
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Serum albumin hydrogels in broad pH and temperature ranges: characterization of their self-assembled structures and nanoscopic and macroscopic properties†
S. Hamidreza
Arabi
 ,
Behdad
Aghelnejad
,
Christian
Schwieger
,
Annette
Meister
,
Andreas
Kerth
and
Dariush
Hinderberger
*
,
Behdad
Aghelnejad
,
Christian
Schwieger
,
Annette
Meister
,
Andreas
Kerth
and
Dariush
Hinderberger
*
Institut für Chemie, Martin-Luther-Universität Halle-Wittenberg, Von-Danckelmann-Platz 4, 06120 Halle, Saale, Germany. E-mail: dariush.hinderberger@chemie.uni-halle.de
First published on 15th February 2018
Abstract
We report extended pH- and temperature-induced preparation procedures and explore the materials and molecular properties of different types of hydrogels made from human and bovine serum albumin, the major transport protein in the blood of mammals. We describe the diverse range of properties of these hydrogels at three levels: (1) their viscoelastic (macroscopic) behavior, (2) protein secondary structure changes during the gelation process (via ATR-FTIR spectroscopy), and (3) the hydrogel fatty acid (FA) binding capacity and derive from this the generalized tertiary structure through CW EPR spectroscopy. We describe the possibility of preparing hydrogels from serum albumin under mild conditions such as low temperatures (notably below albumin's denaturation temperature) and neutral pH value. As such, the proteins retain most of their native secondary structure. We find that all the combined data indicate a two-stage gelation process that is studied in detail. We summarize these findings and the explored dependences of the gels on pH, temperature, concentration, and incubation time by proposing phase diagrams for both HSA and BSA gel-states. As such, it has become possible to prepare gels that have the desired nanoscopic and macroscopic properties, which can, in future, be tested for, e.g., drug delivery applications.
Introduction
Serum albumin is the most abundant protein in the blood plasma of mammals, reaching a concentration of 40 to 50 mg ml−1 in plasma, and it is the primary carrier of various solutes in plasma.1,2 Its high abundance, stability and availability at high purity and low cost make serum albumin a model protein in many physicochemical and biomaterials science studies.3,4 We have, in the past, elucidated the structural versatility and flexibility especially of human (and bovine) serum albumin through an electron paramagnetic resonance (EPR) spectroscopy-based research platform.5–7Concurrently, in the biomedical sciences there is an increasing need to develop new materials which simultaneously have the desired affinities to biological and therapeutic materials and high biocompatibility. Hydrogels made from synthetic or bio-macromolecules have exhibited great potential for biological and medical applications.8–10 Among them, protein gels can be assumed to be cage-like unit structures forming a matrix in which the solvent (water in the case of hydrogels) and potentially releasable drugs and adjuvants are trapped.11
There has been an ongoing endeavor to synthesize hydrogels from serum albumin in such a way that robust, biocompatible hydrogels are delivered while preserving protein functionality, i.e. the capability to bind, retain, and release a variety of molecules in a well-defined manner. So far, very few (considering the abundance and availability of albumin) different synthetic methods have been developed12–15 and so far only hydrogels from bovine serum albumin (BSA), which has a primary sequence identity with human serum albumin (HSA) of ∼76%,16 have been reported. Here, we report, to the best of our knowledge for the first time, gels made from HSA (as well as from BSA, which have been described before), and we particularly focus on (i) systematically studying the conditions (temperature, pH and incubation time) under which gelation can take place, (ii) the characterization of the gels at the macroscopic level (mechanical properties as seen in rheological measurements) as well as (iii) the characterization of the accompanying changes in structure, dynamics, and functionality at the molecular level as seen in ATR-IR and electron paramagnetic resonance (EPR) spectroscopy.
Thermally induced BSA hydrogels (see Fig. 1) are already well established and one finds long-standing work on the mechanism of thermally induced gelation of proteins such as BSA.17–19 Heating-induced gel formation consists of two sequential processes. First, unfolding of polypeptide segments induced by heating causes conformational changes and an altered tertiary structure in the protein. A subsequent phase of protein–protein (secondary structure element) interactions results in a progressive build-up of a network structure leading to the final gel.20 The main disadvantage of this method is the extensive protein denaturation, with the risk of compromising protein functionality and biocompatibility.21 The currently accepted working hypothesis20,22–25 states that in order to obtain thermally induced albumin hydrogels at neutral pH, the temperature must be set above the denaturation temperature of the protein (at pH 7.4 the denaturation temperature of serum albumin is 62 °C (ref. 26)).
 | ||
| Fig. 1 (A) Bovine and human serum albumin hydrogels are formed at 50 °C and pH 7.0 and 7.2, respectively. (B) Superimposed three-dimensional structures of HSA and BSA are shown in blue and red. The protein topologies from pdb IDs 1BM0HSA47 and 3v03BSA.48 (C) Chemical structure of 16-DSA used as a spin probe. | ||
Another preparation method for albumin hydrogels has recently been introduced by Baler et al. (2014),22 which the authors called electrostatically triggered serum albumin hydrogels. In this method, the pH of the precursor solution (solution of serum albumin dissolved in water) is lowered to the value of 3.5 using 2 M HCl; consequently, the net charge on the protein (according to a theoretical PROPKA analysis) changes from −16 at pH 7.4 to +100 at pH 3.5.27 One effect of this charge inversion is that now positively charged protein domains repel each other, which causes partial denaturation of the protein and results in solvent exposure of the buried hydrophobic areas of the albumin. This then triggers intermolecular self-assembly of the proteins into a hydrogel at 37 °C. It has been suggested that hydrophobic interactions and counter-ion binding are the key drivers of protein aggregation in this system.27 The gelation process takes place at much lower temperatures when the pH is adjusted. It has been reported that this mechanism works in the pH range of 3.0 to 4.0 for BSA with the optimum value of 3.5.22
The incubation time ti during gelation, i.e. the time after which the gelation process is quenched, is a critical factor that has remarkably not been studied systematically thus far. As will be shown in the following, by changing ti it is possible to obtain serum albumin hydrogels (from both HSA and BSA) at temperatures far below the denaturation temperature of the proteins at neutral pH. Furthermore, it will be shown that by varying ti, also the pH range in which electrostatically triggered albumin hydrogels form is much broader than reported. Changing the incubation time ti affects the mechanical properties of the hydrogels in a highly complex manner. Some gels harden with longer ti, while others start to decompose again; the rise times of gelation can be very different, ranging from instantaneous to several hours, and many other non-trivial differences are observed. Herein, we report a systematic study of the combined effects of pH, temperature and incubation time on albumin hydrogel formation and its properties and provide explicit phase diagrams for both human and bovine serum albumin. Besides the mechanical properties as studied by rheology, the changes in secondary structure are characterized using their IR-spectroscopic signatures. Our investigation is complemented by a nanoscopic view on the fatty acid (FA) binding capacity in albumin hydrogels as seen by continuous wave (CW) electron paramagnetic resonance (EPR) spectroscopy, which on the one hand is of great importance with respect to the capability of these hydrogels for controlled drug release and on the other hand has been developed into a method of characterizing the proteins’ tertiary structure in an experimental, “coarse-grained” manner.5–7
Materials and methods
Bovine serum albumin (lyophilized powder, essentially fatty acid free, >96%, A6003, Sigma, St Louis, MO, USA) and albumin from human serum (lyophilized powder, essentially fatty acid free, A1887, Sigma, St Louis, MO, USA) were used in the experiments. In ref. 22 and 27, the authors introduced “TBSA” and “PBSA” as nomenclature for thermally and electrostatically (pH) triggered BSA hydrogels, respectively. Since we here extend the work to gels made from HSA and we wish to clearly discriminate the gel-state from the non-gelated precursor solutions, we introduce a notation as follows: gXSAi (Θ,p,t), where g denotes the gel-state, X = B, H clarifies the origin (B = bovine, H = human) and the index i = T, p indicates the preparation method (T = thermally induced or p = electrostatically/pH-induced). Since we present a larger variety of different gels and their physical properties as a function of temperature (Θ), pH (p), and incubation time (t), these parameters can be specified in parentheses (Θ = temperature in °C, p = pH, t = incubation time in minutes). This means that, e.g., gHSAp(65,3.5,60) denotes an HSA-based hydrogel that was electrostatically prepared at pH 3.5, 65 °C for 60 minutes.Thermally induced human/bovine serum albumin hydrogels (gHSAT, gBSAT)
Serum Albumin (HSA/BSA) was added into a flask equipped with a magnetic stirrer. Then, according to the desired concentration of HSA/BSA in water, deionized water was added into the flask with final concentrations in the range of 12 to 20 wt% (1.8 to 3.0 mM). The protein–water solution was stirred at a low rate, 100 rpm, until the dissolution of the albumin precursor solution in water was completed. The pH values of the solutions were 7.0 and 7.2 for BSA and HSA, respectively. The final solution was sterilized with a 0.2 μm nylon syringe filter into vials. The vials were kept at different temperatures (37 to 80 °C) in a thermomixer for different durations in order to yield gHSAT and gBSAT, respectively.pH-Induced human/bovine serum albumin hydrogels (gHSAp, gBSAp)
gHSAp and gBSAp were prepared according to the procedure explained above for the gXSAT analogues with some extra steps. After the preparation of the precursor solution, the pH was lowered or increased by addition of different acids or bases (2 M HCl, 0.1, 1, 2 M H2SO4, 2 M H3PO4, 2 M CH2(COOH)2, 2 M NaOH) to the solution. In contrast to the pH range reported by Baler et al. (pH 3.0–4.0)22 we obtained gBSAp and gHSAp at pH values of 4.3 or lower (down to 1.0) and even above pH 10.6. As shown below, at high pH, we obtained hydrogels at temperatures as low as room temperature. Again, the incubation time needed for the hydrogels to form depends on the temperature, the concentration of the protein solution and the used acid/base and varies from several seconds to several days.Loading FAs into the hydrogels
Three different methods of loading FAs into the hydrogels were tested: (1) 16-DSA (the spin-labeled stearic acid, Fig. 1C) was first dissolved in 0.1 M KOH. Then, according to the desired albumin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :FA molar ratio, the 16-DSA solution was added to the precursor solution of the hydrogel. Finally, the gel was synthesized by changing the temperature and/or pH to obtain FA-loaded hydrogels (gXSAT and gXSAp). (2) The solid powder 16-DSA was added directly to the precursor solution of the hydrogel. (3) Hydrogels were prepared as described above without fatty acid. At the desired stage in the formation process of the hydrogel, a solution of 16-DSA dissolved in 0.1 M KOH was simply poured onto the stable hydrogels.
:FA molar ratio, the 16-DSA solution was added to the precursor solution of the hydrogel. Finally, the gel was synthesized by changing the temperature and/or pH to obtain FA-loaded hydrogels (gXSAT and gXSAp). (2) The solid powder 16-DSA was added directly to the precursor solution of the hydrogel. (3) Hydrogels were prepared as described above without fatty acid. At the desired stage in the formation process of the hydrogel, a solution of 16-DSA dissolved in 0.1 M KOH was simply poured onto the stable hydrogels.
Rheological characterization
The rheological characterization was performed to investigate the gelation kinetics as well as the viscoelastic behavior of the hydrogels. We define the gelation start-time as the time when loss and the storage modulus (GG′′ and G′, respectively) start to deviate, i.e. where the elastic behavior of the material is more pronounced than its viscous properties (G′ > G′′). Additionally, an evaluation of loss and storage modulus during this process has been recorded in order to obtain an insight into the stability of the hydrogels, the time they take to show steady state behavior and more generally the mechanical properties of the gels.The experiments were performed using a Physica MCR 301 rheometer (Anton Paar, Graz, Austria) equipped with a CP50-2/TG cone/plate measurement system. To avoid evaporation of water during the measurements the measuring gap was covered with silicon oil and the closing cap of the bracket was down. 0.5% oscillatory strain was applied throughout the experiment. The frequency of oscillations (ω) was set to 1 rad s−1.
Transmission electron microscopy (TEM)
The samples of albumin precursor solutions (1 wt% (0.15 mM)) were prepared according to different gel preparation methods, explicitly, (1) incubated at 65 °C for 2 hours at neutral pH, (2) incubated at 50 °C for 24 hours at neutral pH, (3) incubated at 37 °C for 12 hours and the pH was set to 3.5 using 2 M HCl. The samples were prepared by spreading 5 μL of the dispersion onto a Cu grid coated with a Formvar-film (PLANO, Wetzlar, Germany). After 1 min, excess liquid was blotted off with filter paper and 5 μL of 1% aqueous uranyl acetate solution were placed onto the grid and drained off after 1 min. The dried specimens were examined with an EM 900 transmission electron microscope (Carl Zeiss Microscopy GmbH, Oberkochen, Germany). Micrographs were taken using an SSCCD SM-1k-120 camera (TRS, Moorenweis, Germany).ATR IR spectroscopy
The secondary structure content of a protein sample can be characterized using vibrational (infrared and Raman) spectroscopy.28 The characteristic IR bands of proteins are the amide I and amide II bands.29,30 While the amide I band is mainly due to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of the amide group, the amide II band is primarily caused by bending vibrations of N–H bonds.30–32
O bonds of the amide group, the amide II band is primarily caused by bending vibrations of N–H bonds.30–32
Attenuated Total Reflection (ATR) Fourier Transform Infrared Spectroscopy (FTIR) was used to examine how the secondary structure of serum albumin changes during hydrogel formation. A Bruker Tensor 27 FT-IR spectrometer equipped with a BioATRCell II and an LN-MCT photovoltaic detector and the OPUS Data Collection Program (all from Bruker, Ettlingen, Germany) were used for these experiments. 30 μL of an XSA precursor solution were pipetted onto the Si-crystal surface before heating the sample to the desired temperature by means of a circulating water bath using a HAAKE C25P thermostat. Spectral recording was started once the desired temperature was reached. The lag time due to heating was approx. 2 min. The spectra were obtained with the following parameters: resolution 4 cm−1, scanning was conducted from 4000 to 900 cm−1 (256 scans recorded), scanning velocity: 20 kHz, zero-filling factor: 4, apodization function: a Blackman-Harris 3-term. spectrometer and an ATR cell were purged with dry air. As a reference, an empty ATR cell was used. Each spectrum was corrected for water contributions by subtracting a spectrum of H2O recorded at the same temperature and pH.
Principal component analysis (PCA) of IR data
PCA is a technique which transforms a number of possibly inter-correlated variables into a smaller number of orthogonal variables called principal components. In general terms, the dimensionality of large datasets would be reduced by using a vector space transform. This way the user could spot trends and patterns in large datasets.33 The goal is to de-correlate the original data by finding the directions along which the variance is maximized and then use these directions to define the new basis.34 The first principal component (PC1) has the largest possible variance. The direction of the second largest variance in the data, which is orthogonal to PC1, is the second principal component (PC2). By using orthogonal projection of the data in the new principal component coordinate system onto PC1 or PC2, we achieve scores on PC1 and scores on PC2, respectively. The scores are plotted over time to assess the kinetics of the transition. Mathematically, the principal components are the eigenvalues of the covariance matrix of the data matrix X, where PC1 is the eigenvector with the highest eigenvalue, PC2 the eigenvector with the second highest eigenvalue, etc. Xi,j contains the spectral intensities, where columns (j) represent the probed wavenumbers and rows (i) the different temperatures of measurement. After data transformation, Xi,j = Si,j·Pj,j where Si,j is the score matrix and Pj,j contains rows of PC1 to PCj. There is an extensive body of literature regarding PCA analysis and multivariate data analysis.34–36As our IR spectra contain several intricately linked and correlated changes in peak intensities,37,38 we evaluated them in a principal component analysis. The PCA was applied to the spectral range of 1600 to 1700 cm−1 after subtraction of a linear baseline and vector normalization of the spectra. Principal component analysis was performed using the princomp function of MATLAB (MathWorks, Inc., USA).
Continuous wave (CW) EPR spectroscopy
CW EPR spectroscopy was used to investigate the fatty acid binding capacity of the hydrogels.39CW EPR spectroscopy as a magnetic resonance technique can be used to monitor and characterize the electronic and molecular structures, self-assembly, and dynamics of natively paramagnetic species and selectively introduced paramagnetic probe molecules under different conditions.40,41 To investigate the FA binding capacity of the serum albumin hydrogels, spin-labeled FAs were introduced into the hydrogels. Albumin–FA interactions can then be monitored by observing immobilization of rotational motion upon FA binding to albumin5,6 and according to changes in environmental polarity.42–44 We use spin-labeled stearic acid derivatives, which we have shown to be bound by albumin (both HSA and BSA) in solution and which report on the availability and chemical nature of long-chain fatty acid binding sites.5,16 The stearic acids are spin-labeled at position 16 from the carboxylic acid headgroup, hence called 16-DSA (16-doxyl stearic acid, free radical, 253596, ALDRICH), bearing a nitroxide doxyl group, Fig. 1C.
CW EPR spectra were recorded using a MiniScope MS400 (Magnettech, Berlin, Germany) spectrometer working at X-band (a microwave frequency of approximately ν = 9.4 GHz and a magnetic field sweep of 15 mT centered at 340 mT). Temperatures were adjusted with a Temperature Controller H03 (Magnettech) with an accuracy of ∼0.2 °C. The exact frequency was recorded using a frequency counter (Racal Dana 2101, Neu-Isenburg, Germany). Absorption spectra were detected as first-derivative spectra through modulation5 and the simulations were performed using the EasySpin program package for MATLAB, in which the Schneider–Freed model of slow and intermediate rotational motion is implemented to solve the Schrödinger equation for slowly tumbling nitroxides.45 The final simulated spectrum is the result of a linear (weighted) combination of three different components representing hydrophilic, hydrophobic and aggregated (micellized) spin probes.45,46
Results & discussion
In general, hydrogels from serum albumin (HSA and BSA) are prepared from a precursor solution of HSA or BSA dissolved in deionized water. This precursor solution is converted into a gel during the incubation period, in which the temperature and/or pH of the solution as well as the incubation time are adjusted to obtain the desired hydrogels at certain stages of equilibration.We found that by changing the pH value of the solution to the acidic range of 1.0 to 4.3 or the basic range of >10.6, the pH induced hydrogels (gXSAp) form at low temperatures. Moreover, a variety of different types of thermally induced hydrogels (gXSAT) form at neutral pH of the precursor solution at different incubation times ti and temperatures, even at temperatures much lower than the denaturation temperature of the respective protein. Fig. 1 shows gXSAT hydrogels, a plot of the crystal structures of HSA and BSA and the molecular structure of the EPR-active FA spin probe 16-DSA used in this study.
Viscoelastic behavior of the hydrogels
The rheological properties of protein solutions arise from intermolecular interactions between proteins that facilitate the formation of a cohesive continuous protein network. In viscoelastic protein solutions as they evolve during gelation, these noncovalent interactions include solvation, hydrophobic, van der Waals, electrostatic and dipolar interactions (like hydrogen bonding). The magnitude and number of these interactions as well as the interaction pattern/distribution determine the extent of conformational changes in the protein.49Fig. 2 depicts time-dependent rheological measurements of storage and loss moduli, G′ and G′′, respectively, during the formation of gXSAp/T from 20 wt% precursor solutions at different temperatures. In Fig. 2(A) and (B), the temperature in HSA (A) and BSA (B) solutions at ∼pH 7 was increased to 65 °C (above the denaturation temperature). The storage modulus increases instantaneously above the loss modulus, suggesting that gelation starts immediately.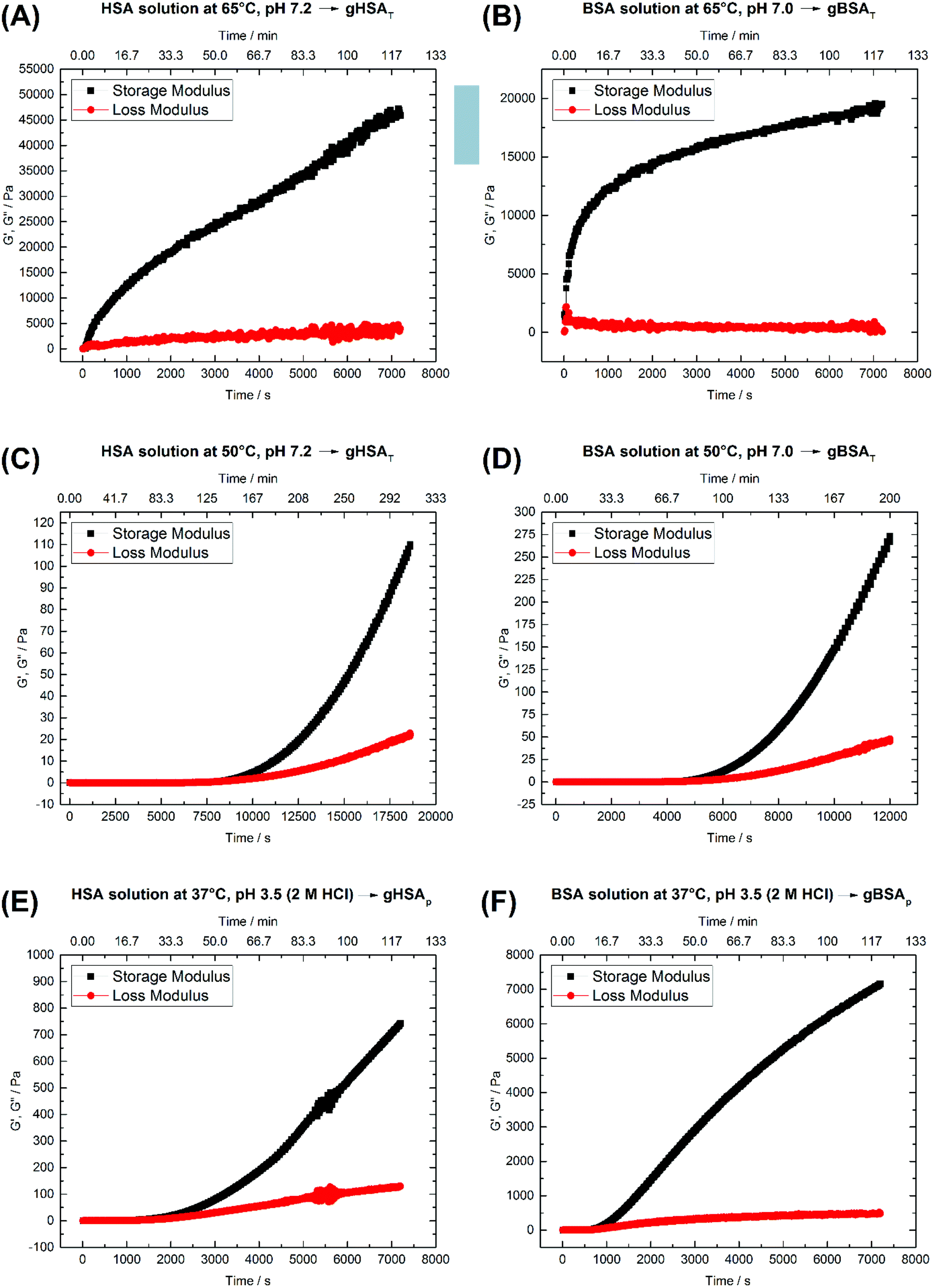 | ||
| Fig. 2 Time-dependent storage (G′) and loss (G′′) moduli of 20 wt% (A) HSA precursor solution at 65 °C, pH 7.2 forming gHSAT(65,7.2,t), (B) BSA precursor solution at 65 °C, pH 7.0 forming gBSAT(65,7.0,t), (C) HSA precursor solution at 50 °C, pH 7.2 forming gHSAT(50,7.2,t), (D) BSA precursor solution at 50 °C, pH 7.2 forming gBSAT(50,7.2,t), (E) HSA precursor solution at 37 °C, pH 3.5 (2 M HCl used as an acid) forming gHSAp(37,3.5,t) and (F) BSA precursor solution at 37 °C, pH 3.5 (2 M HCl used as as acid) forming gBSAp(37,3.5,t). See Table 1 for a comparison of the G′ and G′′ values at different time scales. | ||
After the initially steep increase in the storage modulus curve in the first 15 minutes (900 s), the growth continues in a much slower manner. These results show that the storage modulus G′ increases up to 20000 Pa for BSA and 45000 Pa for HSA after two hours, which are values typical of mechanically robust hydrogels (when the difference between G′ and G′′ is more than 10000 Pa after 1 hour, we assume the hydrogel to be mechanically robust). On the other hand, even longer incubation times do not necessarily lead to better mechanical properties, but in contrast may impair the water-holding capacity.
As one can see in Fig. 2(A) and (B), 45 minutes for the formation of gHSAT(65,7.2,45) and 2 hours for gBSAT(65,7.2,120) are good incubation times to achieve mechanically robust hydrogels at this temperature. In Fig. 2(C) and (D), we present the same experiments performed at 50 °C (12 degrees lower than the denaturation temperature of HSA and BSA). There was no sign of hydrogel formation after two hours, which can be expected under the assumption that gBSAT does not form at temperatures below 62 °C.
However, on increasing the incubation time, the storage modulus increases above the loss modulus curve, indicating (at least weak) hydrogel formation. These low-temperature incubated hydrogels are mechanically much weaker than the ones formed above the denaturation temperature Td of the protein. The storage modulus is two orders of magnitude smaller than that recorded at temperatures above Td, see Fig. 2(A–D), respectively. Moreover, the incubation time needed for gel formation is much longer and the hydrogel properties are strongly time dependent. Nonetheless, hydrogels with noticeable elastic behavior can be obtained at temperatures below Td. Additionally, as the IR results suggest, see Fig. 4, serum albumin retains more of its native secondary structure during the gelation process as the temperature is lower. One may envision potential application of these hydrogels e.g. for fast small molecule (drug) release, but a study of the specific release profiles is outside the scope of this study.
Fig. 2(E) and (F) show the time-dependent results for gHSAp(37,3.5,t) and gBSAp(37,3.5,t) formation. In both cases the pH of the precursor solution of the hydrogel (20 wt%) was adjusted to 3.5 by adding 2 M HCl and time-dependent measurements of G′ and G′′ were performed at 37 °C. When inspecting Fig. 2(E) and (F), it appears that in these cases the deviation of G′ from G′′ commences considerably faster than for gHSAT(50,7.2,t) and gBSAT(50,7.2,t), namely at times below one hour. This is still not as fast as in the case of gHSAT(65,7.2,t) and gBSAT(65,7.2,t), though. Note that the exact time dependence is only valid for specified parameters e.g. the chosen frequency of ω (see Fig. S1†).
For probing the tertiary structure changes in the proteins constituting the hydrogels in an indirect and a “coarse grained manner”, we use our EPR-based platform for studying the capacity to bind long-chain fatty acids.5,6 Hence, we first examined how addition of fatty acids affects the gelation kinetics and the viscoelastic behavior of the hydrogels. It has been reported that SDS (Sodium Dodecyl Sulfate)20 and long-chain fatty acids50 increase the thermal stability of BSA and HSA in solution, respectively.
To analyze the potential effects of stearic acid (SA) on albumin gelation we repeated the gHSAT and gBSAT formation experiments in the presence of SAs at both 50 °C and 65 °C (HSA, BSA) during the formation of gXSAT and also at 37 °C for BSA. The results (see the ESI†) reveal that SAs lead to a delayed onset of the gelation process and weaken the mechanical properties of the gels. Depending on the method of preparation and how the FAs are added to the solutions (see the EPR result part and the ESI†) gel formation is obstructed completely at certain molar ratios of albumin:FAs. It should be noted that 16-DSA and SA are EPR-spectroscopically shown to be interchangeable in their binding to HSA and BSA.5,6
TEM images
With TEM, we can describe the morphological differences of albumin hydrogels prepared with different methods down to the scale of ∼10 nm. Fig. 3 illustrates that in temperature-induced gXSAT formation, protein fibrillation occurs more readily, even at 50 °C (Fig. 3(C)), compared to the pH-induced method, Fig. 3(D). On the other hand, TEM images, alongside other methods, reveal that protein aggregation is highly concentration dependent. To obtain TEM results, we had to dilute the precursor solution to 1 wt%, which is below the minimum concentration required for gel formation. Hence, the results can only give indications about the potential gelation processes taking place at higher precursor solution concentration (20 wt%). At very low concentration, very few protein fibrils are detected. Fig. 3(A) show the 1 wt% HSA solution and (B) 0.3 wt% HSA solution, incubated for 2 hours at 65 °C. While in (A) many smaller fibers in the length range of a few tens of nanometers are observable, in (B) fewer but longer and thicker fibers can be detected. This difference became more prominent when we lowered the temperature or when pH-induced gels and precursor solutions were used. In (C) and (D) it seems that a complex geometry of protein molecules is formed more than individual detectable fibers. However, some fibers (see the arrows in the TEM images) can also be spotted; these fibers are smaller in length and lower in abundance in comparison with (A). Due to the locally high concentration of protein in the samples used in TEM measurements, these results are helpful to obtain a deeper understanding of the results obtained by rheometry where the gXSAT(65,7,t) shows stronger mechanical properties and has a lower minimum concentration threshold for gel formation. At the same time, TEM results show that in gXSAT(65,7,t) fibers are longer and easier to detect. This correlation becomes clearer in the IR part.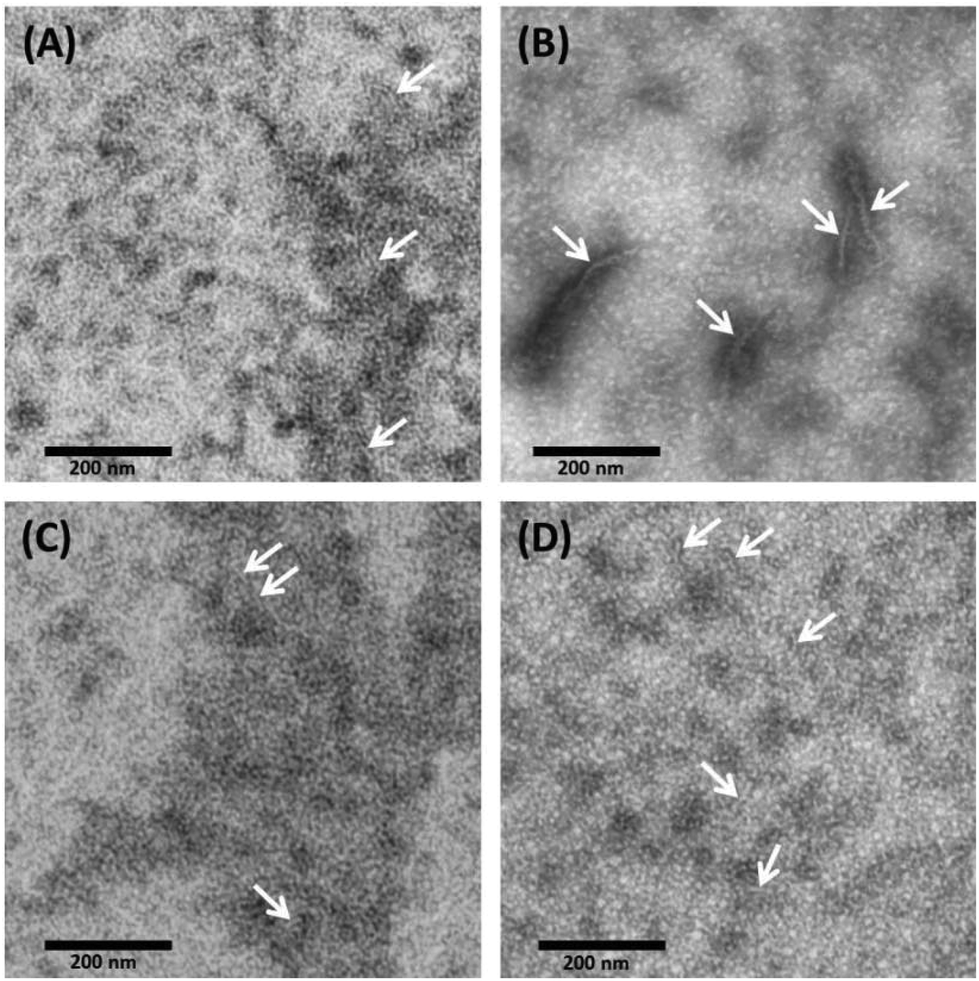 | ||
| Fig. 3 TEM micrograph obtained from (A) 1 wt% HSA solution at neutral pH, incubated for 2 hours at 65 °C (B) 0.3 wt% HSA solution at neutral pH, incubated for 2 hours at 65 °C, (C) 1 wt% HSA solution at neutral pH, incubated for 24 hours at 50 °C and (D) 1 wt% HSA solution at pH 3.5 using 2 M HCl, incubated for 12 hours at 37 °C. | ||
In Fig. 3A, C and D, the average fiber diameter is 5 nm, whereas in B, fiber diameters of 5 and 10 nm were measured. The larger diameter is most probably due to aggregation of individual fibers.
The length of the fibers could not be determined since the flexible fibers are strongly intertwined so that the fiber ends could not be detected.
Secondary structure changes of serum albumin during gelation
According to its crystal structure, BSA contains 67% α-helices next to 10% β-turn and 23% extended chain configuration without any β-sheets. It is well known that changes in temperature, pH, ionic strength and other physical and chemical factors affect the secondary structure content.1,31,51 To study the changes in the secondary structure of the proteins, we performed time-dependent ATR-IR measurements during the gelation process (the hydrogels were formed on the ATR crystal of the spectrometer). As is known, hydrogel formation proceeds through the formation of β-sheets, which can include fibrillar morphologies as seen in TEM from our low concentration solutions.44,53 The bands at 1618 and 1684 cm−1 are attributed to the formation of intermolecular, hydrogen-bonded β-sheet structures that lead to protein aggregation or gel formation.52 As shown in Fig. 4, these bands increase during gelation in all cases, for gXSAp as well as gXSAT formation, at temperatures above or below the denaturation temperature. The β-sheet forms at the cost of native α-helical structure, suggesting that the intramolecular hydrogen bonds involved in α-helix formation become available for intermolecular H-bonding, leading to three-dimensional network formation of the hydrogels. Hence, hydrogel formation and the accompanying secondary structure changes can be quantified through the ratio of α-helical to β-sheet content as reflected in the intensities of both bands (I(α-helix)/I(β-sheet)).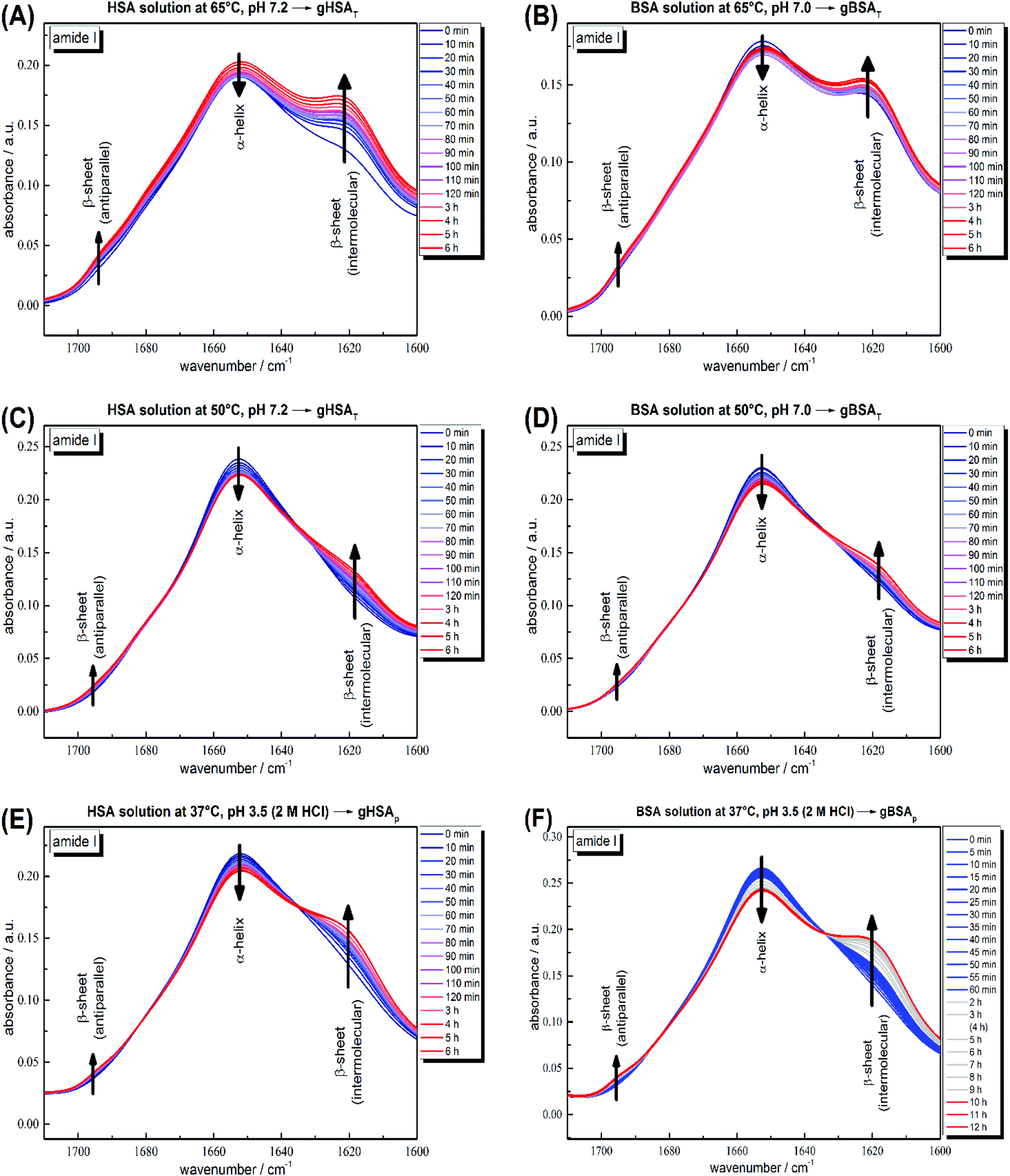 | ||
| Fig. 4 Time-dependent ATR-IR measurements of 20 wt% (A) HSA precursor solution at 65 °C, pH 7.2 forming gHSAT(65,7.2,t), (B) BSA precursor solution at 65 °C, pH 7.0 forming gBSAT(65,7.0,t), (C) HSA precursor solution at 50 °C, pH 7.2 forming gHSAT(50,7.2,t), (D) BSA precursor solution at 50 °C, pH 7.2 forming gBSAT(50,7.2,t), (E) HSA precursor solution at 37 °C, pH 3.5 (2 M HCl used as an acid) forming gHSAp(37,3.5,t) and (F) BSA precursor solution at 37 °C, pH 3.5 (2 M HCl used as an acid) forming gBSAp(37,3.5,t). Band attributions are according to Clark et al.52 | ||
The general trend shows a decrease in α-helix content while a new beta-sheet structure forms. However, the α-helix/β-sheet ratio is different in each case, which can now be used to more quantitatively correlate the structural evolution with the differences in hydrogel properties prepared by the various methods described above. Fig. 4(A) and (B) shows the IR spectra of the gelation processes of gHSAT and gBSAT at 65 °C, respectively. When we compare these spectra with the respective spectra obtained at a gelation temperature of 50 °C (Fig. 4(C) and (D)), we see that the α-helix/β-sheet ratio is much higher for the latter (see the PCA evaluation part, Fig. 5 and the ESI, Fig. S3–S6†). This is an indication that the proteins experience notably less structural change during hydrogel formation when the temperature is below the denaturation temperature. Moreover, there is a dead time of ∼2 minutes before T-equilibration is reached in the ATR-IR spectrometer. This is a critical time for gel formation in the case of gHSAT and gBSAT at 65 °C (see Fig. 2); hence, the first and potentially strongest decrease in the amount of α-helix content is hard to detect at this temperature. By comparing these results with those for the gelation of gHSAp and gBSAp, shown in Fig. 4(E) and (F) alongside the rheological results, we can state the relations between the β-sheet/α-helix ratio, gelation kinetics and mechanical properties, as listed in Table 2. For detailed information see the Discussion and the ESI.†
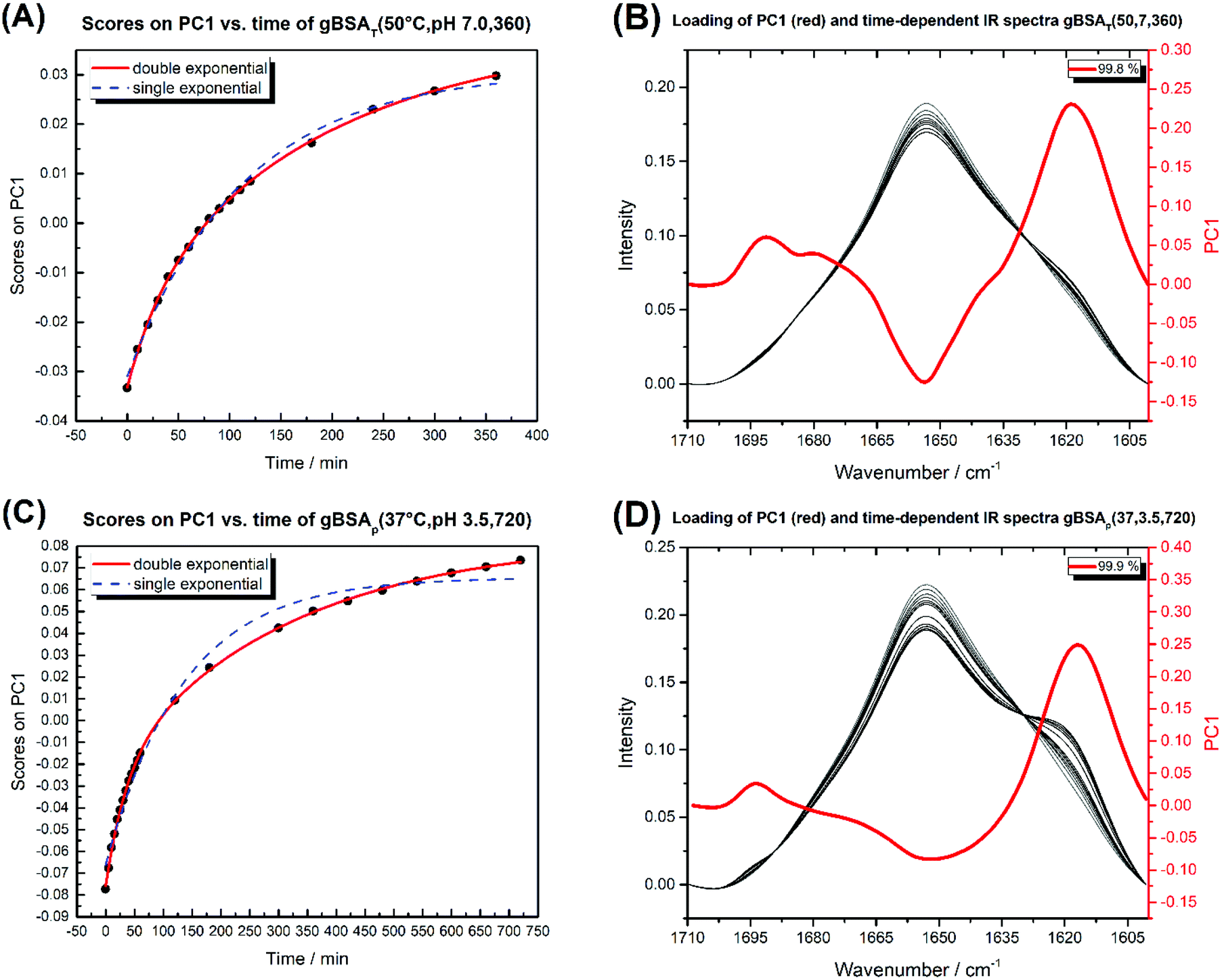 | ||
| Fig. 5 (A) Diagram of PC1 scores against time for gBSAT(50,7,t). (B) The time-dependent IR spectra of gBSAT(50,7,t) (white to black) and the loading of PC1 in red. (C) Diagram of PC1 scores against time for gBSAp(37,3.5,t). (D) The time-dependent IR spectra of gBSAp(37,3.5,t) (white to black) and the loading of PC1 in red. The double and single exponential functions were fitted to PC1 scores vs. time. The obtained time constants are shown in Table 3. | ||
| Type | Temperature [°C] | pH | Fig. 2 | 15 min | 60 min | 120 min | |||
|---|---|---|---|---|---|---|---|---|---|
| G′ [Pa] | G′′ [Pa] | G′ [Pa] | G′′ [Pa] | G′ [Pa] | G′′ [Pa] | ||||
| gHSAT | 65 | 7 | A | 11500 |
1220 | 27300 |
2580 | 45900 |
3980 |
| gBSAT | 65 | 7 | B | 12000 |
715 | 16300 |
309 | 19500 |
568 |
| gHSAT | 50 | 7 | C | 0.00368 | 0.0492 | 0.00797 | 0.00328 | 0.0898 | 0.175 |
| gBSAT | 50 | 7 | D | 0.024 | 0.00865 | 0.0791 | 0.0619 | 34.7 | 7.62 |
| gHSAp | 37 | 3.5 | E | 1.74 | 1.03 | 141 | 46 | 742 | 130 |
| gBSAp | 37 | 3.5 | F | 123 | 46.8 | 3700 | 374 | 7160 | 479 |
| β-Sheet/α-helix ratio |
| Gelation kinetics (from left to right: the incubation time needed shortens) |
| Mechanical properties (from left to right: the storage modulus increases) |
| gXSAT(50,7,t) < gXSAp(37,3.5,t) < gXSAT(65,7,t) |
PCA evaluation
Fig. 5 shows the PC1 scores and the respective IR spectra for (a) gBSAT(50,7) and (b) gBSAp(37,3.5). In all cases (see also Fig. S3–S6†), more than 98% of changes can be comprised in one component (PC1), i.e. are strongly correlated. This principal component (red traces in Fig. 5C and D) describes a decrease in α-helical and random structure content and a concomitant increase in intermolecular β-sheet und turn content. However, PC1 is not identical for the different gels formed under different conditions. This indicates that the difference in ionic strength changes the gelation mechanism to some extent. In gBSAT(50,7) and in general the temperature-induced samples, hydrogel networks form by the formation of intermolecular β-sheets at the cost of a reduction of α-helices (see Fig. S3–S5†). In the pH induced hydrogel formation gXSAp(37,3.5), see also Fig. S6,† PC1 is broader in the wavenumber region of 1640–1660 cm−1 than temperature induced ones. We speculate that this alteration can be explained since, in gXSAp(37,3.5), intermolecular β-sheets not only form by reduction of α-helix, but also by reduction of random coils, and this is why more α-helix content is preserved in the pH induced gelation mechanism.To compare the kinetics of gelation at the molecular level with macroscopic results (see the Rheological characterization part), the PC1 scores vs. time are best fitted through a double exponential equation (1), revealing underlying fast and slow processes. The resulting time constants are summarized in Table 3. We can deduce that in all gelation mechanisms the majority of the changes at the molecular level occur during the fast process. This is decisively different from what one notices macroscopically by rheological measurements, especially in the case of gXSAT(50,7) and gXSAp(37,3.5), where there is a lag time before the transformation from a protein solution into a protein gel is observable. ATR-IR measurements show that in this lag time already secondary structure transitions occur. Precursor structures for the full gel state, such as XSA oligomers, might be formed in this faster process. After the onset of gelation, the secondary structure changes persist, but are slowed down (second time constant), probably due to hindered diffusion within the gel.
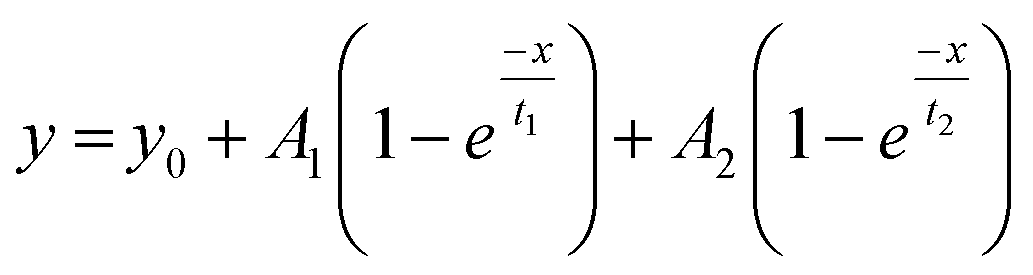 | (1) |
| HSA | BSA | |||||
|---|---|---|---|---|---|---|
| gHSAT(65,7) | gHSAT(50,7) | gHSAp(37,3.5) | gBSAT(65,7) | gBSAT(50,7) | gBSAp(37,3.5) | |
| t 1 | 5.9 | 45 | 18 | 6 | 32 | 35 |
| t 2 | 126 | 251 | 163 | 182 | 192 | 312 |
The time constants in Table 3 show the same trend as we observed in rheological characterization (see Table 2), i.e. both processes are faster at higher temperatures. However, these time constants are not characteristic numbers of the system, since they depend on the incubation time changes (in gBSAp(37,3.5,t)).
To unravel similarities and differences between pH and temperature induced gelation processes we performed a simultaneous PCA on all three BSA as well as on all three HSA datasets. In Fig. 6 we present the results of the gBSA analysis (for the gHSA analysis see Fig. S8†). Scores on PC2 are plotted against scores on PC1. Remarkably, we observe that in gXSAT – regardless of the incubation temperature – PC2 is negligible. The gelation apparently follows the same mechanism at both the investigated temperatures; however, the extent of the structural change described by PC1 is much higher at higher temperatures, as can be concluded from the higher scores on PC1. Interestingly, also gXSAp reaches comparably high scores on PC1, even though the gelation is performed at low temperatures. In addition, PC2 plays a significant role (22% of total variance) in gXSAp, i.e. it describes the differences in the gelation processes at neutral and acidic pH values. The main difference depicted by PC2 is a different content in β-turn structures, which is lower the higher the scores on PC2. This would imply a higher content in parallel β-sheets and unordered structures in the gels formed at low pH value.
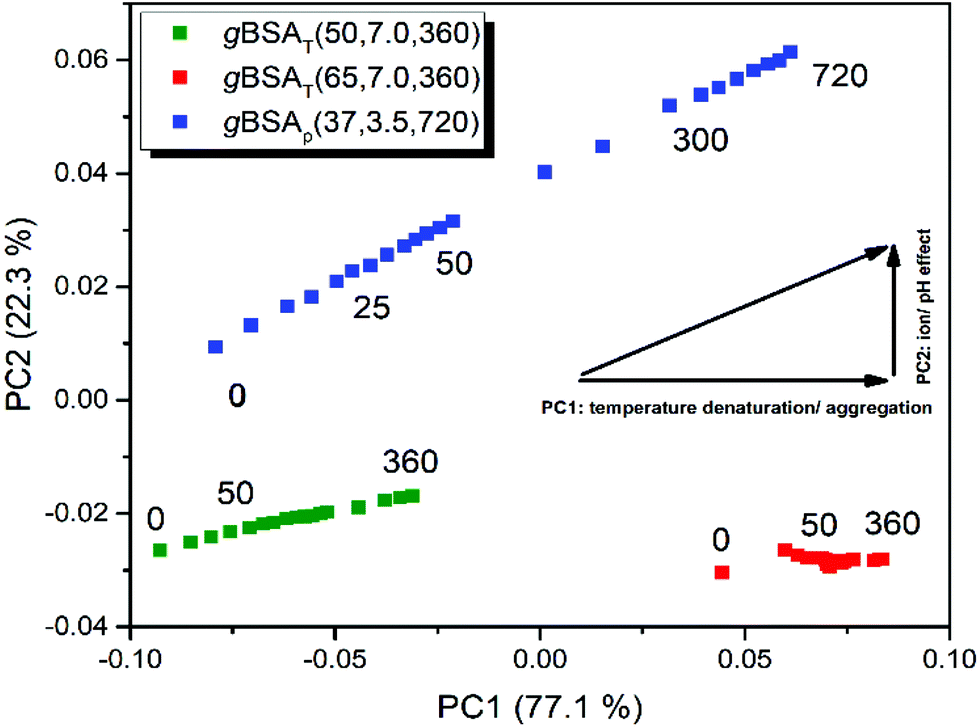 | ||
| Fig. 6 Diagram of PC1 against PC2 scores during the gelation process of BSA. Blue squares depict the process of gBSAp(37,3.5,720). The green and red squares depict the gBSAT(50,7,360) and gBSAT(65,7,360), respectively. All preparations were done on the ATR crystal. The PC2 component becomes relevant only for gBSAp, where the pH was lowered to 3.5 by using 2 M HCl. | ||
The pH value not only has an independent role in the gelation process, but also it is an auxiliary factor for the role which temperature plays in the process; although the gXSAp is being prepared at 37 °C the step changes of PC1 are far larger in gXSAT prepared at 50 °C.
Fatty acid binding capacity of the hydrogels
The FA binding capacity of HSA and BSA in solution, including the dynamics of the bound Fas and also their distribution are known.5,54,55From CW EPR measurements, the long chain FA binding can be monitored quantitatively and even subtleties of the binding process and the bound states can be analyzed (see, e.g., Akdogan et al.16 and Reichenwallner et al.5). BSA and HSA in their native isoform have also been found to feature 7 ± 1 long-chain fatty acid binding sites. In our experiments on the hydrogels and the gelation process, introducing amphiphilic spin-labeled FAs that are amenable to EPR spectroscopy is not as straightforward as for low-concentration solutions. We have tested the dynamics of the FAs for all three methods introduced in the Materials and Methods part for loading FAs into the hydrogel. Our results show that there are strong FA binding sites available inside the hydrogels, with certain differences in the final number of bound FAs. In the following, we describe EPR-spectroscopic results gained from the different methods of FA addition. Please note that the driving force for FA-binding to protein binding sites is the fact that the amphiphilic 16-DSA molecules partition between the more nonpolar/hydrophobic binding pockets and the surrounding aqueous solution with a strong preference for the protein-based pockets when available.
In Fig. 7(A) the spectra of our spin-probe FA, 16-DSA, dissolved in PBS buffer solution at 5 °C and 37 °C are shown; as expected the spectra reflect the freely tumbling FAs without protein. Fig. 7(B) and (C) show the spectral signatures of the individual 16-DSA fractions that contribute to the overall EPR spectrum. Spectra of the spin probes in polar/hydrophilic environments, in hydrophobic environments, and in micelles are shown in Fig. 7(B), while the spectra that show freely tumbling and strongly immobilized spin probes in FA binding sites of albumin are shown in Fig. 7(C). For a quantitative comparison, the simple rotational correlation time τc (the rotational correlation time is the average time it takes for a spin probe to rotate by one radian; in other words the time scale of individual stochastic fluctuations, which is calculated from the simulation parameter diffusion tensor D (rotational correlation time 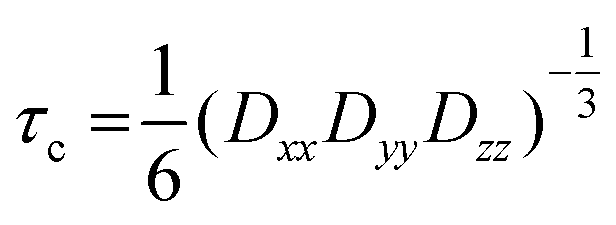 )), can be used to characterize the rotational reorientation, as shown in Table 4. Fig. 7(D–F) display the spectra of 16-DSA that was introduced into the hydrogels through method (I). The results of (E) and (F) are comparable with the rheological characterization and IR spectra, as gHSAT(65,7) shows higher elasticity and a mechanically more robust gel; the FAs are also immobilized more strongly than in gBSAT(65,7); on the other hand, although there is no sign of freely tumbling FAs in gHSAT(65,7); less than 1% of FAs in gBSAT(65,7) are apparently unbound. This can be due to two reasons: smaller changes in the secondary structure of HSA during gelation (see the IR results) and/or the possibility that the hydrophobic regions of HSA are more easily reachable for FAs, as HSA is significantly more hydrophobic than BSA, indicated in the cumulative hydropathy index.16Fig. 7(G) and (H) both show gBSAT(65,7) at 7:1 16-DSA:BSA ratio where the gels are prepared by method (II), and present the importance of mixing time. In Fig. 7(G), after introducing 16-DSA into the gel, the vials were put into the thermomixer for gel preparation; Fig. 7(H) shows the spectrum of 16-DSA in the hydrogel, in which the 16-DSA powder was simply added to the precursor solution, then shaken for 3 days and subsequently incubated in the thermomixer. Remarkably, it shows that almost 60% of 16-DSA shows intermediate rotational motion and only 40% are strongly bound to protein (immobilized). When the solution and FAs are mixed long enough all the FAs are bound to the BSA/HSA. In method (2), which gave the largest amount of immobilized 16-DSA molecules, as quantified from rigorous spectral simulations, the most striking finding spectroscopically is that regardless of the 16-DSA:albumin ratio, the spin-labeled FAs are all either strongly immobilized or at least intermediately immobilized (after a long enough mixing time), there is no sign of freely tumbling or micellized 16-DSA molecules. Both, the strongly and intermediately immobilized FAs, are spectral components that can be observed when studying FA-binding to BSA (or HSA) in low-concentration protein solutions. The strong rotational immobilization in BSA usually takes place in FA binding sites that can be found at the interfaces of α-helices.5,16,56 The intermediate-bound state(s), on the other hand, indicate more rotational freedom, which could stem from more transient attachment to BSA in water-swollen regions of the protein gels and that we could, e.g., also identify in β-sheet structure forming elastin-like polypeptides.44,57 Finally, Fig. 7(J) shows the spectrum of gBSAT(65,7) in which 16-DSA are added into the gels through method (III). Apparently, the 16-DSA molecules diffuse into the hydrogel and almost 67% of them are bound to protein-based binding sites and 32% form micelles (almost 1% are not bound and rotate freely). In addition, the value of the isotropic 14N-hyperfine coupling of a nitroxide spectrum aiso is indicative of the local polarity around the NO-group. High polarities lead to larger hyperfine coupling, while less polar environments shift aiso to lower values.43,57,58 All these quantitative results gained from rigorous simulation of the spectra are shown in Table 4. The preliminary results of EPR tests on the hydrogels using other spin-probes, namely TEMPO (214000 ALDRICH) and 4-hydroxy TEMPO benzoate (371343 ALDRICH), and their potential effect on the mechanical properties of the hydrogels are available in the ESI.†
)), can be used to characterize the rotational reorientation, as shown in Table 4. Fig. 7(D–F) display the spectra of 16-DSA that was introduced into the hydrogels through method (I). The results of (E) and (F) are comparable with the rheological characterization and IR spectra, as gHSAT(65,7) shows higher elasticity and a mechanically more robust gel; the FAs are also immobilized more strongly than in gBSAT(65,7); on the other hand, although there is no sign of freely tumbling FAs in gHSAT(65,7); less than 1% of FAs in gBSAT(65,7) are apparently unbound. This can be due to two reasons: smaller changes in the secondary structure of HSA during gelation (see the IR results) and/or the possibility that the hydrophobic regions of HSA are more easily reachable for FAs, as HSA is significantly more hydrophobic than BSA, indicated in the cumulative hydropathy index.16Fig. 7(G) and (H) both show gBSAT(65,7) at 7:1 16-DSA:BSA ratio where the gels are prepared by method (II), and present the importance of mixing time. In Fig. 7(G), after introducing 16-DSA into the gel, the vials were put into the thermomixer for gel preparation; Fig. 7(H) shows the spectrum of 16-DSA in the hydrogel, in which the 16-DSA powder was simply added to the precursor solution, then shaken for 3 days and subsequently incubated in the thermomixer. Remarkably, it shows that almost 60% of 16-DSA shows intermediate rotational motion and only 40% are strongly bound to protein (immobilized). When the solution and FAs are mixed long enough all the FAs are bound to the BSA/HSA. In method (2), which gave the largest amount of immobilized 16-DSA molecules, as quantified from rigorous spectral simulations, the most striking finding spectroscopically is that regardless of the 16-DSA:albumin ratio, the spin-labeled FAs are all either strongly immobilized or at least intermediately immobilized (after a long enough mixing time), there is no sign of freely tumbling or micellized 16-DSA molecules. Both, the strongly and intermediately immobilized FAs, are spectral components that can be observed when studying FA-binding to BSA (or HSA) in low-concentration protein solutions. The strong rotational immobilization in BSA usually takes place in FA binding sites that can be found at the interfaces of α-helices.5,16,56 The intermediate-bound state(s), on the other hand, indicate more rotational freedom, which could stem from more transient attachment to BSA in water-swollen regions of the protein gels and that we could, e.g., also identify in β-sheet structure forming elastin-like polypeptides.44,57 Finally, Fig. 7(J) shows the spectrum of gBSAT(65,7) in which 16-DSA are added into the gels through method (III). Apparently, the 16-DSA molecules diffuse into the hydrogel and almost 67% of them are bound to protein-based binding sites and 32% form micelles (almost 1% are not bound and rotate freely). In addition, the value of the isotropic 14N-hyperfine coupling of a nitroxide spectrum aiso is indicative of the local polarity around the NO-group. High polarities lead to larger hyperfine coupling, while less polar environments shift aiso to lower values.43,57,58 All these quantitative results gained from rigorous simulation of the spectra are shown in Table 4. The preliminary results of EPR tests on the hydrogels using other spin-probes, namely TEMPO (214000 ALDRICH) and 4-hydroxy TEMPO benzoate (371343 ALDRICH), and their potential effect on the mechanical properties of the hydrogels are available in the ESI.†
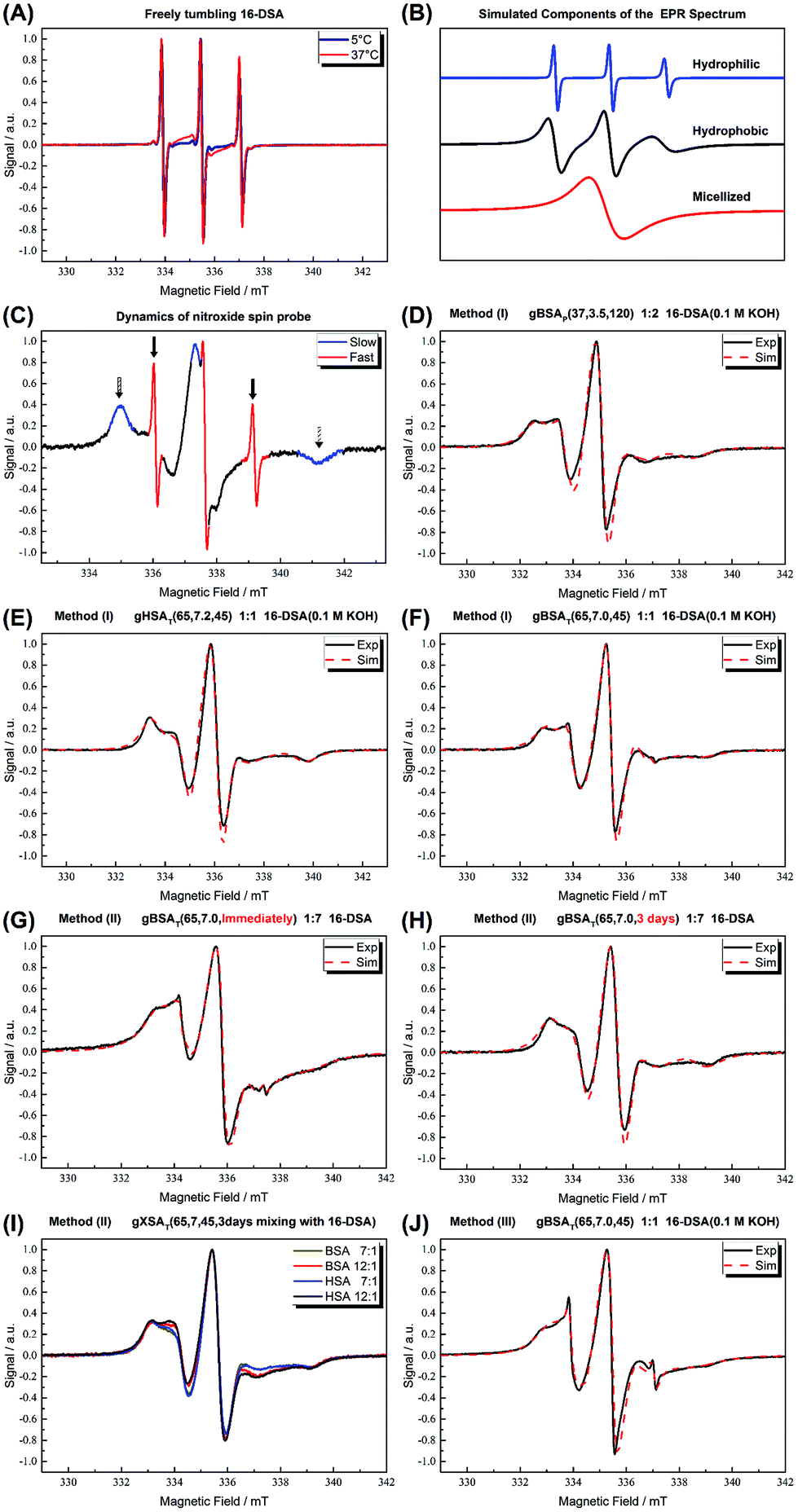 | ||
| Fig. 7 (A) CW EPR spectra of 16-DSA in buffer solution at 5 °C and 37 °C are shown. As shown in ref. 5, the spectra are indicative of freely tumbling and micellized 16-DSA. (B) Simulated components of which the EPR spectra are composed. (C) Signatures of different regions of EPR spectrum for 16-DSA. (D) gBSAp(37,3.5,t) hydrogels at a 2:1 16-DSA:BSA molar ratio (method (I)). (E) gHSAT(65,7.2,t) hydrogels at a 2:1 16-DSA:HSA molar ratio prepared using method (1). (F) gBSAT(65,7.2,t) hydrogels at a 2:1 16-DSA:BSA molar ratio prepared using method (1). (G) gBSAT(65,7.0,t) prepared once after addition of 16-DSA due to method (2), FA:albumin molar ratios of 7:1 (F) gBSAT(65,7.0,t) prepared after letting the added 16-DSA be mixed with the precursor solution for 3 days due to method (2), FA:albumin molar ratios of 7:1. (I) gBSAT(65,7.0,t) and gHSAT(65,7.0,t) hydrogels at high FA:albumin molar ratios of 7:1 and 12:1 (method (2)). (J) gBSAT(65,7.0,t) prepared by method (3) where the 16-DSA solution is poured on the hydrogels prepared without 16-DSA, FA:albumin molar ratios of 1:1. The spectra were measured at 37 °C. | ||
| Figure | Fraction | Correlation time τc (ns) | Hyperfine splitting constant a (MHz) |
|---|---|---|---|
| D | 93.6% | 6.1 | 46.1 |
| 6.4% | 1.7 | 46.9 | |
| E | 100% | 8.9 | 45 |
| F | 99.3% | 8.6 | 45.8 |
| 0.7% | 0.24 | 46.1 | |
| G | 59.8% | 4.5 | 44 |
| 40% | 14 | 45 | |
| 0.2% | 0.17 | 45.8 | |
| H | 100% | 8.7 | 45.5 |
| J | 66.8% | 11.9 | 45 |
| 32% | 4.9 | 42.8 | |
| 1.2% | 0.17 | 46 |
The EPR spectra thus also contain information comparable to those from ATR-IR experiments and rheological characterization, with the additional advantage of delivering these insights from a tracer molecule point of view and reflecting their interaction with the gel scaffolds. This might be of interest and direct use for applications in drug release.
A more detailed description of all these results, the specific conditions for each item, the role of time and mixing, the comparison between the spectra in solution and hydrogel state, and the diffusion of FAs into the hydrogels as observed in the FA addition method (3) are available in the ESI.†
Protein gelation as a process can be described as a phenomenon of protein aggregation in which the attractive and repulsive forces and the interactions between the polymer (protein) and the solvent, itself, and co-solutes are balanced so that a three-dimensional network or matrix forms. This matrix is able to trap or immobilize a large amount of solvent (water).59
Since intermolecular contacts are pivotal for protein gelation, it is concentration dependent. There is a critical concentration for most globular proteins below which a gel network cannot form. This critical concentration is known as the least concentration endpoint (LCE).49 It also includes differences in molecular properties such as net charge, molecular size, conformation and the number density of functional groups available for cross-linking and hence is protein-specific. Moreover, several extrinsic factors such as pH, ionic strength, incubation time, and temperature affect this value.
We have shown that thermally induced albumin hydrogels form at temperatures considerably below the denaturation temperature of serum albumin even at neutral pH values. In Fig. 8, we now summarize the combined macroscopic and nanoscopic insights gained into the gHSAT and gBSAT systems in approximated phase diagrams at different precursor solution concentrations and gHSAp and gBSAp phase diagrams at a fixed concentration, i.e. 20 wt% (3 mM) and different pH values and incubation times. These diagrams reflect the properties at a specific incubation time, which as described above is an important parameter in the parameter space of albumin gelation, of ti = 48 hours. If the incubation time of these temperature-triggered gelation processes is varied, it can affect the shape of the phase diagrams drastically. Different types of gels shown in Fig. 8 are labeled according to their gelation kinetics and viscoelastic properties. In our categorization, gel type I represents gels that need several hours for formation and that have a G′ < 1000 [Pa] even after 5 hours. Gel type II denotes the type of gel formed fast (in less than one hour) but with weak mechanical properties.
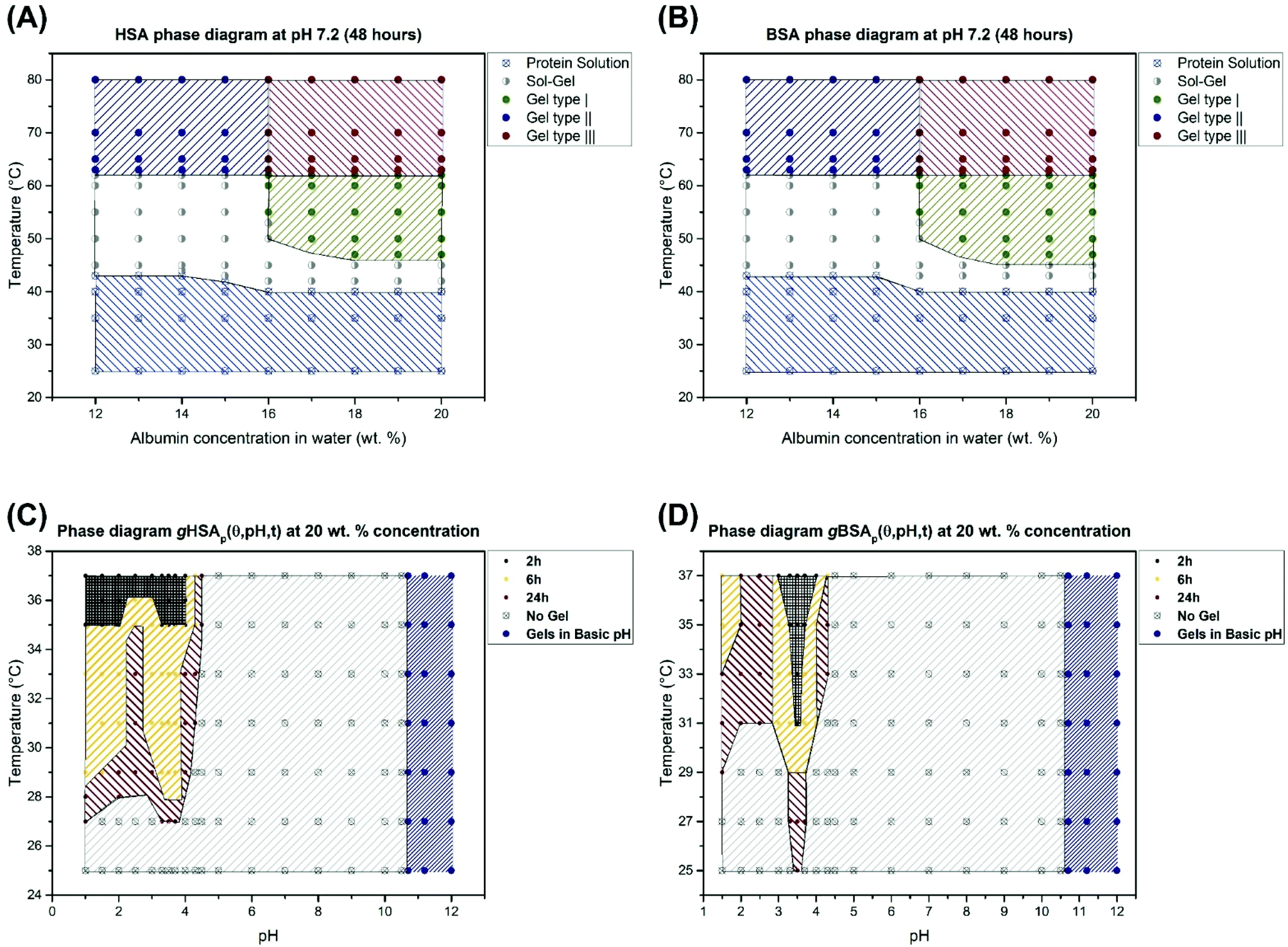 | ||
| Fig. 8 (A) Phase diagram for human serum albumin-based gels after 48 hours based on different concentrations and temperatures. (B) Phase diagram for bovine serum albumin-based gels after 48 hours based on different concentrations and temperatures. These phase diagrams are valid only for the time scale of 48 hours. Changing this time may affect the shape of these phase diagrams. (C) Phase diagram for 20 wt% HSA solution at different pH values and incubation times. 2 h means that for gel formation after a maximum of 2 hours the gels form (according to the temperature and pH value), and the same applies for 6 hours and 24 hours showing the maximum time needed for obtaining hydrogels. Gels at high pH values (pH > 10.6) form in less than 2 hours at RT. (D) The same phase diagram for 20 wt% BSA solution. | ||
Gel type III has very strong mechanical properties and gelation happens very fast and their G′ surpass 10000 Pa after 1 hour. The rheological characterization (Fig. 2) suggests that gelation kinetics are dependent on both, temperature and pH value. We observe that in gXSAp(37,3.5,120) the gelation starts earlier than in gXSAT(50,7.0,120), despite the lower temperature and that it has a much higher storage modulus after the respective time periods. The PCA analysis of the FTIR spectra shows that at the molecular level we can observe the same trend, namely, the rate of change in the secondary structure of the protein follows the same trend as what we see at the macroscopic level (see Fig. 5, Fig. S3–S7 in the ESI†); however, in all cases the changes at the molecular level happen with a steep slope in the beginning of the gelation process and, depending on the preparation method, these changes become observable at the macroscopic level. One can even describe this as a two-stage process, with early fast formation of gel-point contacts (e.g. beta-sheet, or hydrophobic contacts, always from a highly concentrated solution) that have yet little effect on the mechanical, macroscopic properties. The contact formation slows down considerably in the second stage, when the solution has already turned considerably viscous, which slows down the new contact formation in the highly water-swollen sample. Further investigation provided a detailed view on how the pH value affects the gelation process of serum albumin. Interestingly, in the low-pH region of the gXSAp diagrams in Fig. 8(C) and (D) we find a very delicate connection between the incubation time and the minimum temperatures needed to obtain gels. Moreover, pH 4.3 (acidic hydrogels forms below pH 4.3) is the pH value below which an abrupt transition between the so-called N form and F form of albumin occurs.60,61 This can thus be seen as the first step of protein denaturation that is necessary to form the electrostatically derived gels. The gels created at high pH-values (pH > 10.6) are not further investigated for their specific gelation kinetics and will be reported in a subsequent publication.
Considering the process of protein aggregation which leads to gel formation we can define a model process that involves an XSA transition state that is subsequently only then converted into intermolecular β-sheets when a concentration threshold is surpassed. Simplified, the gelation process of serum albumin can be seen as:
| α-helix → [‡] → intermolecular β-sheet |
In this two stage scheme according to PCA analysis and IR results, the changes at the molecular level happen faster in the beginning of the gelation process (these changes are not observable macroscopically) until an intermediate state forms. Then on increasing the incubation time this intermediate state – by some local network formation – transforms into the gel state and from this stage on, we can observe hydrogel formation at the macroscopic level (see the Rheological characterization part).
To further substantiate this postulated process, we used a 5 wt% (0.75 mM) HSA/BSA precursor solution (below the LCE) to repeat all the IR-based experiments described above. Despite an, also in this case, observable reduction in α-helical content, there is no sign of intermolecular β-sheet formation.
The IR results back the speculation that the incubation time can affect the properties of the hydrogels in a non-trivial fashion. In Fig. 4(A) we observe that after two hours the trend of β-sheet formation at the cost of α-helix reduction is reversed; namely, the β-sheet content is reduced and a new structure (whether α-helix or random coil cannot be distinguished at ease) forms. This is even observable at the macroscopic level, in which the hydrogel structure is damaged in such a way that its water-holding capacity is diminished, see Fig. 2(A), and after almost 80 minutes there is a jump in the storage modulus, which is attributable to the loss of water and an increase in the protein concentration.
The TEM micrographs of the (produced from diluted precursor solutions) gel-state samples also support the idea that at the molecular level, β-sheet formation leads to fibrillar morphologies, as is well known from amyloid-forming proteins. pH-Induced gels show much smaller and less fibrillar structures that are rather reminiscent of larger networks.
From a thermodynamics point of view, there is an entropy gain during gel formation. The unfolding and rearrangement of the proteins from intra- to intermolecular interfaces is an entropy-driven process, in which the transfer of the nonpolar residues from the aqueous phase to the non-aqueous phase and the consequent release of water molecules from the hydration shells of these groups increase the overall entropy.49 This process is affected by the intramolecular constraints and extensive segment–segment interactions in the folded state and particularly by covalent linkages such as the eight pertinent disulfide bonds in both BSA and HSA.62 In low-concentration solutions, one would summarize the conformational changes that happen even at human body- or room temperature under protein degradation – it is apparent that above a certain concentration threshold of the serum albumin solution, the equilibrium during the degradation is shifted to protein aggregation and/or gel formation, which can therefore take place at low temperatures, e.g. 37 °C when the gelation incubation time increases, to e.g. 72 hours. Other factors like the pH value are auxiliary to this process, and may affect the gelation kinetics, LCE, etc.
The investigation of the FA binding capacity of the hydrogels shows that the FAs are bound inside the hydrogels. Depending on the method one uses to introduce the FAs into the hydrogels (see Fig. 7) the amount of FAs that can be bound by the hydrogels and also the strength of the binding sites differ. In method (2) in which the solid powder 16-DSA is added directly to the precursor solution, the largest amount of FAs is trapped inside the hydrogels. When one dissolves the FAs into the KOH and then adds this solution to the precursor solution of the hydrogel, at high albumin:FA ratios, first of all the overall concentration of the protein decreases, leading to mechanically weaker hydrogels until no hydrogels form at all. As described above, besides the 16-DSA molecules that are strongly immobilized, the intermediately immobilized FAs with more residual rotational mobility in the hydrogels are indicative of water-swollen nanoscopic regions, as evidenced by the higher aiso values. The mesh size of the hydrogels and the nanoscopic water pools also play an important role. In method (2) we observe that the FAs do not form micellar aggregates when the mixing time is sufficient (see Fig. 7(G, H, I)), which indicates that the FAs are homogenously spread all over the gel and there is just a few of them in each hydrophilic part. That is the reason why method (2) gives the possibility of loading even more than 7 FAs per protein in the hydrogels, information that again in the light of potential uses in controlled drug release is of high interest.
Conclusions
We have presented different synthetic methods for the preparation of HSA (for the first time) and BSA hydrogels and characterized and correlated their macroscopic and nanoscopic features. Our results considerably expand the range of thermally induced albumin hydrogels. We have shown that hydrogels can be formed at incubation temperatures significantly below the denaturation temperature of the protein. The critical role of the gelation incubation time in protein aggregation and gelation phenomena is discussed explicitly and the phase spaces of thermally induced and electrostatically triggered gels are explored in phase diagrams for HSA and BSA, based on temperature, concentration, pH and time.Additionally, we were able to provide more details building on the pioneering work of Baler et al.22 on electrostatically triggered BSA hydrogels and their gelation mechanism. Changing the charge distribution on the BSA by changing the pH causes partial denaturation of the protein. Newly exposed hydrophobic areas and the presence of counter-ions facilitate the hydrogel formation of BSA at low temperature. We showed that the pH range is significantly broader than what is suggested and extended this research to HSA. The preliminary results of our experiments also suggest that the counter-ion not only affects the minimum protein concentration required for gelation, but also affects the stability of the hydrogels, gelation kinetics, mechanical properties and, notably, the fatty acid (FA) binding capacity of the gels. On the other hand, regardless of the acids or bases used to change the pH of the protein solution, the pH range in which electrostatically induced hydrogels can, in principle, be formed is bound to below pH 4.3 and above pH 10.6.
Rheological characterization, a study of secondary structure changes of proteins during gelation via ATR-IR spectroscopy and an investigation of the FA binding capacity of the hydrogels by CW EPR spectroscopy together indicate a two-stage gelation process of hydrogel formation from this family of abundant biomacromolecules. We hope that this initial study, which connects the nanoscopic properties with the macroscopic features of the hydrogels leading to phase diagrams, will pave the way also for their exploration in applications such as controlled release of (bio-)molecules.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Annekatrin Rother for recording the electron micrographs and Franziska Zeuner and Heike Schimm for technical support. This work was supported by the European Regional Development Fund (ERDF) and the Federal State of Saxony-Anhalt.References
- T. Peters Jr., in All About Albumin, Academic Press, San Diego, 1995, pp. 9–II, DOI:10.1016/B978-012552110-9/50004-0.
- Y. S. Day and D. G. Myszka, J. Pharm. Sci., 2003, 92, 333–343 CrossRef CAS PubMed.
- M. L. Ferrer, R. Duchowicz, B. Carrasco, J. G. de la Torre and A. U. Acuna, Biophys. J., 2001, 80, 2422–2430 CrossRef CAS PubMed.
- E. Gianazza, M. Galliano and I. Miller, Electrophoresis, 1997, 18, 695–700 CrossRef CAS PubMed.
- J. Reichenwallner and D. Hinderberger, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5382–5393 CrossRef CAS PubMed.
- M. J. N. Junk, H. W. Spiess and D. Hinderberger, Angew. Chem., Int. Ed., 2010, 49, 8755–8759 CrossRef CAS PubMed.
- M. J. N. Junk, H. W. Spiess and D. Hinderberger, J. Magn. Reson., 2011, 210, 210–217 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- I. M. El-Sherbiny and M. H. Yacoub, Global Cardiol. Sci. Pract., 2013, 2013, 316–342 Search PubMed.
- E. A. Foegeding, Am. Oil Chem. Soc., 1989, 58, 185–194 Search PubMed.
- A. H. Clark, G. M. Kavanagh and S. B. Ross-Murphy, Food Hydrocolloids, 2001, 15, 383–400 CrossRef CAS.
- D. Seliktar and L. Oss-Ronen, US20110238000A1, 2011 Search PubMed.
- Y. Sun and Y. Huang, J. Mater. Chem. B, 2016, 4, 2768–2775 RSC.
- F. Iemma, U. G. Spizzirri, F. Puoci, R. Muzzalupo, S. Trombino and N. Picci, Drug Delivery, 2005, 12, 229–234 CrossRef CAS PubMed.
- Y. Akdogan, J. Reichenwallner and D. Hinderberger, PLoS One, 2012, 7, e45681 CAS.
- J. D. Ferry, in Advances in protein chemistry, ed. M. L. Anson and T. E. John, Academic Press, 1948, vol. 4, pp. 1–78 Search PubMed.
- H. Jian, Y. L. Xiong, F. Guo, X. Huang, B. Adhikari and J. Chen, Int. J. Food Sci. Technol., 2014, 49, 2529–2537 CrossRef CAS.
- X. D. Sun and R. A. Holley, Compr. Rev. Food Sci. Food Saf., 2011, 10, 33–51 CrossRef CAS.
- J. I. Boye, I. Alli and A. A. Ismail, J. Agric. Food Chem., 1996, 44, 996–1004 CrossRef CAS.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153–203 CrossRef CAS PubMed.
- K. Baler, R. Michael, I. Szleifer and G. A. Ameer, Biomacromolecules, 2014, 15, 3625–3633 CrossRef CAS PubMed.
- S. Damodaran, Interrelationship of molecular and functional properties of food proteins Food Proteins, 1989 Search PubMed.
- N. Matsudomi, D. Rector and J. Kinsella, Food Chem., 1991, 40, 55–69 CrossRef CAS.
- M. Paulsson, P. O. Hegg and H. B. Castberg, J. Food Sci., 1986, 51, 87–90 CrossRef CAS.
- M. Yamasaki, H. Yano and K. Aoki, Int. J. Biol. Macromol., 1990, 12, 263–268 CrossRef CAS PubMed.
- K. Baler, O. A. Martin, M. A. Carignano, G. A. Ameer, J. A. Vila and I. Szleifer, J. Phys. Chem. B, 2014, 118, 921–930 CrossRef CAS PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- A. Bouhekka and T. Bürgi, Appl. Surf. Sci., 2012, 261, 369–374 CrossRef CAS.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS PubMed.
- R. Lu, W.-W. Li, A. Katzir, Y. Raichlin, H.-Q. Yu and B. Mizaikoff, Analyst, 2015, 140, 765–770 RSC.
- S. Krimm and J. Bandekar, Adv. Protein Chem., 1986, 38, 181–364 CrossRef CAS PubMed.
- H. Abdi and L. J. Williams, Wiley Interdiscip. Rev.: Comput. Stat., 2010, 2, 433–459 CrossRef.
- J. Shlens, 2014, arXiv preprint arXiv:1404.1100.
- S. Dray, Comput. Stat. Data Anal., 2008, 52, 2228–2237 CrossRef.
- Principal Component Analysis, ed. I. T. Jolliffe, Springer New York, New York, NY, 2002, pp. 150–166, DOI:10.1007/0-387-22440-8_7.
- P. Robert, M. F. Devaux and D. Bertrand, J. Near Infrared Spectrosc., 1996, 4, 75–84 CrossRef.
- C. Schwieger and M.-H. Ropers, Food Hydrocolloids, 2013, 30, 241–248 CrossRef CAS.
- M. Kaupp, M. Bühl and V. G. Malkin, in Calculation of NMR and EPR Parameters, Wiley-VCH Verlag GmbH & Co. KGaA, 2004, pp. 1–5, DOI:10.1002/3527601678.ch1.
- D. Hinderberger, H. W. Spiess and G. Jeschke, J. Phys. Chem. B, 2004, 108, 3698–3704 CrossRef.
- N. M. Atherton, Principles of electron spin resonance, Ellis Horwood, New York, revised edition, 1993 Search PubMed.
- M. J. N. Junk, W. Li, A. D. Schlüter, G. Wegner, H. W. Spiess, A. Zhang and D. Hinderberger, Angew. Chem., Int. Ed., 2010, 49, 5683–5687 CrossRef PubMed.
- D. Kurzbach, M. J. N. Junk and D. Hinderberger, Macromol. Rapid Commun., 2013, 34, 119–134 CrossRef CAS PubMed.
- K. Widder, S. R. MacEwan, E. Garanger, V. Nunez, S. Lecommandoux, A. Chilkoti and D. Hinderberger, Soft Matter, 2017, 13, 1816–1822 RSC.
- D. J. Schneider and J. H. Freed, in Spin Labeling: Theory and Applications, ed. L. J. Berliner and J. Reuben, Springer US, Boston, MA, 1989, pp. 1–76, DOI:10.1007/978-1-4613-0743-3_1.
- K. A. Earle and D. E. Budil, in Advanced ESR Methods in Polymer Research, John Wiley & Sons, Inc., 2006, pp. 53–83, DOI:10.1002/047005350X.ch3.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439–446 CrossRef CAS PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174–182 CrossRef CAS PubMed.
- S. Damodaran, IFT Basic Symp. Ser., 1994, 9, 1–37 CAS.
- P. D. Ross and A. Shrake, J. Biol. Chem., 1988, 263, 11196–11202 CAS.
- D. Charbonneau, M. Beauregard and H.-A. Tajmir-Riahi, J. Phys. Chem. B, 2009, 113, 1777–1784 CrossRef CAS PubMed.
- A. H. Clark, D. H. Saunderson and A. Suggett, Int. J. Pept. Protein Res., 1981, 17, 353–364 CrossRef PubMed.
- D. Kurzbach, V. S. Wilms, H. Frey and D. Hinderberger, ACS Macro Lett., 2013, 2, 128–131 CrossRef CAS.
- S. Curry, P. Brick and N. P. Franks, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1999, 1441, 131–140 CrossRef CAS.
- V. A. Livshits and D. Marsh, Biochim. Biophys. Acta, Biomembr., 2000, 1466, 350–360 CrossRef CAS.
- A. A. Bhattacharya, T. Grüne and S. Curry, J. Mol. Biol., 2000, 303, 721–732 CrossRef CAS PubMed.
- D. Kurzbach, W. Hassouneh, J. R. McDaniel, E. A. Jaumann, A. Chilkoti and D. Hinderberger, J. Am. Chem. Soc., 2013, 135, 11299–11308 CrossRef CAS PubMed.
- D. R. Kattnig and D. Hinderberger, Chem. – Asian J., 2012, 7, 1000–1008 CrossRef CAS PubMed.
- R. H. Schmidt, Gelation and coagulation Protein Functionality in Foods, 1981 Search PubMed.
- W. Qiu, L. Zhang, O. Okobiah, Y. Yang, L. Wang, D. Zhong and A. H. Zewail, J. Phys. Chem. B, 2006, 110, 10540–10549 CrossRef CAS PubMed.
- J. Reichenwallner, C. Schwieger and D. Hinderberger, Polymers, 2017, 9, 324 CrossRef.
- S.-H. Chong and S. Ham, Chem. Phys. Lett., 2012, 535, 152–156 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7bm00820a |
| This journal is © The Royal Society of Chemistry 2018 |