
Polymeric micro- and nanoparticles for immune modulation
Elana
Ben-Akiva†
ab,
Savannah
Est Witte†
ab,
Randall A.
Meyer†
ab,
Kelly R.
Rhodes†
ab and
Jordan J.
Green
*abcd
aDepartment of Biomedical Engineering and Institute for NanoBioTechnology, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA. E-mail: green@jhu.edu
bTranslational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA
cDepartment of Oncology, the Sidney Kimmel Comprehensive Cancer, and the Bloomberg∼Kimmel Institute for Cancer Immunotherapy, Johns Hopkins School of Medicine, Baltimore, Maryland 21231, USA
dDepartment of Ophthalmology, Department of Neurosurgery, Department of Materials Science and Engineering, and Department of Chemical and Biomolecular Engineering, Johns Hopkins University, Baltimore, Maryland 21218, USA
First published on 1st November 2018
Abstract
New advances in biomaterial-based approaches to modulate the immune system are being applied to treat cancer, infectious diseases, and autoimmunity. Particulate systems are especially well-suited to deliver immunomodulatory factors to immune cells since their small size allows them to engage cell surface receptors or deliver cargo intracellularly after internalization. Biodegradable polymeric particles are a particularly versatile platform for the delivery of signals to the immune system because they can be easily surface-modified to target specific receptors and engineered to release encapsulated cargo in a precise, sustained manner. Micro- and nanoscale systems have been used to deliver a variety of therapeutic agents including monoclonal antibodies, peptides, and small molecule drugs that function to activate the immune system against cancer or infectious disease, or suppress the immune system to combat autoimmune diseases and transplant rejection. This review provides an overview of recent advances in the development of polymeric micro- and nanoparticulate systems for the presentation and delivery of immunomodulatory agents targeted to a variety of immune cell types including APCs, T cells, B cells, and NK cells.
Introduction
Novel biomaterials approaches for immune system modulation have recently been of great interest for treatments of diseases such as cancer,1 infectious disease,2 autoimmune disorders,3 and regenerative medicine.4 Biocompatible materials such as biodegradable and bioeliminable polymers have proven well suited to deliver signals to the immune system in order to direct an activating immune response against a detrimental disease or suppress an unwanted response against one's self antigens.5 From a reductionist standpoint, two primary approaches have been considered based on length scales as well as the specific immune cell type targeted to promote health and prevent disease. These two classes are macro-scale implantable scaffolds for controlled drug release, and regenerative medicine6 as well as nano- to micro-scale particulate delivery systems for drug delivery.7 Although both have been successful in immunoengineering applications, this review will highlight recent advances in particulate systems for biologic delivery (Table 1).| Cell type | Modulation | Material | Biologic | Size | Result | Ref. |
|---|---|---|---|---|---|---|
| a iLN = intra-lymph node. | ||||||
| APC | Activating | PLGA with PEGylated lipid bilayer | OVA peptide and MPLA adjuvant | 200 nm vs. 2–3 μm | Nanoparticles induced a greater antigen-specific immune response and both sizes were effective at very low doses | 34 |
| Activating | PBAE | IL-12 | 30–40 nm | Repolarized TAMs from M2 to M1 phenotype and induced significant tumor burden decrease in melanoma model compared to soluble IL-12 | 53 | |
| Suppressive | Polystyrene beads, PLGA particles | MOG35–55 peptide | 500 nm | Prevented disease onset and reduced severity of paralysis in an EAE model | 64 | |
| Suppressive | PLGA | MOG35–55 peptide and rapamycin | 3–4 μm | Single dose administered iLNa at the peak of disease resulted in 75% recovery from EAE-induced paralysis | 80 | |
| T-cells | Activating | PLGA | Immune signal 1 and 2 and anti-PD1 | 4–5 μm | Combination therapy resulted in 40% B16-F10 tumor burden decrease | 90 |
| Activating | Iron Oxide | Anti-PDL1 and anti-41BBL | 80 nm | Redirection of tumor signals to result in 50% B16-F10 tumor burden decrease | 95 | |
| Suppressive | PLGA | H2 kb, PD-L1-Fc, and CD47-Fc, TGF-β | 5 μm | Induced CD4+FOXP3+ Tregs 1.5-fold band deleted myelin autoreactive CD4 and CD8+ T-cells by 2-fold | 109 | |
| Suppressive | PLGA | H2 kb dimer, anti-Fas, PD-L1, CD47-Fc, TGF-β | 200 nm | Inhibited alloreactive CD8+ T-cells and exprended Tregs for doubled survival rate in alloskin transplant | 118 | |
| Suppressive | Carboxylated polystyrenebeads | HLA 02![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :01 epitopes :01 epitopes |
500 nm | Promote antigen specific tolerance for type 1 diabetes by doubling the amount of CD4+ CD25+ Tregs and 10-fold depletion of autoantigen specific T lymphocytes | 116 | |
| Suppressive | PLGA | Dby and Uty peptide, PD1 | 450 nm | Dby with PD1 induced tolerance to mismatched bone marrow transplantation comparable to positive control, where the level of donor cells was 49% by week 20 | 111 | |
| B-cells | Activating | Calcium phosphate | HEL antigen | 200–400 nm | 100-Fold increase in B-cell activation compared to soluble antigen | 131 |
| Suppressive | DSPC liposomes | CD22 glycan ligands and multiple antigens (HEL, OVA, MOG, FVIII) | 100 -130 ± 30 nm | 10-Fold decrease in antigen specific igG antibodies for multiple antigens after treatment | 139 | |
| Suppressive | DSPC and cholesterol liposomes | CD22 glycan ligand, OVA antigen, and rapamycin | 160 ± 30 nm | Coadministration of STAL-NPs with RAPA results in 5-fold decrease in OVA-specific antibody compared to STAL-NPs in naïve mice | 141 | |
Micro- and nanoparticle systems are an important class of many biomaterials-based drug delivery systems. These technologies possess key advantages for modulation of immune activity. Microparticles and nanoparticles are on the same size scale as cells and subcellular components, making them an ideal vehicle for a variety of applications such as stimulation of surface receptors or internalization and intracellular cargo release.8 In many cases, the release of therapeutics from particle cores can be controlled on a desired time scale.9 Fragile biological cargoes such as peptide or protein antigens can be encapsulated in the cores of particulate materials and reach their targets without being exposed to harsh physiological conditions.8 In addition, these technologies can be administered both locally and systemically for optimal pharmacokinetics. Finally, these particles can be made stealthy through incorporation of a surface feature such as polyethylene glycol (PEG) for “shielded” particles or a more natural approach such as mimicking the “do not eat me” signal of red blood cells.10 Taken together, drug delivery at the micron and nanoscale is an important foundation for biomaterials based immune modulation.
Some of the most successful classes of existing standalone drugs that can be delivered using micro- and nanoparticles are protein biologics and small molecule. For many conditions, these drugs have been successful in the clinic for targeted treatment in standalone and targeted treatment strategies. One example of the protein class of drugs are monoclonal antibodies (mAbs). mAbs have been applied to many diseases with a viable molecular target, such as an immune checkpoint in cancer11 or an excess of macrophages in rheumatoid arthritis.12 Another example of these protein biologics are peptide antigens which are used in many vaccine formulations. These antigens can be synthetically made such as in the case of hepatitis B13 or delivered as part of an inactivated pathogen such as the common forms of the flu vaccine.14 There also exist a plethora of small molecule drugs that can be used for immune modulation such as rapamycin for non-specific immune suppression,15 and inhibitors of indoleamine 2,3-dioxygenase for cancer immunotherapy.16 These immunomodulatory protein biologics and small molecules make ideal candidates for delivery using current micro- and nanoparticle strategies as they can synergize with several previously mentioned advantages of these systems to amplify their therapeutic effect for their intended purpose.
In this review, the major recent advances in the development of micro- and nanoparticulate materials for the delivery and presentation of immunomodulatory protein biologics and small molecules will be covered. The review will not cover other important areas in drug delivery for immunomodulation such as gene delivery (to which the reader is directed to a comprehensive review on the topic).17 Both activation of the immune system, such as against cancer cells or infectious diseases, and suppression of the immune system, such as in the case of autoimmune disorders and transplant tolerance, will be described. As each particulate system is designed based on the specific immune cell type that it modulates, this review is structured based on these cell types: antigen presenting cells (APCs) including dendritic cells and macrophages, T cells, B cells, and NK cells. Continued research into the development of these biomaterials based biologic delivery strategies will unlock the full potential of immunomodulatory proteins and peptides for next generation immunotherapies.
Antigen presenting cells (APC)
Dendritic cells and macrophages are the major professional APCs of the immune system that have the potential to process and present antigen to activate T cells against cancer and infectious diseases. APCs can also adopt a tolerogenic phenotype and suppress T cells that are reactive to self-antigen, as is the case in autoimmune disease. APCs present antigen in the context of the major histocompatibility complex (MHC) to T cells, specifically using MHC I for presentation to CD8+ T cells or MHC II for presentation to CD4+ T cells. APCs also display a costimulatory signal, referred to as signal 2, that directs the T cell response. Positive costimulatory signals stimulate T cells to mount a response against cells displaying the antigen of interest. On the other hand, a lack of positive costimulation and negative costimulatory signals result in the suppression of effector T cells and direct T cells to adopt a regulatory phenotype.APCs for immunostimulation
While APCs can be tuned broadly for immunostimulation against diverse antigens, utilizing particles to engineer APCs for cancer immunotherapy is an area of particularly rapidly growing interest. For further information on polymeric particle-based vaccines to fight infectious diseases, a reader is directed elsewhere.18,19 Antigen processing and presentation is often dysregulated or inefficient during cancer.20 As a result, extensive research has been performed to engineer particle-based cancer vaccines that direct APCs to present tumor antigen to T cells in an immunostimulatory manner. Additionally, tumor-associated macrophages (TAMs) play an important role in the tumor microenvironment. Tumor cells stimulate TAMs to adopt an immunosuppressive phenotype, which impairs their ability to present antigen and perform other immunostimulatory anti-tumor activities.21 Researchers are investigating methods to modulate TAMs to make them more immunostimulatory or deplete them altogether.Many polymeric particulate systems have been engineered to deliver antigen and adjuvant to APCs, namely dendritic cells, for cancer vaccination. Both micro- and nanoparticles synthesized from poly(lactic-co-glycolic acid (PLGA), a biodegradable polymer that has been used for multiple FDA-approved devices, have been extensively studied for cancer vaccine delivery by encapsulating peptide antigen and adjuvant.27–32 PLGA particles encapsulating peptide antigen and adjuvant have been found to induce a much stronger anti-tumor immune response compared to soluble antigen and adjuvant.30,33 For example, PLGA nanoparticles encapsulating a melanoma-associated antigen and the adjuvant monophosphoryl lipid A (MPLA) stimulated dendritic cells to induce approximately 10-fold higher CD8+ T cell proliferation compared to free antigen and adjuvant.30 To overcome obstacles associated with this conventional approach, such as low antigen encapsulation efficiency, Bershteyn et al. engineered PLGA particles coated with a PEGylated phospholipid bilayer with incorporated lipophilic adjuvants attached protein antigen.34 Conventional polymeric particles for cancer vaccination encapsulate a single peptide antigen. Fang et al. engineered PLGA nanoparticles coated with melanoma cancer cell membranes in order to deliver the full range of cancer antigens to antigen presenting cells, as opposed to vaccinating against just one or a few tumor-associated antigens, and found that the particles successfully delivered tumor antigen to dendritic cells.35 In addition to PLGA, other biodegradable polymers, such as polylactide (PLA), poly(amino acids) and polysaccharides, have been explored for cancer vaccine delivery.36,37
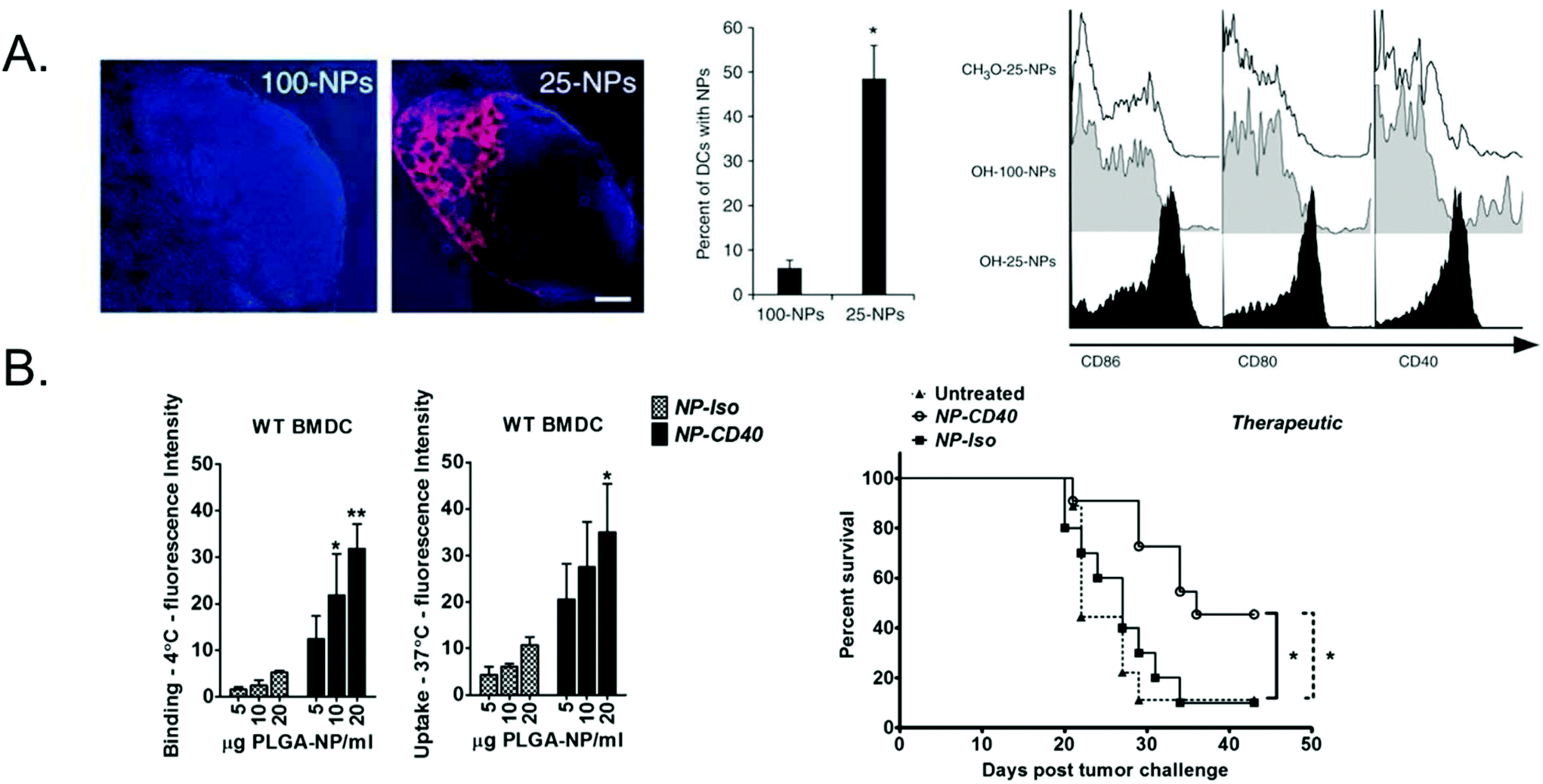 | ||
| Fig. 1 Passive vs. active targeting of polymeric particle vaccines. (A) 25 or 100 nm PPS nanoparticles encapsulating OVA and LPS were injected intradermally and 25 nm were more efficiently transported to and retained in the draining lymph node and taken up by dendritic cells. 25 nm particles also more effectively activated dendritic cells, as measured by upregulation in maturation markers. Subfigure A adapted with permission from Nature Biotechnology, Copyright Nature Publishing Group 2007.23 (B) PLGA nanoparticles encapsulating OVA peptide antigen and adjuvants were surface functionalized with anti-CD40. Dendritic cells had significantly higher binding to and uptake of CD40-targeted NPs (NP-CD40) compared to an isotype control (NP-iso) and induced prolonged survival in a therapeutic in vivo melanoma model compared to the non-targeted nanoparticles. Subfigure B adapted with permission from Biomaterials, Copyright Elsevier 2015.45 | ||
One strategy that has been investigated for TAM-targeted cancer therapies is depletion of TAMs. Ernsting et al. synthesized a nanoparticle composed of PEG and carboxymethylcellulose to deliver docetaxel, a chemotherapy agent.49 These nanoparticles selectively killed stromal cells and macrophages in the tumor microenvironment of a pancreatic cancer mouse model. However, this depletion of TAMs was transient, and the TAMs had fully repopulated within two weeks. Niu et al. fabricated an acid-sensitive sheddable PEGylated PLGA nanoparticle encapsulating doxorubicin and functionalized with mannose to target the mannose receptor overexpressed on macrophages and selectively deplete TAMs.50,51 The acid sensitive nature of the particle is designed to avoid off-target macrophages and specifically target TAMs. Under normal physiological conditions, the PEG chains shield the mannose on the particle surface, but in the slightly acidic tumor microenvironment the PEG chains “shed” from the particle, exposing the macrophage-targeting mannose.
An alternative strategy to TAM depletion is to re-polarize TAMs from the M2 phenotype to the M1 phenotype. This approach has the added benefit of introducing the anti-tumor activity of M1 macrophages while still reducing the protumorigenic M2 phenotype. Fuchs et al. found that polystyrene nanoparticles functionalized with cationic or anionic surface molecules stimulated M2 macrophage re-education into M1 macrophages.52 Wang et al. designed pH-sensitive poly(β-amino ester) (PBAE) nanoparticles encapsulating IL-12, a cytokine that has been shown to re-polarize macrophages towards an M1 phenotype.53 These polymeric nanoparticles were administered in an in vivo melanoma model and led to macrophage re-polarization and tumor regression with minimal side-effects due to the tumor microenvironment-responsive nature of the particles. A major challenge of the TAM re-polarization approach is inducing long-term TAM re-education into M1 macrophages, as macrophages are highly plastic may return to the M2 phenotype once the drug is no longer present due to the immunosuppressive tumor microenvironment.
APC for immunosuppression
The majority of particulate tolerance therapies rely on passive targeting strategies since large nanoparticles and microparticles are efficiently taken up by APC in the spleen and liver, which is often desirable for tolerance induction.58 These particle-based platforms for antigenic delivery to APC promote tolerance by exploiting natural tolerogenic processes or by providing tolerogenic cues by co-delivering immunomodulatory molecules.
Particle-based platforms can be engineered to exploit apoptotic clearance pathways without the limitations of cell-based therapies. It was discovered that polystyrene beads and PLGA nano and microparticles with a zeta potential of −40 to −70 mV are preferentially taken up by a subset of macrophages in the spleen and liver expressing the Macrophage Receptor with Collagenous Structure (MARCO), a scavenger receptor involved in apoptotic debris clearance, thereby bypassing the need for codelivery of apoptotic cell debris.58,63 Polystyrene and PLGA particle-based platforms delivering antigenic peptides have been used to induce tolerance in a variety of models including experimental autoimmune encephalomyelitis (EAE) and transplantation,64–66 and uptake by MARCO-expressing macrophages is crucial for the tolerogenic effects of the therapies.64
To more closely mimic apoptotic cells, particles bearing apoptotic markers have also been engineered to induce tolerance. Liposomes containing phosphatidylserine (PS) and carrying antigenic material have been shown to promote tolerance in several models including hemophilia and Type I Diabetes (T1D).67,68 The use of polymeric particles instead of liposomes allows for manipulation of particle shape to enhance biomimicry and the pharmacokinetic properties of the carrier. Interestingly, 80 × 320 nm rod-shaped PLGA particles decorated with PS were shown to be more effective at inducing tolerance in an EAE model compared to 1 μm cylindrical PS-decorated PLGA particles and PS-containing liposomes.69
In addition to MARCO-expressing macrophages, APC in other sites including the liver, Peyer's patches, and the oral and digestive tract have been shown to mediate tolerance.70,71 Delivery of antigen to these tolerogenic environments can be accomplished with polymeric particle-based carriers.
Other biodegradable polymeric particle drug delivery platforms have been designed to deliver a range of broad immunosuppressants in a localized, controlled fashion. Rapamycin, a mammalian target of rapamycin (mTOR inhibitor), is a powerful immunosuppressant, but systemic delivery is problematic due to poor solubility and bioavailability, and nonspecific suppression of immune cells.76 When encapsulated in PLGA micro and nanoparticles, rapamycin has been shown to inhibit DC functioning following particle uptake compared to soluble rapamycin.77,78 PLGA microcarriers co-delivering encephalitogenic peptides and rapamycin substantially reduced the onset and severity of EAE (Fig. 2A and B), and led to recovery in 75% of mice when administered at the peak of disease (Fig. 2C). PLGA nanocarriers loaded with the same contents decreased the severity and onset of paralysis when administered prophylactically (Fig. 2D) and prevented relapse in a Relapsing-Remitting EAE model when administered as a single dose at the peak of disease (Fig. 2E).79,80 Inclusion of rapamycin was essential for enhanced tolerogenic effects, as administration of peptide-loaded particles only conferred partial disease protection. Rapamycin-loaded nanoparticles co-administered with clinically approved biologics to treat a variety of diseases including Pompe disease, hemophilia, and inflammatory arthritis reduces the formation of antidrug antibodies and improves tolerance to the biologic.79,81–83 Antiproliferative agents such as mycophenolic acid (MPA) also have negative effects when administered systemically that can be ameliorated through delivery in particle carriers. Injections of MPA-loaded nanoparticles significantly extended graft survival compared to a 1000-fold higher dose of free MPA in a murine skin transfer model, and no drug toxicity was observed for the nanoparticle treatment.66 The nanoparticles were found to be taken up by DC, leading to upregulation of programmed death ligand 1 (PDL-1) which helped promote graft survival. Steroids are also commonly used as immunosuppressive agents, but are associated with unpleasant side effects when administered systemically. Treatment with dexamethasone and MOG peptide-loaded acetylated dextran microparticles resulted in a significant decrease in EAE clinical score compared to soluble drug, empty particles, and particles loaded with either peptide or drug alone.84
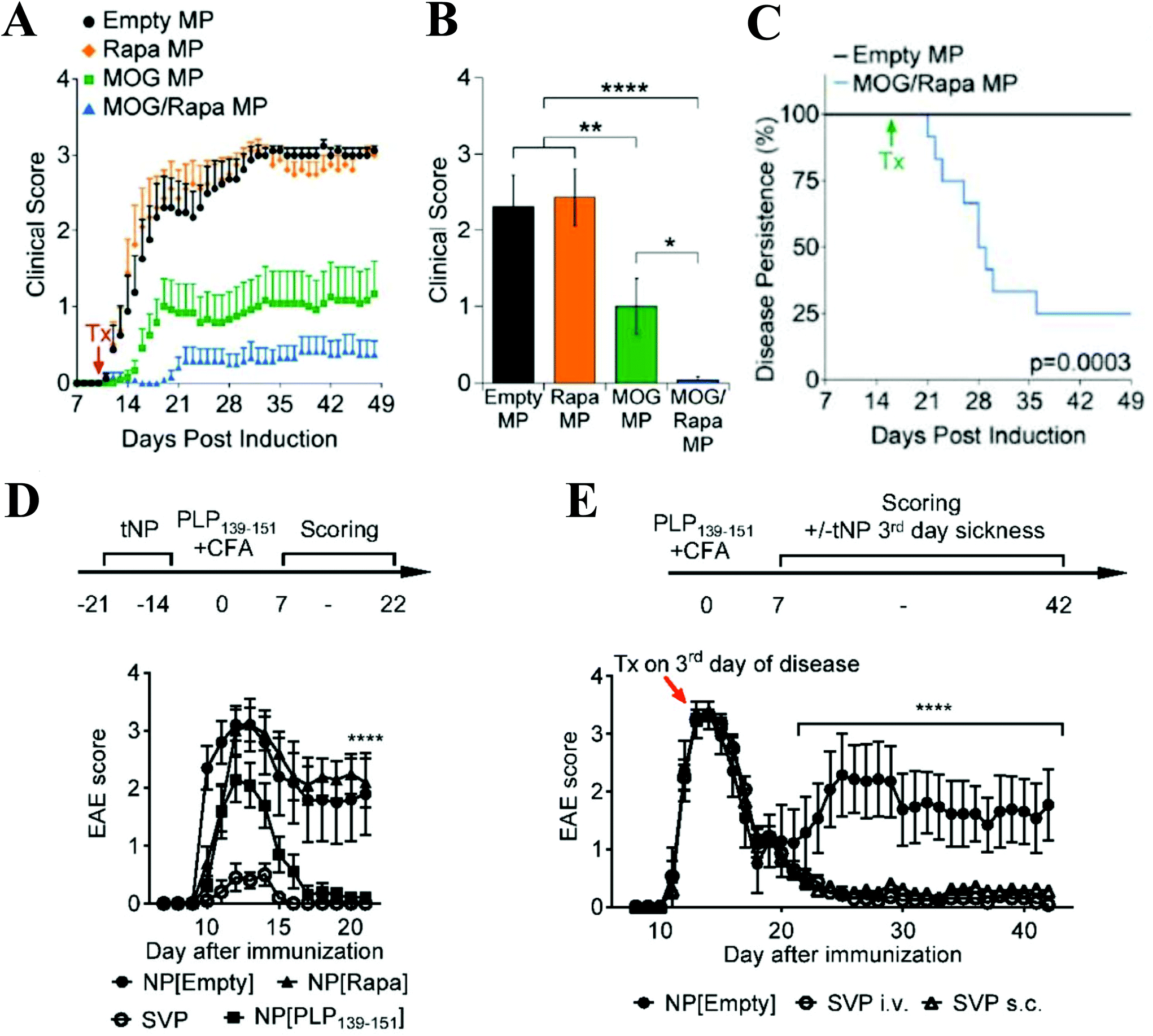 | ||
| Fig. 2 (A) Prophylactic treatment with microparticles coloaded with MOG and rapamycin delayed onset and reduced severity of EAE symptoms compared to empty, MOG-loaded, and rapamycin-loaded particles. (B) MOG + rapamycin-loaded particles mediated a significant decrease in EAE clinical score on Day 19 post-EAE induction. (C) When administered at the peak of disease, MOG + rapamycin-loaded particles facilitated recovery in 75% of mice. (D) Prophylactic treatment with nanoparticles coloaded with PLP and rapamycin (SVP) delayed onset and reduced severity of symptoms in Relapsing-Remitting EAE model. Nanoparticles loaded with PLP only conferred partial protection from disease symptoms. (E) A single dose of SVP particles administered intravenously or subcutaneously at the peak of disease prevented relapse. Subfigures A–C reprinted with permission from Cell Rep, Copyright Elsevier 2016.80 Subfigures D-E reprinted with permission from PNAS, Copyright National Academy of Sciences 2015.79 | ||
T-cells
T-cells, as the major effector arm of the immune system with respect to cancer immunotherapy and infectious disease, have been the subject of extensive investigation with respect to delivery of biomedical therapeutics from particulate systems.85 Direct delivery to T-cells can be thought of as “short-circuiting” the typical immune response, bypassing the need of an antigen presenting cell to relay the information. Broadly, two strategies that have been used in the particle-based delivery of therapeutics to T-cells are (1) The presentation of immobilized biologics from particulate surfaces, such as through the use of artificial antigen presenting cells (aAPCs) and (2) The extracellular or intracellular delivery of soluble biological factors via release from particles.T cells for immunostimulation
aAPC particulate systems made from materials such as biodegradable polymers, iron oxide nanoparticles, and lipid based materials85 have typically been used as a standalone therapy.88,89 However, new evidence suggests that they would benefit from coadministration of other biologics. Recently their activity has been shown to be augmented by the presence of other immunostimulatory cytokines and monoclonal antibodies. Kosmides et al. investigated the use of aAPCs with systemic administration of anti-PD1 (Fig. 3A).90 The aAPCs were found to be effective stimulators of T-cell activation and the anti-PD1 was found to protect the expanded cells from exhaustion. This combination therapy resulted in a 50% reduction in tumor burden (Fig. 3b) and an increase in median survival from day 17 to day 21 for the dual treatment strategy compared to either therapy on its own or a control.90
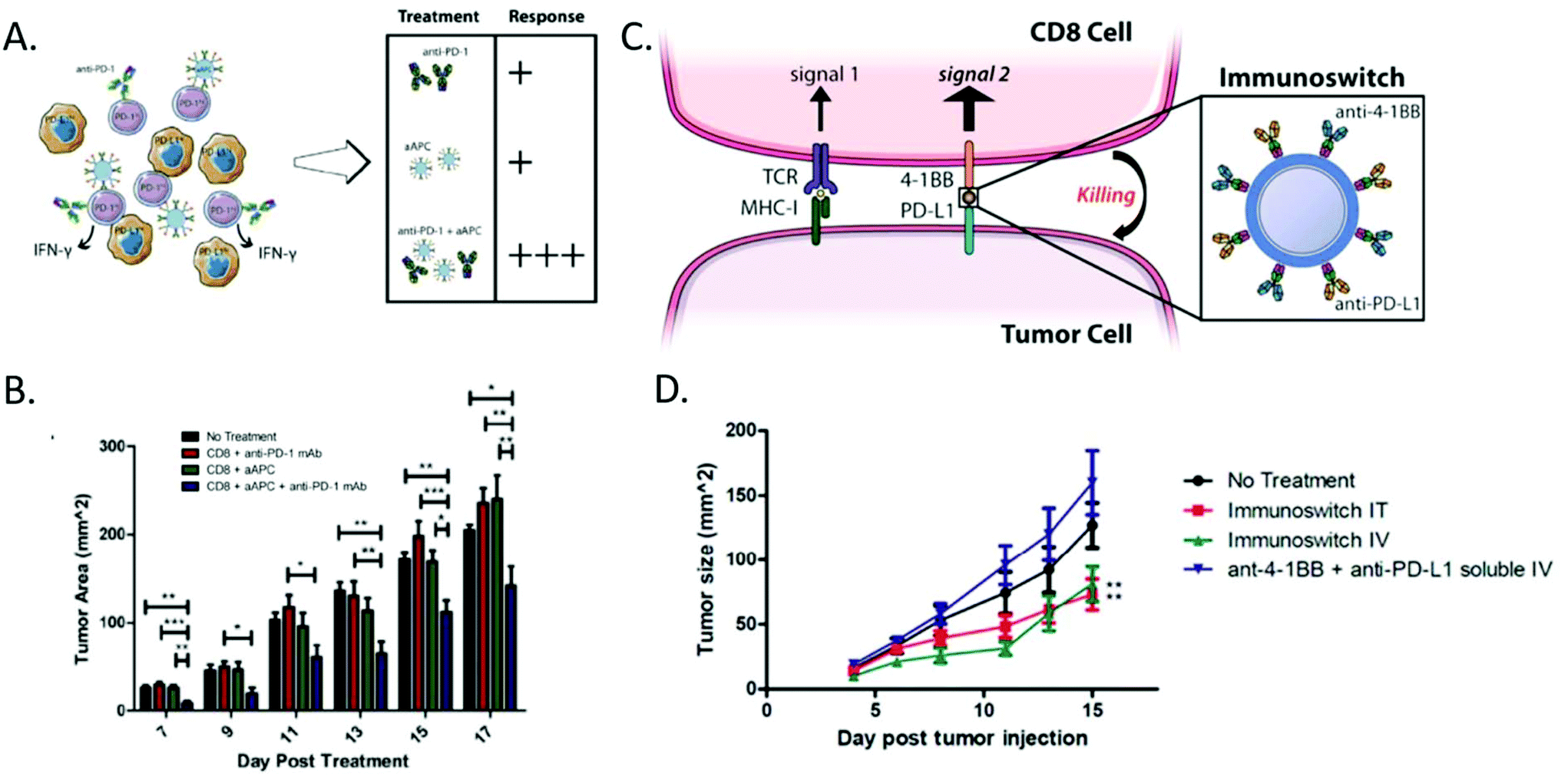 | ||
| Fig. 3 Artificial antigen presenting cell strategies can be combined with biologic delivery for enhanced effect. (A) Anti-PD1 in combination with artificial antigen presenting cells were found to yield the most significant anti-tumor immune response compared to either therapy on its own. (B) The combination therapy was able to mediate a 40% reduction in tumor burden compared to the no treatment and single treatment groups. (Reprinted with permission from Biomaterials, Copyright Elsevier 2017).90 (C) Signal 2 only particles can be combined with immune checkpoint blockades on the surface of a particle to act as an immunoswitch to redirect the cancer cell to activate a CD8+ T-Cell, rather than suppress it. (D) This approach led to a 50% tumor reduction in various B16 models without the need for antigen specificity. (Reprinted with permission from ACS Nano, Copyright American Chemical Society 2017).95 | ||
The delivery of therapeutics directly from aAPCs has also been investigated as a platform to enhance T-cell activation. Steenblock et al. developed an aAPC system that encapsulated the T-cell growth factor IL-2.91 The group found that the particles encapsulating IL-2 were able to outperform empty particles with soluble IL-2. Furthermore, this was later found to be the result of the confined space between the T-cell and the aAPC, which allowed for accumulation of the cytokine for a higher apparent concentration.92 This local release of IL-2 was subsequently appropriated in the context of a high surface carbon nanotube platform for T-cell stimulation for ex vivo stimulation of T-cells for cancer immunotherapy.93 It was determined that the local release of IL-2 resulted in T-cell activity comparable to a 1000 fold increase in soluble IL-2 in an adoptive immunotherapy model. Taken together these results suggest the paracrine delivery of biologics from aAPCs is a promising therapeutic platform for cancer immunotherapy.
aAPCs can also be used to deliver surface bound signals in a targeted fashion. Schütz et al. investigated the use of an antigen loaded MHC Class I dimer conjugated to a particle that also had tethered to the surface an monoclonal antibody for CD19.94 The resultant particle was found to be successful at directing antigen specific T-cells to attack CD19+ leukemia cells. The therapy was found to mediate a near 50% reduction in tumor burden for cognate redirection particles compared to the non-cognate controls.94 Kosmides et al. utilized a nanoparticle loaded with an antibody bearing the Signal 2 molecule anti-41BB and the immune checkpoint blockade anti-PDL1 (Fig. 3c).95 The resultant nanoparticle redirected the normally immunosuppressive surface of a melanoma cell to be immunostimulatory. The therapy also was able mediate a near 50% reduction in tumor burden compared to a control in a B16-F10 melanoma model (Fig. 3d).95
With respect to stimulus responsive particles, a broad area of research that has been investigated is photothermal triggered release of cytokines or other immunomodulatory molecules. Photothermal ablation of tumor tissue, mediated by plasmonic resonation of gold nanoparticles in response to laser irradiation, has gained significant popularity as a treatment paradigm for tumors due to the creation of antigenic material in an immune stimulatory fashion. The generated heat can subsequently promote drug release from a polymeric particle. Luo et al. used photothermal ablation in the context of a PLGA particle releasing a peptide designed to block PD-1 on the surface of T-cells.96 It was found that these particles could mediate a 75% reduction in tumor in a 4T1 cancer model compared to a single dose of the free peptide. Similar responses were found with another immune checkpoint blockade antibody, anti-CTLA4.97
Other studies have used the T-cell itself to serve as the propagator of the stimulus for drug release. Tang et al. used a 50 nm bioreducible protein gel to deliver the immunostimulatory cytokine IL-15.98 Taking advantage of the increase in reduction potential of the exterior of the cell membrane upon TCR stimulation, the authors achieved TCR engagement dependent stimulation of the immune stimulatory cytokine. Utilizing CAR T-cells directed against B16-F10 melanoma, it was determined that the bioreducible responsive protein gels were able to mediate a near 20 day increase in median survival compared to the control of free IL-15.98 The release of cytotoxic granules from CD8+ cells upon engagement with a target has also been shown to result in triggered drug release. Jones et al. encapsulated IL-15 in lipid nanoparticles that were then tethered to the surface of CD8+ T-cells through maleimide–thiol chemistry.99 Upon engagement with a target, the cytotoxic compounds released from the T-cell would then cause the lipid nanoparticle to also release IL-15 to augment the T-cell activity. The nanocarriers bound to the T-cells mediated a 4-fold reduction in HIV+ CD4+ T-cells compared to an empty nanocarrier control in an HIV infection model.99
Non-stimulus controlled drug delivery has also resulted in augmentation of biomaterials impact on T-cell activity and remains a promising strategy for the treatment of many conditions. Many of these platforms involve the controlled release of therapeutics typically administered systemically for immune modulation. Rhines et al. demonstrated that IL-2 loaded gelatin based polymeric microparticles could allow for the controlled release of IL-2 at the local site a glioma in a rodent model.100 This local release of IL-2 increased the typical survival time with BCNU treatment from 32.5 days to 45.5 days. A monoclonal antibody against OX40 has also been delivered using polymeric PLGA nanoparticles and it has been found that this resulted in a 2-fold increase in T-cell proliferation compared to soluble anti-OX40.101
T-cells have also been actively targeted for drug delivery in an non-antigen specific manner for the treatment of MC38 colon cancer.102 Schmid et al. designed PLGA nanoparticles with a monoclonal antibody against PD-1 on the surface of an exhausted T-cell and loaded with an inhibitor of TGFβ signaling. The result was the targeted delivery of the regulatory-breaking drug to lymphocytes that were vulnerable to PD-1 suppression. This resulted in a 60% increase in median survival of mice compared to non-particle bound controls of anti-PD1 and the TGFβ inhibitor.102
T cells for immunosuppression
While the majority of tolerogenic, particle-based immunotherapies have been targeted at APCs to indirectly induce T-cell-mediated responses, an attractive alternative is to stimulate T-cells to induce tolerance directly, thus bypassing the need for processing and manipulating of antigen presenting cells. T-cell mediated tolerance is primarily mediated through regulatory T-cells that include natural CD4+ CD25+ FOXP3+ cells, induced Th3 CD4+ CD25+ FOXP3+ from Tr1 CD4+ CD25− FOXP3− T-cells, and CD8+ Tregs.103,104 FOXP3+ Tregs target APCs and T effector cells with an end result of suppression of autoreactive T-cells while Tr1 Tregs target T effector cells to initiate mucosal immunity and modulate the inflammatory response.105 CD8+ regulatory T-cells are present in lower amounts than their CD4+ counterparts and little is known about their mechanisms other than they suppress effector lymphocytes through cytokine signaling and negative receptor signaling in inflammatory environments, and they may be generated from low avidity CD8+ autoreactive cells.104,106,107 Polymeric strategies to directly induce T-cell-mediated tolerance can be roughly separated into the following categories: 1. Artificial antigen presentation for induction of regulatory T-cells 2. Targeted Drug Delivery to T-cells 3. Depletion of effector T-cells.Tolerogenic aAPCs have been designed to present signal 1 and antigen by a variety of methods. Most similarly to biological APCs, antigen is coupled to the surface with MHC class I or II in the form of peptide-MHC complexes (pMHC), which gives the added advantage of CD8+ vs. CD4+ T-cell specificity, depending on the selection of peptide loaded MHC class I or II (Fig. 4a–c).109,114,115 A challenge moving forward with this pMHC strategy is the diversity in human HLA haplotypes as opposed to murine MHC. In order to avoid this hurdle, Xu et al. conjugated 5 distinct immunodominant HLA- A*02:01-restricted epitopes specific for Type I diabetes to 500 nm polystyrene beads and found that certain combinations resulted in a 10-fold decrease in antigen specific CD8+ T-cell activation.116 A simpler approach of conjugating CD47-Fc to the particle surface has been explored by Wan et al. and Shahzad et al. in response to the recent finding that CD47-Fc can act as a self-marker and prevent macrophage uptake in aAPCs109,117,118 although this approach lacks T-cell specificity and does not classically present antigen on MHC.109,118 Lastly, there are some strategies that aim to create antigen specificity by encapsulating antigen within particles and using coupled targeting ligands to release antigen to the T-cell in a controlled manner. Coupled surface antigen was directly compared to encapsulated antigen by Hlavaty et al. and found that surface coupling Dby CD4+ T-cell specific antigen was 5× more efficient than encapsulating antigen and induced tolerance to mismatched bone marrow transplantation comparable to positive control, where the level of donor cells was 49% by Week 20.111
 | ||
| Fig. 4 Strategies for targeted expansion of CD4+ T-Cells for immunosuppression. (A) pMHCII-NP loaded with MOG antigen for Experimental Autoimmune Encephalitis (EAE) expands cognate disease-suppressing TR1-like CD4+ T-Cells in vivo. Percentage of pMHCII tetramer in CD4+ cells is increased in pMOG coated PEGylated iron oxide NPs compared to uncoated NPs in both spleen and blood. (B) pMOG-NP therapy dampens disease progression when given on day 14 after immunization. Clinical EAE score of pMOG coated NP injected mice is significantly improved compared to uncoated NPs. (C) pMOG-NP therapy restores motor function in paralytic mice when given on day 21. Subfigures A, B, and C adapted with permission from Nature, Copyright Springer Nature 2016.114 (D)–(F) Targeted drug delivery to CD4+ T-Cells using encapsulated TGF-β and IL-2 induces significant expansion of FOXP3+ CD25+ regulatory T-Cells compared to soluble cytokines. (D) Percentage of FOXP3+ CD25+ Regulatory T-Cells in response to targeted nanoparticle cytokine delivery is increased in nanoparticles compared to soluble cytokine (E) Percentage of Regulatory CD4+ T-Cells Dose responses of nano-encapsulated (green), and soluble (blue) cytokine. (F) Proliferation of IL-2 dependent CTRL-1842 cells after dosing with nano-encapsulated and free IL-2. Subfigures D, E and F adapted with permission from Biomaterials, Copyright Elsevier, 2015.123 | ||
In addition to signal 1 and antigen, aAPCs are often designed to present negative signal 2 costimulatory ligands and/or deliver soluble cytokines to reduce effector T-cell stimulation and promote expansion of Tregs. Negative costimulatory ligands have included anti-FAS and PD-L1, or a complete absence of signal 2, as absence of signal 2 altogether has been shown to induce T-cell anergy or induction of regulatory T-cells.119 Costimulation was shown to have a 50% increase of cell engraftment in a model of bone marrow allotransplantation in Hlavaty et al. when soluble PD1 was added to the Dby CD4+ T-cell specific aAPCS compared to an anti-PD1 blockade group.120 Likewise, in the combinatorial approach of Wan et al., it was difficult to separate the impacts of costimulation of PDL1 and release of TGF-β, but both were implicated in long term maintenance of Experimental Autoimmune Encephalitus (EAE) amelioration as opposed to the short-term effects of the anti-FAS costimulatory ligand.109
Currently all targeted T-cell drug delivery for tolerance induction is directed at CD4+ T-cells for the induction of FOXP3+ regulatory T-cells through incorporation of an anti-CD4 surface ligand, although there is potential for expansion to other T-cell types. Park et al. showed initially that CD4 targeted, encapsulated Leukemia inhibitory factor (LIF) in PLGA nanoparticles expanded FOXP3+ CD4+ Tregs in vitro.125 More recently, McHugh et al. demonstrated dramatic expansion of FOXP3+ Tregs and increased, lasting immune suppression after in vivo treatment of encapsulated IL-2 and TGF-β loaded PLGA nanoparticles compared to soluble cytokines (Fig. 4d–f).123
B-cells
B-cells act as the major producer of antibodies that assist in the mediation of a humoral as opposed to cellular immune response. As opposed to T-cells which require antigen presented on the surface of APCs to become active, B-cells have the capability to recognize antigen that is free in solution.113 Although B-cells can recognize free antigen in solution, multiple studies have confirmed that B-cells require a degree of crosslinking of the antigen on the surface of the membrane to achieve activation.129 Furthermore, it has been found that in vivo, B-cells primarily recognize antigens that have been sequestered and presented on the surface of antigen presenting cells.130 These physiological characteristics have driven the evolution of new biomaterials technologies that are aimed at the presentation of antigen to B-cells as well as technologies to protect antigens and deliver them in tact to B-cells in vivo.Antigen presentation based stimulation
The presentation of antigen on a surface may be the most directly mimetic way to stimulate B-cells based on their aforementioned physiology. As such, many biomaterials based strategies to deliver antigen to B-cell to elicit production of antibodies, rely on the presentation of antigen on a particle surface. Temchura et al. developed a calcium phosphate nanoparticle with HEL antigen conjugated to the surface.131 These nanoparticles were able to elicit an antigen specific B-cell response that was comparable to non-specific stimulation with lipopolysaccharide. Furthermore, this response was found to be 100-fold stronger than soluble antigen.131 Similar to this platform, Ingale et al. developed a liposome based presentation system with well-ordered array of HIV-1 antigen on the surface.132 This resulted in a doubling of antibody titers compared to soluble antigen in a rabbit immunization model. These antigen presentation effects can also be amplified through extensive polymerization of the antigen as shown in Bennett et al., where a 300-mer antigen nanoconstruct resulted in twice the BCR stimulation of a 50-mer antigen nanoconstruct.133Other antigen delivery strategies
Aside from antigen presentation, a major goal of delivery to B-cells is the protection of fragile antigens in the body prior to interaction with B-cells. Polymeric particles serve as the ideal candidate for this application due to their ability to encapsulate the antigen and protect it from a harsh exterior environment. One targeted application by Sicard et al. delivered antigens to B-cells using polystyrene nanoparticles that were targeted to the B-cell receptor by a monoclonal antibody.134 Despite targeting the B-cells, the authors ultimately elicited a cellular immune response through natural B-cell antigen presentation to CD4+ cells. The activated B-cells were able to serve as antigen presenting cells to the CD4+ T-cells and expand rare antigen T-cells nearly 2-fold compared to an inactive control.134 Other platforms for targeted delivery to B-cells take advantage of the C3 complement receptor.135 This strategy could deliver antigen to both B-cells and antigen presenting cells that have similar receptors.Non-targeted strategies focus on antigen protection from harsh physiological environments before delivery to B-cells. One antigen that can be difficult to deliver due to its lack of thermal stability is the polio antigen IPV. Tzeng et al. developed a single injection microparticle formulation that consisted of a PLGA matrix with various cationic polymers in the core to stabilize the antigen.136 The particle formulation was found to have a two burst drug release profile, consistent with an initial vaccination and a follow up booster. The particles elicited similar antibody titers in a host with a single injection of particles as the traditional dual injection strategy, thus obviating the need for a second injection. This could be especially useful in developing countries where access to regular medical care is difficult.136 PLGA encapsulation has also been shown to protect Salmonella typhi antigens from long-term degradation, and provided comparable mucosal immunity to bolus injections of the antigen.137
B cells for immunosuppression
While delivery to APCs and T-cells for induction of tolerance may seem a more obvious approach to treat autoimmune disorders and transplant rejection, there is ongoing work harnessing the power of humoral tolerance through particle-mediated manipulation of B cells. B cells are able to directly mediate tolerance by deleting or suppressing autoreactive B cell receptors, removing autoantigen, and releasing tolerogenic cytokines.138 Current particle-based strategies for B cell tolerance induction focus on deletion of autoreactive B cells either on its own or coupled with targeted delivery of the systemic immunosuppressant rapamycin.139–141 B cells are targeted through the incorporation of a CD22 ligand, surface antigen, and incorporate SIGLEC ligands as costimulatory apoptotic signals. Currently, the materials for delivery have been limited to spherical DSPC nanoliposomes (100–160 nm), and the literature is sparse with respect to particle-mediated tolerizing strategies. However, there is much research in the area of B cell mediated tolerance. A unique strategy that has not been investigated using particle-based methods is to induce tolerance by engineering B cells to produce antibody to harmful self-antigens. Chakerian and Kirnbauer et al. have employed this strategy using virus particles as an immunogenic delivery vector of self-antigen to autoreactive B cells to induce expansion of B cells specific to TNF-α, a mediator of collagen type II arthritis.142,143 It was demonstrated that particle delivery aliased B cell recognition of self vs. nonself and allowed the survival and expansion of mature autoreactive B cells. In a mouse model, vaccination with the particles prevented development of collagen type II arthritis.143NK cells
Natural killer cells are similarly important effector cell of the innate immune system. These cells perform a cytotoxic role in cellular immunity, however they do not respond to antigen presented in MHC Class I as CD8+ T-cells do. In the absence of antigen specificity, NK cells can selectively eliminate cancerous cells and infected cells based on stress markers such as the downregulation of MHC Class I.144 Furthermore it has been shown that depletion of NK cells can prevent biomaterials based anticancer therapeutics from performing at their optimal level.145 Drug delivery to natural killer cells is a largely unexplored field in biomaterials research. Related platforms could shed some light on potential strategies for NK cell therapeutic delivery. Transfection modified cancerous cells have been used to deliver surface bound cytokines (such as IL-15 and IL-21) as well as activating signals (41BBL) to natural killer cells to enhance their activation and proliferation.146 These approaches led to therapeutically active NK cells in clinically relevant numbers. Such a strategy could be adapted to a particulate based system for acellular activation of NK cells through biologic delivery. It is advised that these particle systems would be most effective if designed similar to artificial antigen presenting cells for T-cells in order to mimic the micron scale radius of curvature found on these cellularly based NK cell stimulators.Sinusoidal endothelial liver cells
Liver sinusoidal endothelial cells (LSECs) are liver resident scavengers that have been shown to contribute to hepatic tolerance through induction of CD4+ FOXP3+ regulatory T-cells.147 LSECs are able to interact with circulating T-cells and present antigen as well as signal 2 and cytokine release including immunosuppressive molecules PGE2, PD-L1, FasL, and IL-10. To date, the tolerogenic potential of LSECs for treating autoimmune disease has remained largely untapped by tolerogenic particle-based immunomodulation strategies. The only group to investigate this tolerogenic avenue is Carambia et al., who employed 10–20 nm superparamagnetic iron oxide nanocrystals or CdSe/CdS/ZnS-core–shell–shell quantum dots encapsulated into an amphiphilic polymer (poly(maleic anhydride-alt-1-octadecene)). They were then coupled to MBP and MOG peptides to target the SLE's to induce Tregs for Experimental Autoimmune Encephalitis (EAE). They established that administration of LSEC-targeting autoantigen peptide-loaded nanoparticles could prevent or ameliorate the onset of EAE in mice by significant expansion of antigen specific Tregs.148Conclusion
Polymeric micro- and nanoparticles have been extensively explored for immune modulation via biologic and small molecule drug delivery. In this review, major progress in engineering such polymeric particles to suppress the immune system for autoimmune disease and transplant tolerance or to stimulate the immune system to treat cancer were described. Although suppressive and stimulatory particles have opposing effects, these particles share many design principles. Polymeric micro- and nanoparticles have been designed to target immune cells, such as T cells, dendritic cells, macrophages, B cells, and NK cells, to activate them, suppress them, or guide them toward a desired phenotype. Polymeric particles enable flexibility when designing immunotherapies as many properties, such as size, shape, biodegradability, pharmacokinetics, and targeting capability can be modulated to optimize efficacy and safety. Increasing knowledge in recent years of these cellular subsets of the immune system combined with the development of biocompatible polymeric particles has led to a rapid expansion in the field of particles for immune modulation. Particles provide a modular platform for delivery of various small molecules and biologics and enable cell or organ targeting via surface functionalization or size, surface charge, and material modulation. However, many questions still remain about how polymeric particles affect the immune response. For example, there have been conflicting reports on the effect of particle size on the adjuvanticity of particles.149 Systematic and controlled studies will help elucidate the role of various particle properties so that particles can be tuned to achieve the desired therapeutic immune response. Future research into particles engineered for immune suppression and activation will likely yield more intelligently designed particle-based immunotherapies.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
EBA (DGE-1746891), SEW (DGE-1746891), and KRR (DGE-1232825) thank the NSF Graduate Research Fellowship program for support. RM thanks the Achievement Rewards for College Scientists and the Siebel Foundation for support. The authors thank the NIH for support (R01EB022148 and R01EB016721). The authors also thank the Bloomberg∼Kimmel Institute for Cancer Immunotherapy and the Research to Prevent Blindness James and Carole Free Catalyst Award for support in part.References
- C. Wang, Y. Ye, Q. Hu, A. Bellotti and Z. Gu, Tailoring biomaterials for cancer immunotherapy: Emerging trends and future outlook, Adv. Mater., 2017, 29, 1606036 CrossRef PubMed.
- R. Klippstein and D. Pozo, Nanotechnology-based manipulation of dendritic cells for enhanced immunotherapy strategies, Nanomedicine, 2010, 6, 523–529 CrossRef CAS PubMed.
- A. K. Shakya and K. S. Nandakumar, Biomaterials for Induction and Treatment of Autoimmunity, in Advances in Biomaterials for Biomedical Applications, Springer, 2017, pp. 167–184 Search PubMed.
- M. Rahmati, C. P. Pennisi, A. Mobasheri and M. Mozafari, Bioengineered Scaffolds for Stem Cell Applications in Tissue Engineering and Regenerative Medicine, 2018 Search PubMed.
- A. B. Gardner, S. K. Lee, E. C. Woods and A. P. Acharya, Biomaterials-based modulation of the immune system, BioMed Res. Int., 2013, 2013, 732182 Search PubMed.
- S. Franz, S. Rammelt, D. Scharnweber and J. C. Simon, Immune responses to implants–a review of the implications for the design of immunomodulatory biomaterials, Biomaterials, 2011, 32, 6692–6709 CrossRef CAS PubMed.
- J. J. Moon, B. Huang and D. J. Irvine, Engineering nano- and microparticles to tune immunity, Adv. Mater., 2012, 24, 3724–3746 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol., 2015, 33, 941 CrossRef CAS PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- R. A. Meyer, J. C. Sunshine and J. J. Green, Biomimetic particles as therapeutics, Trends Biotechnol., 2015, 33, 514–524 CrossRef CAS PubMed.
- J. Sunshine and J. M. Taube, PD-1/PD-L1 inhibitors, Curr. Opin. Pharmacol., 2015, 23, 32–38 CrossRef CAS PubMed.
- G. R. Burmester, et al., Mavrilimumab, a Fully Human Granulocyte–Macrophage Colony-Stimulating Factor Receptor α Monoclonal Antibody: Long-Term Safety and Efficacy in Patients With Rheumatoid Arthritis, Arthritis Rheumatol., 2018, 70, 679–689 CrossRef CAS PubMed.
- F. DeStefano, E. S. Weintraub and R. T. Chen, Recombinant hepatitis B vaccine and the risk of multiple sclerosis: a prospective study, 2018 Search PubMed.
- H. Caspard, et al., Effectiveness of live attenuated influenza vaccine and inactivated influenza vaccine in children 2–17 years of age in 2013–2014 in the United States, Vaccine, 2016, 34, 77–82 CrossRef CAS PubMed.
- J. Li, S. G. Kim and J. Blenis, Rapamycin: one drug, many effects, Cell Metab., 2014, 19, 373–379 CrossRef CAS PubMed.
- J. L. Adams, J. Smothers, R. Srinivasan and A. Hoos, Big opportunities for small molecules in immuno-oncology, Nat. Rev. Drug Discovery, 2015, 14, 603 CrossRef CAS PubMed.
- S. Y. Tzeng and J. J. Green, Polymeric Nucleic Acid Delivery for Immunoengineering, Curr. Opin. Biomed. Eng., 2018 Search PubMed , in press.
- C.-Y. Lin, et al., Biodegradable polymeric microsphere-based vaccines and their applications in infectious diseases, Hum. Vaccines Immunother., 2015, 11, 650–656 CrossRef PubMed.
- A. Bolhassani, et al., Polymeric nanoparticles, Hum. Vaccines Immunother., 2014, 10, 321–332 CrossRef CAS PubMed.
- D. S. Vinay, et al., Immune evasion in cancer: Mechanistic basis and therapeutic strategies, Semin. Cancer Biol., 2015, 35, S185–S198 CrossRef PubMed.
- T. Chanmee, P. Ontong, K. Konno and N. Itano, Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment, Cancers, 2014, 6, 1670–1690 CrossRef CAS PubMed.
- K. K. Ahmed, S. M. Geary and A. K. Salem, Applying biodegradable particles to enhance cancer vaccine efficacy, Immunol. Res., 2014, 59, 220–228 CrossRef CAS PubMed.
- S. T. Reddy, et al., Exploiting lymphatic transport and complement activation in nanoparticle vaccines, Nat. Biotechnol., 2007, 25, 1159 CrossRef CAS PubMed.
- I. Melero, et al., Therapeutic vaccines for cancer: an overview of clinical trials, Nat. Rev. Clin. Oncol., 2014, 11, 509 CrossRef CAS PubMed.
- C. Mesa and L. E. Fernández, Challenges facing adjuvants for cancer immunotherapy, Immunol. Cell Biol., 2004, 82, 644–650 CrossRef CAS PubMed.
- S. Awate, L. Babiuk and G. Mutwiri, Mechanisms of Action of Adjuvants, Front. Immunol., 2013, 4, 114 Search PubMed.
- K. K. Ahmed, S. M. Geary and A. K. Salem, Development and Evaluation of Biodegradable Particles Co-loaded with Antigen and the Toll-like Receptor Agonist, Pentaerythritol Lipid A, as a Cancer Vaccine, J. Pharm. Sci., 2016, 105, 1173–1179 CrossRef CAS PubMed.
- S. Hamdy, et al., Enhanced antigen-specific primary CD4+ and CD8+ responses by codelivery of ovalbumin and toll-like receptor ligand monophosphoryl lipid A in poly(D,L-lactic-co-glycolic acid) nanoparticles, J. Biomed. Mater. Res., Part A, 2007, 81A, 652–662 CrossRef CAS PubMed.
- S. Hamdy, et al., Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity, Vaccine, 2008, 26, 5046–5057 CrossRef CAS PubMed.
- Z. Zhang, et al., Induction of anti-tumor cytotoxic T cell responses through PLGA-nanoparticle mediated antigen delivery, Biomaterials, 2011, 32, 3666–3678 CrossRef CAS PubMed.
- P. Elamanchili, C. M. E. Lutsiak, S. Hamdy, M. Diwan and J. Samuel, “Pathogen-Mimicking” Nanoparticles for Vaccine Delivery to Dendritic Cells, J. Immunother., 2007, 30, 378–395 CrossRef CAS PubMed.
- C. M. Jewell, S. C. Bustamante López and D. J. Irvine, In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles, Proc. Natl. Acad. Sci., 2011, 108, 15745–15750 CrossRef CAS PubMed.
- X. Q. Zhang, C. E. Dahle, G. J. Weiner and A. K. Salem, A comparative study of the antigen-specific immune response induced by co-delivery of CpG ODN and antigen using fusion molecules or biodegradable microparticles**Xue-Qing Zhang and Christopher E. Dahle contributed equally to this work, J. Pharm. Sci., 2007, 96, 3283–3292 CrossRef CAS PubMed.
- A. Bershteyn, et al., Robust IgG responses to nanograms of antigen using a biomimetic lipid-coated particle vaccine, J. Controlled Release, 2012, 157, 354–365 CrossRef CAS PubMed.
- R. H. Fang, et al., Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS PubMed.
- Z. Luo, et al., Cationic polypeptide micelle-based antigen delivery system: A simple and robust adjuvant to improve vaccine efficacy, J. Controlled Release, 2013, 170, 259–267 CrossRef CAS PubMed.
- H. Kim, T. Uto, T. Akagi, M. Baba and M. Akashi, Amphiphilic Poly(Amino Acid) Nanoparticles Induce Size-Dependent Dendritic Cell Maturation, Adv. Funct. Mater., 2010, 20, 3925–3931 CrossRef CAS.
- V. B. Joshi, S. M. Geary and A. K. Salem, Biodegradable Particles as Vaccine Delivery Systems: Size Matters, AAPS J., 2013, 15, 85–94 CrossRef CAS PubMed.
- S. T. Reddy, A. Rehor, H. G. Schmoekel, J. A. Hubbell and M. A. Swartz, In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles, J. Controlled Release, 2006, 112, 26–34 CrossRef CAS PubMed.
- V. Manolova, et al., Nanoparticles target distinct dendritic cell populations according to their size, Eur. J. Immunol., 2008, 38, 1404–1413 CrossRef CAS PubMed.
- S. N. Thomas, E. Vokali, A. W. Lund, J. A. Hubbell and M. A. Swartz, Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response, Biomaterials, 2014, 35, 814–824 CrossRef CAS PubMed.
- Y. Guo, et al., Erythrocyte Membrane-Enveloped Polymeric Nanoparticles as Nanovaccine for Induction of Antitumor Immunity against Melanoma, ACS Nano, 2015, 9, 6918–6933 CrossRef CAS PubMed.
- L. J. Cruz, et al., Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro, J. Controlled Release, 2010, 144, 118–126 CrossRef CAS PubMed.
- S. S. Saluja, et al., Targeting human dendritic cells via DEC-205 using PLGA nanoparticles leads to enhanced cross-presentation of a melanoma-associated antigen, Int. J. Nanomed., 2014, 9, 5231–5246 Search PubMed.
- R. A. Rosalia, et al., CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses, Biomaterials, 2015, 40, 88–97 CrossRef CAS PubMed.
- L. J. Cruz, et al., Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8+ T cell response: A comparative study, J. Controlled Release, 2014, 192, 209–218 CrossRef CAS PubMed.
- T. Chanmee, P. Ontong, K. Konno and N. Itano, Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment, Cancers, 2014, 6, 1670–1690 CrossRef CAS PubMed.
- R. Noy and J. W. Pollard, Tumor-Associated Macrophages: From Mechanisms to Therapy, Immunity, 2014, 41, 49–61 CrossRef CAS PubMed.
- M. J. Ernsting, et al., Targeting of metastasis-promoting tumor-associated fibroblasts and modulation of pancreatic tumor-associated stroma with a carboxymethylcellulose-docetaxel nanoparticle, J. Controlled Release, 2015, 206, 122–130 CrossRef CAS PubMed.
- M. Niu, et al., Biodistribution and in Vivo Activities of Tumor-Associated Macrophage-Targeting Nanoparticles Incorporated with Doxorubicin, Mol. Pharm., 2014, 11, 4425–4436 CrossRef CAS PubMed.
- M. Niu, S. Valdes, Y. W. Naguib, S. D. Hursting and Z. Cui, Tumor-Associated Macrophage-Mediated Targeted Therapy of Triple-Negative Breast Cancer, Mol. Pharm., 2016, 13, 1833–1842 CrossRef CAS PubMed.
- A.-K. Fuchs, et al., Carboxyl- and amino-functionalized polystyrene nanoparticles differentially affect the polarization profile of M1 and M2 macrophage subsets, Biomaterials, 2016, 85, 78–87 CrossRef CAS PubMed.
- Y. Wang, et al., Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment, Biomaterials, 2017, 112, 153–163 CrossRef CAS PubMed.
- Y. J. Kwon, E. James, N. Shastri and J. M. Frechet, In vivo targeting of dendritic cells for activation of cellular immunity using vaccine carriers based on pH-responsive microparticles, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18264–18268 CrossRef CAS PubMed.
- A. Bandyopadhyay, R. L. Fine, S. Demento, L. K. Bockenstedt and T. M. Fahmy, The impact of nanoparticle ligand density on dendritic-cell targeted vaccines, Biomaterials, 2011, 32, 3094–3105 CrossRef CAS PubMed.
- G. Schreibelt, et al., The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells, Blood, 2012, 119, 2284–2292 CrossRef CAS PubMed.
- O. P. Joffre, D. Sancho, S. Zelenay, A. M. Keller and C. Reis e Sousa, Efficient and versatile manipulation of the peripheral CD4+ T-cell compartment by antigen targeting to DNGR-1/CLEC9A, Eur. J. Immunol., 2010, 40, 1255–1265 CrossRef CAS PubMed.
- S. Kanno, A. Furuyama and S. Hirano, A murine scavenger receptor MARCO recognizes polystyrene nanoparticles, Toxicol. Sci., 2007, 97, 398–406 CrossRef CAS PubMed.
- S. Prasad, D. Xu and S. D. Miller, Tolerance strategies employing antigen-coupled apoptotic cells and carboxylated PLG nanoparticles for the treatment of type 1 diabetes, Rev. Diabetes Stud., 2012, 9, 319–327 CrossRef PubMed.
- S. D. Miller, R. P. Wetzig and H. N. Claman, The induction of cell-mediated immunity and tolerance with protein antigens coupled to syngeneic lymphoid cells, J. Exp. Med., 1979, 149, 758–773 CrossRef CAS PubMed.
- A. Lutterotti, et al., Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis, Sci. Transl. Med., 2013, 5, 188ra175 Search PubMed.
- S. Kontos, I. C. Kourtis, K. Y. Dane and J. A. Hubbell, Engineering antigens for in situ erythrocyte binding induces T-cell deletion, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E60–E68 CrossRef CAS PubMed.
- T. K. Kishimoto and R. A. Maldonado, Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance, Front. Immunol., 2018, 9, 230 CrossRef PubMed.
- D. R. Getts, et al., Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis, Nat. Biotechnol., 2012, 30, 1217–1224 CrossRef CAS PubMed.
- Z. Hunter, et al., A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease, ACS Nano, 2014, 8, 2148–2160 CrossRef CAS PubMed.
- A. C. Shirali, et al., Nanoparticle delivery of mycophenolic acid upregulates PD-L1 on dendritic cells to prolong murine allograft survival, Am. J. Transplant., 2011, 11, 2582–2592 CrossRef CAS PubMed.
- K. Ramani, et al., Phosphatidylserine containing liposomes reduce immunogenicity of recombinant human factor VIII (rFVIII) in a murine model of hemophilia A, J. Pharm. Sci., 2008, 97, 1386–1398 CrossRef CAS PubMed.
- I. Pujol-Autonell, et al., Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes, PLoS One, 2015, 10, e0127057 CrossRef PubMed.
- R. A. Roberts, et al., Towards programming immune tolerance through geometric manipulation of phosphatidylserine, Biomaterials, 2015, 72, 1–10 CrossRef CAS PubMed.
- W. U. Kim, et al., Suppression of collagen-induced arthritis by single administration of poly(lactic-co-glycolic acid) nanoparticles entrapping type II collagen: a novel treatment strategy for induction of oral tolerance, Arthritis Rheum., 2002, 46, 1109–1120 CrossRef CAS PubMed.
- S. Y. Min, et al., Antigen-induced, tolerogenic CD11c+, CD11b+ dendritic cells are abundant in Peyer's patches during the induction of oral tolerance to type II collagen and suppress experimental collagen-induced arthritis, Arthritis Rheum., 2006, 54, 887–898 CrossRef CAS PubMed.
- G. Cappellano, et al., Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis, Vaccine, 2014, 32, 5681–5689 CrossRef CAS PubMed.
- J. S. Lewis, et al., A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice, Clin. Immunol., 2015, 160, 90–102 CrossRef CAS PubMed.
- J. J. Cho, et al., An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis, Biomaterials, 2017, 143, 79–92 CrossRef CAS PubMed.
- J. M. Gammon, L. H. Tostanoski, A. R. Adapa, Y. C. Chiu and C. M. Jewell, Controlled delivery of a metabolic modulator promotes regulatory T cells and restrains autoimmunity, J. Controlled Release, 2015, 210, 169–178 CrossRef CAS PubMed.
- K. A. Hlavaty, X. Luo, L. D. Shea and S. D. Miller, Cellular and molecular targeting for nanotherapeutics in transplantation tolerance, Clin. Immunol., 2015, 160, 14–23 CrossRef CAS PubMed.
- S. Jhunjhunwala, G. Raimondi, A. W. Thomson and S. R. Little, Delivery of rapamycin to dendritic cells using degradable microparticles, J. Controlled Release, 2009, 133, 191–197 CrossRef CAS PubMed.
- A. Haddadi, et al., Delivery of rapamycin by PLGA nanoparticles enhances its suppressive activity on dendritic cells, J. Biomed. Mater. Res., Part A, 2008, 84, 885–898 CrossRef PubMed.
- R. A. Maldonado, et al., Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E156–E165 CrossRef CAS PubMed.
- L. H. Tostanoski, et al., Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific, Cell Rep., 2016, 16, 2940–2952 CrossRef CAS PubMed.
- H. H. Lim, et al., A pilot study on using rapamycin-carrying synthetic vaccine particles (SVP) in conjunction with enzyme replacement therapy to induce immune tolerance in Pompe disease, Mol. Genet. Metab. Rep., 2017, 13, 18–22 CrossRef CAS PubMed.
- A. H. Zhang, R. J. Rossi, J. Yoon, H. Wang and D. W. Scott, Tolerogenic nanoparticles to induce immunologic tolerance: Prevention and reversal of FVIII inhibitor formation, Cell. Immunol., 2016, 301, 74–81 CrossRef CAS PubMed.
- T. K. Kishimoto, et al., Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles, Nat. Nanotechnol., 2016, 11, 890–899 CrossRef CAS PubMed.
- K. J. Peine, et al., Treatment of experimental autoimmune encephalomyelitis by codelivery of disease associated Peptide and dexamethasone in acetalated dextran microparticles, Mol. Pharm., 2014, 11, 828–835 CrossRef CAS PubMed.
- R. A. Meyer and J. J. Green, Artificial Antigen-Presenting Cells: Biomimetic Strategies for Directing the Immune Response, in Biomaterials in Regenerative Medicine and the Immune System, Springer, 2015, pp. 257–277 Search PubMed.
- M. Oelke, et al., Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig–coated artificial antigen-presenting cells, Nat. Med., 2003, 9, 619 CrossRef CAS PubMed.
- J. W. Hickey, F. P. Vicente, G. P. Howard, H.-Q. Mao and J. P. Schneck, Biologically inspired design of nanoparticle artificial antigen-presenting cells for immunomodulation, Nano Lett., 2017, 17, 7045–7054 CrossRef CAS PubMed.
- J. C. Sunshine, K. Perica, J. P. Schneck and J. J. Green, Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells, Biomaterials, 2014, 35, 269–277 CrossRef CAS PubMed.
- R. A. Meyer, et al., Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation, Small, 2015, 11, 1519–1525 CrossRef CAS PubMed.
- A. Kosmides, et al., Biomimetic biodegradable artificial antigen presenting cells synergize with PD-1 blockade to treat melanoma, Biomaterials, 2017, 118, 16–26 CrossRef CAS PubMed.
- E. R. Steenblock and T. M. Fahmy, A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells, Mol. Ther., 2008, 16, 765–772 CrossRef CAS PubMed.
- E. R. Steenblock, T. Fadel, M. Labowsky, J. S. Pober and T. M. Fahmy, An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response, J. Biol. Chem., 2011, M111, 276329 Search PubMed.
- T. R. Fadel, et al., A carbon nanotube–polymer composite for T-cell therapy, Nat. Nanotechnol., 2014, 9, 639–647 CrossRef CAS PubMed.
- C. Schütz, et al., Antigen-specific T cell Redirectors: a nanoparticle based approach for redirecting T cells, Oncotarget, 2016, 7, 68503 CrossRef PubMed.
- A. K. Kosmides, J.-W. Sidhom, A. Fraser, C. A. Bessell and J. P. Schneck, Dual targeting nanoparticle stimulates the immune system to inhibit tumor growth, ACS Nano, 2017, 11, 5417–5429 CrossRef CAS PubMed.
- L. Luo, et al., Sustained release of anti-PD-1 peptide for perdurable immunotherapy together with photothermal ablation against primary and distant tumors, J. Controlled Release, 2018, 278, 87–99 CrossRef CAS PubMed.
- J. Cano-Mejia, et al., Prussian blue nanoparticle-based photothermal therapy combined with checkpoint inhibition for photothermal immunotherapy of neuroblastoma. Nanomedicine: Nanotechnology, Biol. Med., 2017, 13, 771–781 CAS.
- L. Tang, et al., Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery, Nat. Biotechnol., 2018, 36, 707 CAS.
- R. B. Jones, et al., Antigen recognition-triggered drug delivery mediated by nanocapsule-functionalized cytotoxic T-cells, Biomaterials, 2017, 117, 44–53 CrossRef CAS PubMed.
- L. D. Rhines, et al., Local immunotherapy with interleukin-2 delivered from biodegradable polymer microspheres combined with interstitial chemotherapy: a novel treatment for experimental malignant glioma, Neurosurgery, 2003, 52, 872–880 CrossRef PubMed.
- M. Chen, H. Ouyang, S. Zhou, J. Li and Y. Ye, PLGA-nanoparticle mediated delivery of anti-OX40 monoclonal antibody enhances anti-tumor cytotoxic T cell responses, Cell. Immunol., 2014, 287, 91–99 CrossRef CAS PubMed.
- D. Schmid, et al., T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity, Nat. Commun., 2017, 8, 1747 CrossRef PubMed.
- H. Jonuleit and E. Schmitt, The regulatory T cell family: distinct subsets and their interrelations, J. Immunol., 2003, 171, 6323–6327 CrossRef CAS.
- M. R. Vieyra-Lobato, J. Vela-Ojeda, L. Montiel-Cervantes, R. López-Santiago and M. C. Moreno-Lafont, Description of CD8+ Regulatory T Lymphocytes and Their Specific Intervention in Graft-versus-Host and Infectious Diseases, Autoimmunity, and Cancer, J. Immunol. Res., 2018, 2018, 3758713 Search PubMed.
- L. Chatenoud and J.-F. Bach, Adaptive human regulatory T cells: myth or reality?, J. Clin. Invest., 2006, 116, 2325–2327 CrossRef CAS PubMed.
- Z. Chen, et al., CD11c(high)CD8+ regulatory T cell feedback inhibits CD4 T cell immune response via Fas ligand-Fas pathway, J. Immunol., 2013, 190, 6145–6154 CrossRef CAS PubMed.
- P. Zaccone and A. Cooke, Harnessing CD8+ Regulatory T Cells: Therapy for Type 1 Diabetes?, Immunity, 2010, 32, 504–506 CrossRef CAS PubMed.
- A. Moore, J. Grimm, B. Han and P. Santamaria, Tracking the Recruitment of Diabetogenic CD8 T-Cells to the Pancreas in Real Time, Diabetes, 2004, 53, 1459–1466 CrossRef CAS PubMed.
- X. Wan, et al., A Tolerogenic Artificial APC Durably Ameliorates Experimental Autoimmune Encephalomyelitis by Directly and Selectively Modulating Myelin Peptide-Autoreactive CD4+ and CD8+ T Cells., J. Immunol., 2018, 201, 1194–1210 CrossRef CAS PubMed.
- W. Wang, et al., A biodegradable killer microparticle to selectively deplete antigen-specific T cells in vitro and in vivo, Oncotarget, 2016, 7, 12176–12190 Search PubMed.
- K. A. Hlavaty, et al., Tolerance induction using nanoparticles bearing HY peptides in bone marrow transplantation, Biomaterials, 2016, 76, 1–10 CrossRef CAS PubMed.
- J. C. Sunshine, K. Perica, J. P. Schneck and J. J. Green, Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells, Biomaterials, 2014, 35, 269–277 CrossRef CAS PubMed.
- E. Ben-Akiva, R. A. Meyer, D. R. Wilson and J. J. Green, Surface engineering for lymphocyte programming, Adv. Drug Delivery Rev., 2017, 114, 102–115 CrossRef CAS PubMed.
- X. Clemente-Casares, et al., Expanding antigen-specific regulatory networks to treat autoimmunity, Nature, 2016, 530, 434–440 CrossRef CAS PubMed.
- S. Tsai, et al., Reversal of Autoimmunity by Boosting Memory-like Autoregulatory T Cells, Immunity, 2010, 32, 568–580 CrossRef CAS PubMed.
- X. Xu, et al., Multipeptide-coupled nanoparticles induce tolerance in ‘humanised’ HLA-transgenic mice and inhibit diabetogenic CD8+ T cell responses in type 1 diabetes, Diabetologia, 2017, 2418–2431 CrossRef CAS PubMed.
- H. Bruns, et al., CD47 Enhances In Vivo Functionality of Artificial Antigen-Presenting Cells, Clin. Cancer Res., 2015, 21, 2075–2083 CrossRef CAS PubMed.
- K. A. Shahzad, et al., On-target and direct modulation of alloreactive T cells by a nanoparticle carrying MHC alloantigen, regulatory molecules and CD47 in a murine model of alloskin transplantation, Drug Delivery, 2018, 25, 703–715 CrossRef PubMed.
- B. M. Hall, H. Waldmann, T. K. Kishimoto and R. A. Maldonado, Nanoparticles for the induction of Antigen-Specific immunological Tolerance, Front. Immunol., 2018, 9, 1 CrossRef PubMed.
- K. A. Hlavaty, et al., Tolerance induction using nanoparticles bearing HY peptides in bone marrow transplantation, Biomaterials, 2016, 76, 1–10 CrossRef CAS PubMed.
- M. Look, et al., Nanogel-based delivery of mycophenolic acid ameliorates systemic lupus erythematosus in mice, J. Clin. Invest., 2013, 123, 1741–1749 CrossRef CAS PubMed.
- D. R. Getts, L. D. Shea, S. D. Miller and N. J. C. King, Harnessing nanoparticles for immune modulation, Trends Immunol., 2015, 36, 419–427 CrossRef CAS PubMed.
- M. D. McHugh, et al., Paracrine co-delivery of TGF-beta and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells, Biomaterials, 2015, 59, 172–181 CrossRef CAS PubMed.
- T. M. Fahmy, Nanoparticle-Mediated Delivery of Cytokines for Maintenance of the Regulatory T cell Phenotype, Yale University, US Pat., 0361265 A1, 2016 Search PubMed.
- J. Park, et al., Modulation of CD4+ T Lymphocyte Lineage Outcomes with Targeted, Nanoparticle-Mediated Cytokine Delivery, Mol. Pharm., 2011, 8, 143–152 CrossRef CAS PubMed.
- C. Schütz, et al., Killer artificial antigen-presenting cells: a novel strategy to delete specific T cells, Blood, 2008, 111, 3546–3552 CrossRef PubMed.
- C. Shen, et al., Killer artificial antigen-presenting cells deplete alloantigen-specific T cells in a murine model of alloskin transplantation, Immunol. Lett., 2011, 138, 144–155 CrossRef CAS PubMed.
- W. Wang, et al., An Antigen-Presenting and Apoptosis-Inducing Polymer Microparticle Prolongs Alloskin Graft Survival by Selectively and Markedly Depleting Alloreactive CD8+ T Cells, Front. Immunol., 2017, 8, 657 CrossRef PubMed.
- S. V. Desiderio, B-cell activation, Curr. Opin. Immunol., 1992, 4, 252–256 CrossRef CAS PubMed.
- N. E. Harwood and F. D. Batista, Early events in B cell activation, Annu. Rev. Immunol., 2009, 28, 185–210 CrossRef PubMed.
- V. V. Temchura, D. Kozlova, V. Sokolova, K. Überla and M. Epple, Targeting and activation of antigen-specific B-cells by calcium phosphate nanoparticles loaded with protein antigen, Biomaterials, 2014, 35, 6098–6105 CrossRef CAS PubMed.
- J. Ingale, et al., High-density array of well-ordered HIV-1 spikes on synthetic liposomal nanoparticles efficiently activate B cells, Cell Rep., 2016, 15, 1986–1999 CrossRef CAS PubMed.
- N. R. Bennett, D. B. Zwick, A. H. Courtney and L. L. Kiessling, Multivalent antigens for promoting B and T cell activation, ACS Chem. Biol., 2015, 10, 1817–1824 CrossRef CAS PubMed.
- A. Sicard, et al., B cells loaded with synthetic particulate antigens: a versatile platform to generate antigen-specific helper T cells for cell therapy, Nano Lett., 2015, 16, 297–308 CrossRef PubMed.
- A. Francian, K. Mann and M. Kullberg, Complement C3-dependent uptake of targeted liposomes into human macrophages, B cells, dendritic cells, neutrophils, and MDSCs, Int. J. Nanomed., 2017, 12, 5149 CrossRef CAS PubMed.
- S. Y. Tzeng, et al., Stabilized single-injection inactivated polio vaccine elicits a strong neutralizing immune response, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5269–E5278 CrossRef CAS PubMed.
- J. M. Carreño, et al., PLGA-microencapsulation protects Salmonella typhi outer membrane proteins from acidic degradation and increases their mucosal immunogenicity, Vaccine, 2016, 34, 4263–4269 CrossRef PubMed.
- N. Manjarrez-Orduño, T. D. Quách and I. Sanz, B cells and immunological tolerance, J. Invest. Dermatol., 2009, 129, 278–288 CrossRef PubMed.
- M. S. Macauley, et al., Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis, J. Clin. Invest., 2013, 123, 3074–3083 CrossRef CAS PubMed.
- B. H. Duong, et al., Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo, J. Exp. Med., 2010, 207, 173–187 CrossRef CAS PubMed.
- L. Pang, M. S. Macauley, B. M. Arlian, C. M. Nycholat and J. C. Paulson, Encapsulating an Immunosuppressant Enhances Tolerance Induction by Siglec-Engaging Tolerogenic Liposomes, ChemBioChem, 2017, 18, 1226–1233 CrossRef CAS PubMed.
- R. Kirnbauer, F. Booy, N. Cheng, D. R. Lowy and J. T. Schiller, Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 12180–12184 CrossRef CAS.
- B. Chackerian, D. R. Lowy and J. T. Schiller, Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies, J. Clin. Invest., 2001, 108, 415–423 CrossRef CAS PubMed.
- D. R. Shook and D. Campana, Natural killer cell engineering for cellular therapy of cancer, Tissue Antigens, 2011, 78, 409–415 CrossRef CAS PubMed.
- K. D. Moynihan, et al., Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses, Nat. Med., 2016, 22, 1402 CrossRef CAS PubMed.
- C. J. Denman, et al., Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells, PLoS One, 2012, 7, e30264 CrossRef CAS PubMed.
- X. Xu, et al., Liver sinusoidal endothelial cells induce tolerance of autoreactive CD4+ recent thymic emigrants, Sci. Rep., 2016, 6, 19861 CrossRef CAS PubMed.
- A. Carambia, et al., Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice, 2015 Search PubMed.
- M. O. Oyewumi, A. Kumar and Z. Cui, Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses, Expert Rev. Vaccines, 2010, 9, 1095–1107 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |