique used as a detection method for many chemical, biological, and medical applications.9,10 Spectrofluorimetry offers advantages such as low detection limits, sometimes down to parts per billion or lower, as well as target specificity since simultaneous light absorption and emission at particular wavelengths provides a more discriminative route in comparison to absorption spectroscopy. Indeed, many HPLC-based protocols in TDM rely on fluorimetric quantification of the target drug.11 Fluorimetry is also commonly used for bioimaging applications.12 To this end, the use of pH-sensitive fluorescent probes offers a powerful route to spatially resolve the intracellular pH for the study of cell signalling and to identify diseases.13,14 In reverse, it is reasonable to assume that a deliberate and controlled manipulation of the pH may facilitate the quantification of drugs that exhibit strong pH-dependent properties.
In this work, we present a route to make use of the distinct pH-dependent fluorescence properties of the chemotherapeutic prodrug Irinotecan and its active metabolite SN-38 to selectively quantify both compounds at clinically relevant concentrations. The method relies on analytical discrimination through a pH-induced bathochromic shift in the emission spectra. This is, to the best of our knowledge, the first example of a pH-mediated molecular differentiation for the rapid fluorimetric quantification of a mixture of two drugs in human plasma at clinically relevant concentrations.
Irinotecan (7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyl-oxycamptothecin, CPT-11) is commonly used as an antitumor drug, in particular for colon cancer,15 and to a less extent for lung cancer.16 The drug is considered a prodrug since it undergoes cleavage of the bispiperidino-side chain by carboxyesterase to form SN-38 (7-ethyl-10-hydroxycampto-thecin), an active metabolite that has shown to be up to 1000 more potent at inhibiting topoisomerase I than the parent Irinotecan.17 Some of the main challenges for the simultaneous quantification of Irinotecan and SN-38 reside in the fact that their absorption and fluorescence properties are almost identical under physiological conditions, and the fact that the concentration of SN-38 can be over 30 times lower than that of Irinotecan.18
The pH of the media in which molecules are dissolved can play a major role not only for their solubility but also their optical performance. For Irinotecan and SN-38, this was investigated by characterising the absorbance as well as the emission behaviour at excitation wavelengths of 370 nm and 430 nm under acidic and basic conditions, respectively. As shown in Fig. 1 and Table 1, Irinotecan exhibited near identical optical properties for both absorbance and fluorescence under acidic and basic conditions, while SN-38 displayed a pronounced shift for absorbance as well as emission when basifying. Control over the pH, in particular a reliable basification, was of crucial importance for this work. An acidic solution was made by adding 0.05 vol% trifluoroacetic acid to MeOH (hereafter TFAM, pH = 1.4). For basification, a buffer was prepared by dissolving 98 mg of NaOH and 372 mg of KCl in 100 ml of MeOH, resulting in a pH of 12.3 (thereafter PSB – potassium chloride and sodium hydroxide buffer). The common route for basification presented herein, an equal addition in volume of PSB to TFAM (Vt = 8 ml), resulted in a pH value of 12.1. Volumetric variations of ±1 ml in the added amount of PSB had virtually no effect on the pH of the media (see Fig. S4 in the ESI†). All pH values were determined by a pH meter with aqueous calibration.
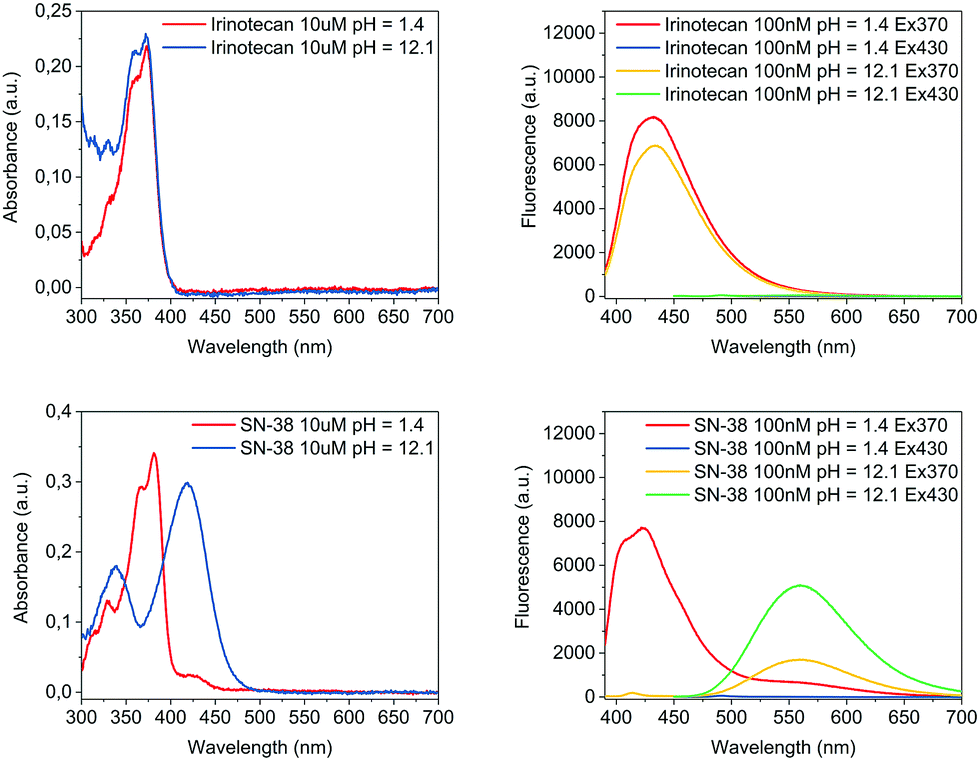 | ||
| Fig. 1 Absorbance spectra (left) and fluorescence spectra (right) of Irinotecan (top), and SN-38 (bottom). Compounds were dissolved at the indicated concentration in TFAM (pH = 1.4) for characterisation under acidic conditions before an equal volume of PSB was added for basification (pH = 12.1). | ||
| Compound | λ max abs acidic pH (nm) | λ max abs basic pH (nm) | λ max em acidic pH (nm) | λ max em basic pH (nm) |
|---|---|---|---|---|
| Irinotecan | 370 | 370 | 432 (λex = 370 nm) | 434 (λex = 370 nm) |
| SN-38 | 380 | 420 | 422 (λex = 370 nm) | 559 (λex = 430 nm) |
A rationalisation of the molecular structure under acidic and basic conditions is depicted in Scheme 1. The difference between the chemical structure of Irinotecan and SN-38 lies in the substitution of the bis-piperidino alkyl chain present in Irinotecan for an alcohol group in SN-38. For Irinotecan, the electronic conjugation of the camptothecin core therefore remains similar in acidic and basic pH (Scheme 1, top). This is not the case for SN-38, where the aromatic alcohol is deprotonated at basic pH, forming the alkoxy ion (Scheme 1, bottom). The alkoxy ion pushes the electronic density towards the camptothecin core, thus shifting the absorbance and fluorescence spectra of SN-38 to longer wavelengths. Please see Fig. S1–S3 in the ESI† for the corresponding 1H NMR spectra of Irinotecan and SN-38. Fig. S3 (ESI†) shows how the aromatic alcohol proton at 10.4 ppm disappears after basification for SN-38 but not for Irinotecan. It is worth noting that both Irinotecan and SN-38 exhibit a pH-dependent lactone/carboxylate equilibrium.19 However, this did not affect the results presented in this work, as the lactone/carboxylate moiety is not conjugated with the camptothecin core.
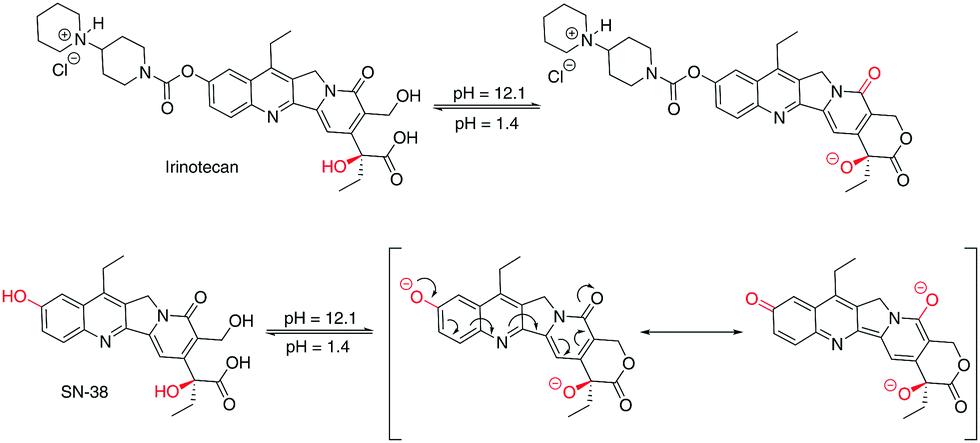 | ||
| Scheme 1 Effect of pH in the chemical structure of Irinotecan (top), and SN-38 (bottom). | ||
Under basic conditions, but with a pH below 11, the aromatic alcohol groups were found to be only partially deprotonated and, consequently, the pH had a very strong effect on the intensity of the fluorescence spectrum (see Fig. S4 in the ESI†). Above a pH of 11, the deprotonation of the alcohol group was almost completed which translated to a stable fluorescence spectrum at higher pH values. The fluorescence of SN-38 was only investigated at pH below 13 due to concerns regarding the stability of the molecule. At a pH of 12 the drug was found to be very stable for prolonged periods, with no appreciable signs of degradation being observed within 24 hours.
The viability of using a pH-mediated fluorimetric quantification of both compounds was further investigated by measuring the fluorescence of Irinotecan and SN-38 in MeOH over a range of relevant concentrations. Values previously reported in patients ranged from 10 to 10000 ng ml−1 (17 to 17000 nM) for Irinotecan and 1 to 500 ng ml−1 (2.5 to 1270 nM) for SN-38.20 Consequently, a spectrum from 2 to 2500 nM was studied for Irinotecan and 0.5 to 300 nM for SN-38. While the fluorescence of Irinotecan was measured in acidic conditions (pH = 1.4) using an excitation wavelength of 370 nm, the fluorescence of SN-38 was determined in basic conditions (pH = 12.1) with an excitation wavelength of 430 nm. As shown in the ESI† (Fig. S5), the fluorescence intensity of both compounds was found to be linear (R2 = 0.999 for both Irinotecan and SN-38) across the range of investigated concentrations. Thus, the observed sensitivity for fluorimetric detection greatly exceeded previously reported requirements.20
For the processing of human plasma samples, solid phase extraction (SPE) is a viable alternative to conventional protocols based on liquid–liquid extraction, offering benefits such as faster, less labour-intensive sample processing, reduced solvent use and higher concentration factors.21 In the herein presented approach, we developed a SPE protocol using 0.05 vol% TFA in MeOH and 0.05 vol% TFA in H2O (hereafter TFAH) as solvents. An overview of the individual steps is summarised in Table S1, ESI.† The drug recoveries were found to be 57.7 ± 2.4% for Irinotecan, and 98.8 ± 2.8% for SN-38. The extraction efficiencies were calculated by comparing the fluorescence obtained after SPE of either Irinotecan or SN-38 with the expected fluorescence based on the corresponding calibration curve.
The quantification of the drugs was carried out using the following procedure. First, a set of representative patient sample compositions of Irinotecan and SN-38 at various stages after intravenous administration was defined based on data found in literature (Table 2).22–25 Samples of pooled human plasma from healthy donors where then spiked with in total seven different concentrations of Irinotecan and SN-38 across the clinical relevant range, i.e. 103 to 2200 nM Irinotecan and 19 to 188 nM SN-38. After SPE, the extracted fractions (Vt = 1 ml) were diluted with an equal volume of TFAM to facilitate sample processing, and the fluorescence of the solution was measured at 432 nm (λex = 370 nm) to determine the overall concentration of the drug mixture (Irinotecan and SN-38). Subsequently, another dilution with an equal volume of PSB was carried out to basify the solution, and the fluorescence was measured at 559 nm (λex = 430 nm) to quantify the amount of SN-38 present in the sample.
| Sample no | 0 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|
| Concentration Irinotecan (nM) | Human plasma | 2200 | 1581 | 1136 | 756 | 327 | 103 |
| Concentration SN38 (nM) | Human plasma | 188 | 126 | 57 | 23 | 33 | 19 |
Once the amount of SN-38 was established, it was subtracted from the overall concentration of both compounds measured in acidic conditions to determine the concentration of Irinotecan. In all the cases, the concentration of the drugs was calculated by comparing the fluorescence after SPE, or SPE and basification, with the expected fluorescence from the corresponding calibration curves for Irinotecan or SN-38 (ESI,† Fig. S5). The sample processing translated to an overall dilution of the concentration of Irinotecan by a factor of 6.9× in the aliquot where fluorimetric quantification was carried out. The dilution factors for the individual processing steps were 2× for the SPE dilution, 2× for the dilution with TFAM, and 1.7× to account for the Irinotecan recovery efficiency during SPE. This compared to a dilution factor of 8× for SN-38 when quantified under basic conditions, which was based on individual factors of 2× for the SPE dilution, 2× for the dilution with TFAM and a final 2× for the basification with PSB. Due to the close-to-unity extraction efficiency, no SPE loss factor was necessary for SN-38. Alongside, the entire protocol was carried out with a reference sample that contained only pooled plasma in order to account for the parasitic fluorescence of the plasma (Fig. S6, see ESI†).
The results are summarised in Fig. 2. In total seven samples were run in triplicate, six spiked and one reference sample. For Irinotecan, 17 out of 18 obtained data points were within 15% error (94%). In the case of SN-38, 16 out of 18 data points were within this tolerance (89%). An overview of the fluorimetric signal intensity and errors obtained across the range of samples can be found in the ESI† (Tables S1 and S2). We note that for samples with concentrations below 100 nM for Irinotecan and 15 nM for SN-38, the measured background signal of the reference sample became significant, resulting in higher errors below this level, which is at the bottom end of the relevant clinical range.
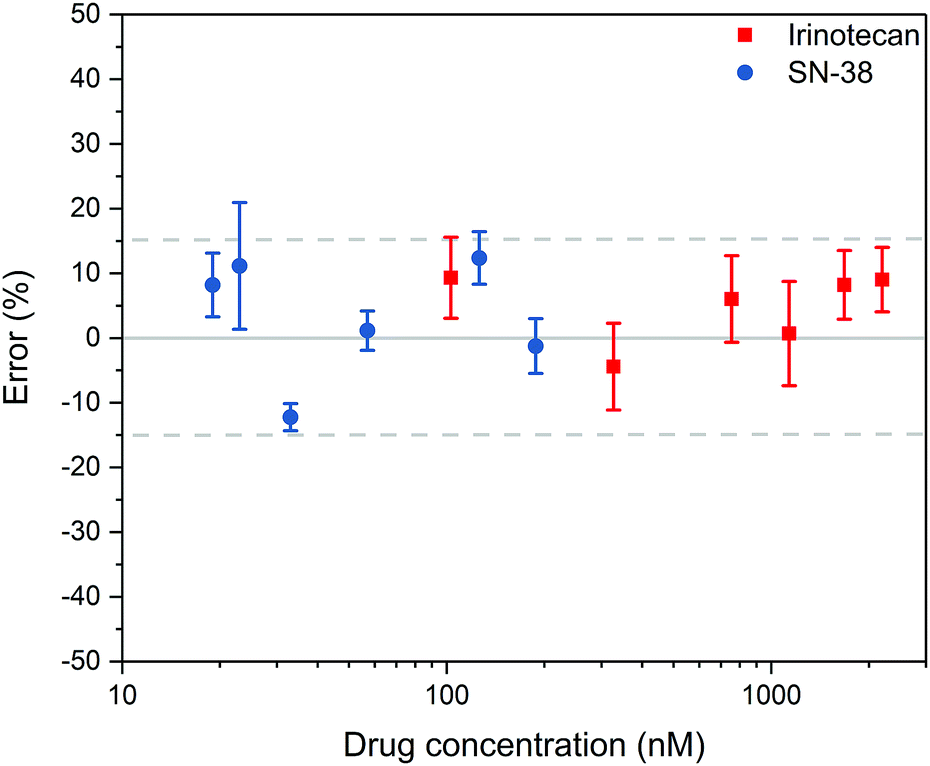 | ||
| Fig. 2 Quantification of Irinotecan and SN-38 from spiked plasma (n = 3). The graph shows the average value and the standard deviation of the measurements. | ||
We want to point out that our method is based on fluorimetric quantification and thus relies on an adequate manipulability of the emission properties of target compounds. In order to show the applicability to other commonly used chemotherapeutic drugs with similar molecular structure, we compared the pH-dependent optical properties of SN-38 to Epirubicin and Methotrexate.26–28 With the former containing an alcohol and the latter an amine group attached to the aromatic core, a basic pH is likely to cause deprotonation resulting in a change of the optoelectronic properties of the molecules. The absorbance and fluorescence spectra of the three compounds were recorded in acidic (pH = 1.4) and basic (pH = 12.1) conditions (Table S3 and Fig. S10, see ESI†). In all cases, a change from acidic to basic conditions resulted in a modification in the optical properties of the drugs, with the absorption spectrum shifting to longer wavelengths, from 30 nm for Methotrexate to almost 85 nm for Epirubicin, which compared to 40 nm for SN-38. The bathochromic shift in the absorbance spectra of the molecules in basic conditions was determined to be 15 nm in the case of Methotrexate and 97 nm for Epirubicin in comparison to 137 nm for SN-38. Further studies are now geared towards translating the protocol to the quantification of a range of anthracyclines and other suitable chemotherapeutic drugs.
In conclusion, we have developed a fast and reliable method to quantify the amount of the chemotherapeutic prodrug Irinotecan and its active metabolite SN-38 spiked in human plasma at clinically relevant concentrations based on pH-mediated fluorescence spectroscopy. We want to emphasize on the simplicity of the processing steps and the minimal time requirement of less than 30 min from sample withdrawal to the analytical result. The herein reported pH-mediated molecular differentiation method allowed to quantify Irinotecan and SN-38, two otherwise similar compounds down to nanomolar concentrations and molar ratios of up to 30:1. We anticipate the method to be equally effective in isolating other target compounds with applicable molecular design rules from a complex environment by utilising a bathochromic shift.
This project received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 633635 (DIACHEMO). YY acknowledges University College London for an Overseas Research Scholarship. The authors want to express their gratitude to E. Marangon and G. Toffoli for useful discussions and for providing pooled plasma samples from healthy donors.
Conflicts of interest
There are no conflicts to declare.References
- A. Dasgupta, Therapeutic Drug Monitoring: Newer Drugs and Biomarkers, Academic Elsevier Science, 2012 Search PubMed.
- E. Salvati, F. Stellacci and S. Krol, Nanomedicine, 2015, 10, 3495–3512 CrossRef CAS PubMed.
- P. L. Bonate and D. R. Howard, Pharmacokinetics in Drug Development, Springer International Publishing, Cham, 2016 Search PubMed.
- N. B. La Thangue and D. J. Kerr, Nat. Rev. Clin. Oncol., 2011, 8, 587–596 CrossRef CAS PubMed.
- M. Ramesh, P. Ahlawat and N. R. Srinivas, Biomed. Chromatogr., 2010, 24, 104–123 CrossRef PubMed.
- A. Dasgupta, Advances in Chromatographic Techniques for Therapeutic Drug Monitoring, CRC Press, 2009 Search PubMed.
- C. Pistos and J. T. Stewart, J. Pharm. Biomed. Anal., 2003, 33, 1135–1142 CrossRef CAS PubMed.
- N. E. Kontny, G. Hempel, J. Boos, A. V. Boddy and M. Krischke, Ther. Drug Monit., 2011, 33, 766–771 CrossRef CAS PubMed.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- J. Yao, M. Yang and Y. Duan, Chem. Rev., 2014, 114, 6130–6178 CrossRef CAS PubMed.
- B. Martinc, R. Roškar, I. Grabnar and T. Vovk, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 962, 82–88 CrossRef CAS PubMed.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- J. R. Casey, S. Grinstein and J. Orlowski, Nat. Rev. Mol. Cell Biol., 2010, 11, 50–61 CrossRef CAS PubMed.
- Y. Urano, D. Asanuma, Y. Hama, Y. Koyama, T. Barrett, M. Kamiya, T. Nagano, T. Watanabe, A. Hasegawa, P. L. Choyke and H. Kobayashi, Nat. Med., 2009, 15, 104–109 CrossRef CAS PubMed.
- G. Folprecht and C.-H. Köhne, Nat. Clin. Pract. Oncol., 2005, 2, 578–587 CrossRef CAS PubMed.
- D. R. Spigel, F. A. Greco, J. D. Zubkus, P. B. Murphy, R. A. Saez, C. Farley, D. A. Yardley, H. A. Burris and J. D. Hainsworth, J. Thorac. Oncol., 2009, 4, 1555–1560 CrossRef PubMed.
- Y. Liu, H. Y. Piao, Y. Gao, C. H. Xu, Y. Tian, L. H. Wang, J. W. Liu, B. Tang, M. J. Zou and G. Cheng, Int. J. Nanomed., 2015, 10, 2295–2311 CAS.
- X. Liu and A. B. Hummon, J. Am. Soc. Mass Spectrom, 2015, 26, 577–586 CrossRef CAS PubMed.
- M. N. Tallman, J. K. Ritter and P. C. Smith, Drug Metab. Dispos., 2005, 33, 977–983 CrossRef CAS PubMed.
- E. Marangon, B. Posocco, E. Mazzega and G. Toffoli, PLoS One, 2015, 10, e0118194 Search PubMed.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- G. Toffoli, M. R. Sharma, E. Marangon, B. Posocco, E. Gray, Q. Mai, A. Buonadonna, B. N. Polite, G. Miolo, G. Tabaro and F. Innocenti, Clin. Cancer Res., 2017, 23, 918–924 CrossRef CAS PubMed.
- X. Chen, C. J. Peer, R. Alfaro, T. Tian, S. D. Spencer and W. D. Figg, J. Pharm. Biomed. Anal., 2012, 62, 140–148 CrossRef CAS PubMed.
- W. Zhang, G. E. Dutschman, X. Li, M. Ye and Y.-C. Cheng, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3038–3044 CrossRef CAS PubMed.
- J. G. Slatter, L. J. Schaaf, J. P. Sams, K. L. Feenstra, M. G. Johnson, P. A. Bombardt, K. S. Cathcart, M. T. Verburg, L. K. Pearson, L. D. Compton, L. L. Miller, D. S. Baker, C. V. Pesheck and R. S. Lord, Drug Metab. Dispos., 2000, 28, 423–433 CAS.
- I. Imaz, M. Rubio-Martínez, L. García-Fernández, F. García, D. Ruiz-Molina, J. Hernando, V. Puntes and D. Maspoch, Chem. Commun., 2010, 46, 4737 RSC.
- M. Florescu, L. S. Magda, O. A. Enescu, D. Jinga and D. Vinereanu, J. Am. Soc. Echocardiogr., 2014, 27, 83–92 CrossRef PubMed.
- J. Bluett, I. Riba-Garcia, K. Hollywood, S. M. M. Verstappen, A. Barton and R. D. Unwin, Analyst, 2015, 140, 1981–1987 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc07668a |
| ‡ Current address: Children's Hospital of Philadelphia, Civic Center Blvd, Philadelphia, PA 19104, USA. |
| This journal is © The Royal Society of Chemistry 2018 |