
Recellularization of decellularized adipose tissue-derived stem cells: role of the cell-secreted extracellular matrix in cellular differentiation†
V.
Guneta
a,
Z.
Zhou
b,
N. S.
Tan
 cde,
S.
Sugii
b,
M. T. C.
Wong
f and
C.
Choong
*a
cde,
S.
Sugii
b,
M. T. C.
Wong
f and
C.
Choong
*a
aSchool of Materials Science and Engineering, Nanyang Technological University, Singapore. E-mail: cleochoong@ntu.edu.sg; Fax: +65 6790 9081; Tel: +65 6513 8166
bSingapore Bioimaging Consortium, Duke-NUS Graduate Medical School, Singapore
cSchool of Biological Science, Nanyang Technological University, Singapore
dInstitute of Molecular and Cell Biology, Agency for Science Technology & Research (A*STAR), Biopolis, Singapore
eKK Research Centre, KK Women's and Children Hospital, Singapore
fPlastic, Reconstructive and Aesthetic Surgery Section, Tan Tock Seng Hospital (TTSH), Singapore
First published on 25th October 2017
Abstract
Adipose-derived stem cells (ASCs) are found in a location within the adipose tissue known as the stem cell niche. The ASCs in the niche are maintained in the quiescent state, and upon exposure to various microenvironmental triggers are prompted to undergo proliferation or differentiation. These microenvironmental triggers also modulate the extracellular matrix (ECM), which interacts with the cells through the cytoskeleton and induces downstream events inside the cells that bring about a change in cell behaviour. In response to these changes, the cells remodel the ECM, which will differ according to the type of tissue being formed by the cells. As the ECM itself plays an important role in the regulation of cellular differentiation, this study aims to explore the role of the cell-secreted ECM at various stages of differentiation of stem cells in triggering the differentiation of ASCs. To this end, the ASCs cultured in proliferation, osteogenic and adipogenic media were decellularized and the secreted ECM was characterized. Overall, it was found that osteo-differentiated ASCs produced higher amounts of collagen and glycosaminoglycans (GAG) compared to the undifferentiated and adipo-differentiated ASCs. The two types of differentiated ECMs were subsequently shown to trigger initial but not terminal differentiation of ASCs into osteo- and adipo-lineages respectively, as indicated by the upregulation of lineage specific markers. In addition, integrin subunits alpha (α) 6 and integrin beta (β) 1 were found to be produced by ASCs cultured on cell-secreted ECM-coated substrates, suggesting that the integrins α6 and β1 play an instrumental role in cell–ECM interactions. Taken together, this study demonstrates the importance of the ECM in cellular fate decisions and how ECM-coated substrates can potentially be used for various tissue engineering applications.
Introduction
Adipose tissue can be obtained by minimally invasive procedures such as liposuction, and is shown to have a higher yield of mesenchymal stem cell (MSC)-like cells than the bone marrow.1,2 Also, owing to their self-renewal and multipotential differentiation characteristics, adipose tissue-derived stem cells (ASCs) have been well characterized and studied for their applications in tissue engineering and regenerative medicine.3–8Stem cells, such as ASCs, are found within a stem cell niche, which is a specialized microenvironment that supports stem cell quiescence under normal conditions and promotes proliferation and differentiation in response to local and systemic cues. Within this niche, the stem cells are in communion with various supporting cells, membrane proteins, secreted soluble factors, cytokines and the insoluble extracellular matrix (ECM).9 The ECM is responsible for providing the niche with a structure and also for maintaining the homeostasis of stem cells. As such, the ECM provides a platform for interaction between the cells and the niche through signaling via soluble and matrix bound factors.10 The stem cells adhere to the ECM and are affected by the variation in composition and physical properties such as elasticity and topography of the ECM.7,10 The changes in the ECM bring about a modulation in the shape of the cell by modifying the actin cytoskeleton structure of the cells.11 This further triggers downstream intracellular events that alter the transcriptomic profile and eventually regulate stem cell behaviour.12,13 In response to the ECM, the stem cells themselves secrete ECM proteins that are specific to the type of tissue in terms of quantity and composition.13
The plastic adherence properties of ASCs allow for routine culturing in tissue culture plastic (TCP) flasks.14–16 However, for tissue engineering approaches involving biomaterials, a bioactive coating of an ECM protein, such as fibronectin (FN), laminin (LM) or collagen (Col), is often used, either individually or in combination, for improving the cell–material interactions.17–19 In order to overcome the lack of tissue-specific complexity offered by these purified proteins, nowadays decellularized tissues are also being used as bioactive coatings or as scaffolding material itself.6,20,21 As ASCs are known to secrete generous amounts of ECM material while in culture in vitro, we hypothesize that the ECM produced by these cells at the various stages of differentiation would be unique to that stage.
In this study, ASCs are cultured in vitro on TCP while maintaining their undifferentiated state. Differentiation into osteogenic and adipogenic lineages was triggered by the use of differentiation media supplements, and the ECM secreted by the ASCs onto TCP was subsequently characterized. The presence of ECM proteins such as fibronectin, laminin, Col I and Col IV was confirmed along with quantitative analysis of total protein, collagen and GAG secreted by the cells. Subsequently the effect of the cell-secreted ECM on the proliferation and differentiation state of ASCs was investigated by seeding ASCs directly onto the ECM-coated TCP surfaces.
Results and discussion
Confirmation of ASC differentiation
The ASCs cultured in vitro were differentiated into the osteogenic and adipogenic lineages using appropriate differentiation media supplements and stained with Alizarin Red and Oil Red O respectively (Fig. 1). Alizarin Red staining showed positive staining for the calcium deposited by the osteo-differentiated cells (Fig. 1a(i)), which was absent in undifferentiated ASCs (Fig. 1a(ii)). From our observations, we could confirm that the ASCs were successfully differentiated into osteoblasts, since terminally differentiated osteoblasts secrete mineralized ECM.22 On the other hand, positive Oil Red O staining for lipid droplets accumulated within the adipo-differentiated cells was observed (red colour: yellow arrows, Fig. 1b(i)), which was absent in undifferentiated ASCs (Fig. 1b(ii)). Since lipid accumulation is characteristic of mature adipocytes,23 the adipogenic media components used in this study were therefore successful in bringing about differentiation of ASCs into the adipogenic lineage.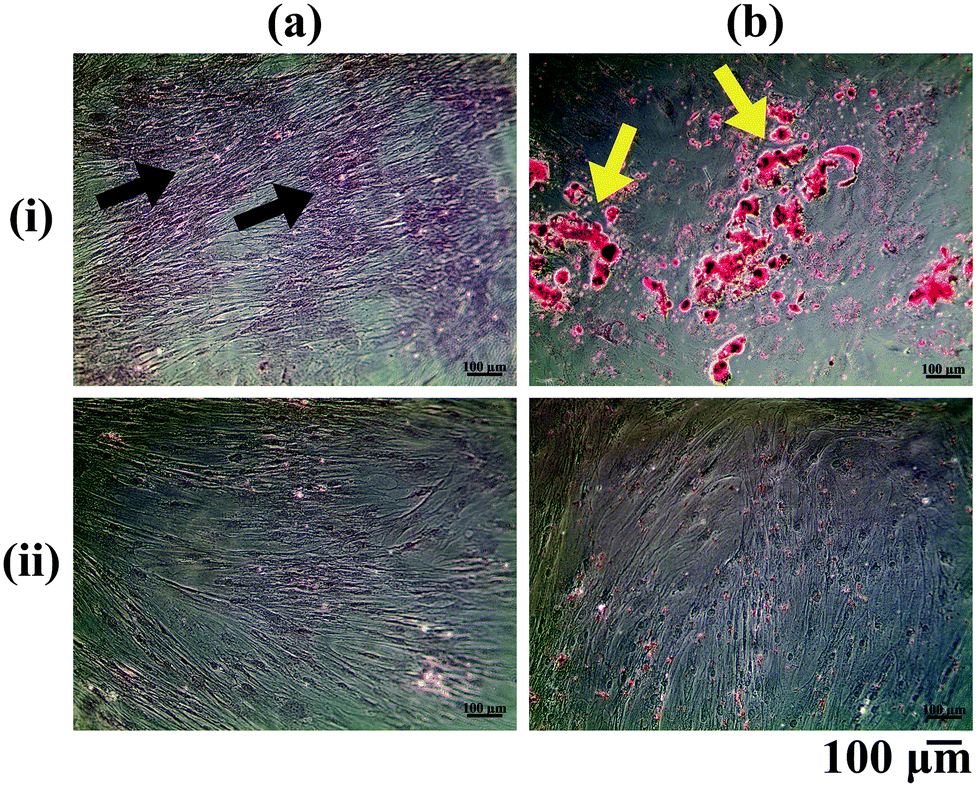 | ||
| Fig. 1 Microscopic images of ASCs cultured in vitro in (i) differentiation and (ii) proliferation media stained with (a) Alizarin Red and (b) Oil Red O stains (black arrows = mineralization produced by osteo-differentiated cells and yellow arrows = lipid droplets accumulated within the adipo-differentiated cells). | ||
Confirmation of decellularization
ASCs cultured on TCP well plates were subsequently decellularized and the removal of cellular material was confirmed by staining of the nucleus of the cells and quantifying the DNA content in the well plates. Positive DAPI staining was observed for the cells in the non-decellularized wells, but not for the wells after decellularization (Fig. 2a). The amount of DNA present in the well plates was also found to be significantly lower after decellularization (p < 0.05) (Fig. 2b). 2.97 ± 1.80, 3.00 ± 0.68 and 2.62 ± 1.14 ng mL−1 of DNA was present in the ECM secreted by the cells cultured in proliferation media (P-ECM), osteogenic media (O-ECM) and adipogenic media (A-ECM), respectively, as compared to 305.44 ± 15.19, 423.02 ± 59.53 and 292.32 ± 24.86 ng mL−1 of DNA present before decellularization. The values after decellularization were all less than 50 ng mL−1, which is the acceptable range for clinical applications.24 Taken together, these results suggest that ASC cell cultures were successfully decellularized and all cellular material was removed leaving behind only the cell-secreted ECM on the TCP surface.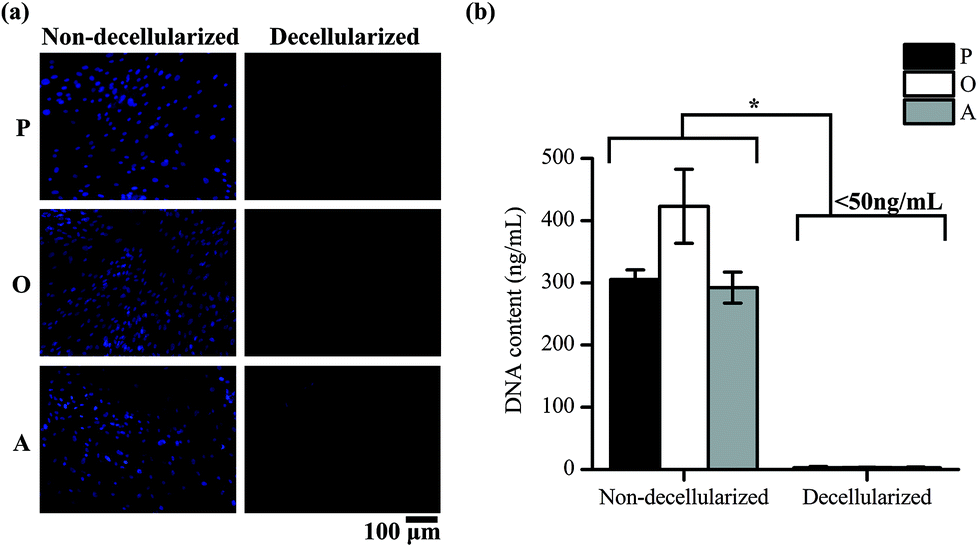 | ||
| Fig. 2 Decellularization of ASC cell culture confirmed by (a) the absence of cells (i.e. no DAPI staining) and (b) reduction in DNA content for cells cultured in proliferation (P), osteogenic (O) and adipogenic (A) media after decellularization (*p < 0.05). | ||
Characterization of ECM
P-ECM, O-ECM and A-ECM secreted by the ASCs cultured in proliferation, osteo- and adipo-differentiation media, respectively, were characterized for the presence of ECM proteins FN, LM, Col I and Col IV before and after decellularization. As shown in Fig. 3, all four proteins were secreted by the ASCs in undifferentiated, osteo-differentiated and adipo-differentiated states. Counterstaining with DAPI (blue colour) for the cell nucleus was visible in the non-decellularized plates and was observed to be absent after decellularization. After removal of the cellular material, the ECM proteins (FN, LM, Col I, and Col IV) secreted by the ASCs (red colour) continue to be present on the TCP surface. This is in concurrence with previous studies that have shown ECM retention after decellularization of in vitro cultures of other types of cells.25,26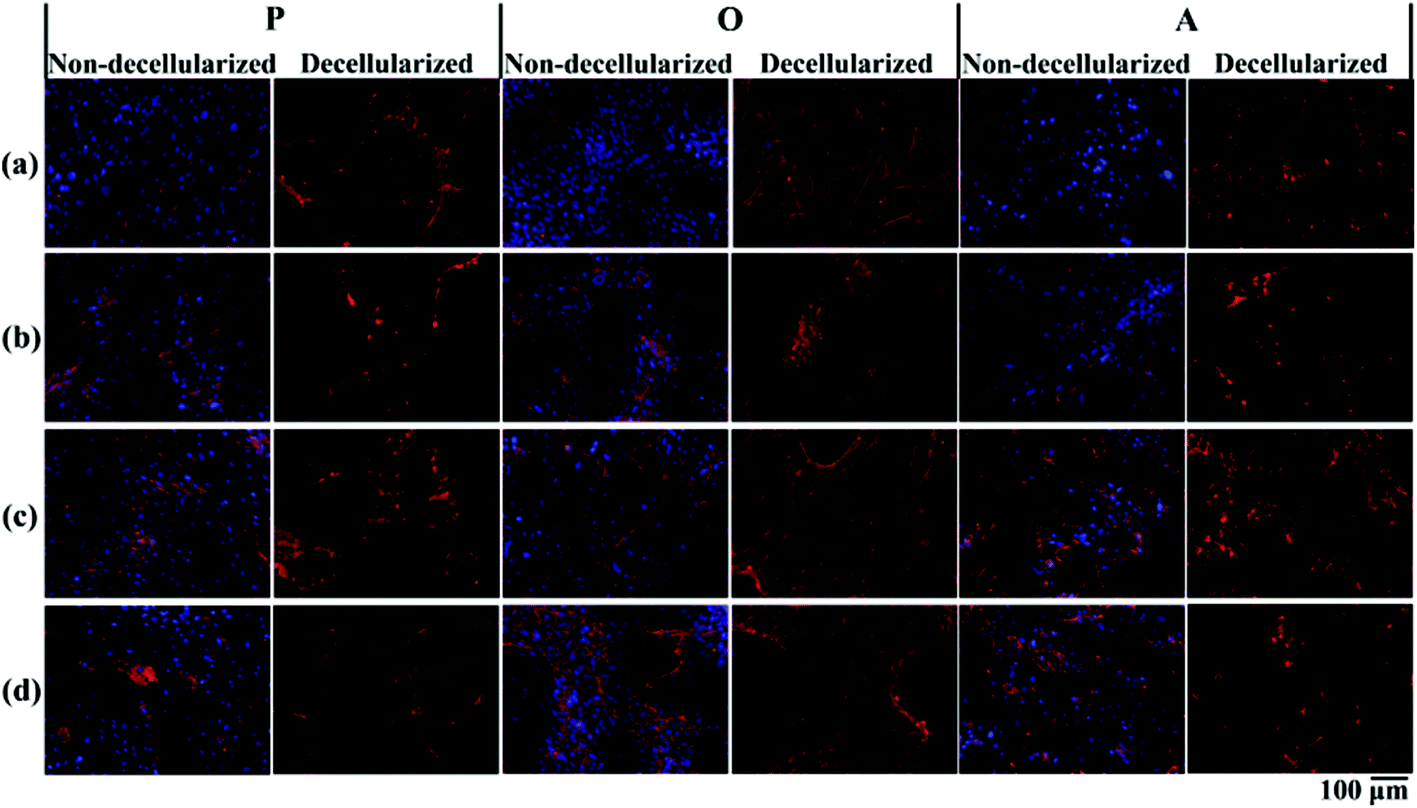 | ||
| Fig. 3 Immunostaining for the presence of ECM proteins: (a) fibronectin (FN), (b) laminin (LM), (c) collagen type 1 (Col I) and (d) collagen type IV (Col IV), before and after decellularization for ASCs cultured in proliferation (P), osteogenic (O) and adipogenic (A) media. | ||
Further quantification of the total protein secreted by the ASCs (Fig. 4a) showed that there was no significant difference between the amount of proteins secreted by both undifferentiated and differentiated ASCs before decellularization, as well as between osteo- and adipo-differentiated ASCs. However, after decellularization, significantly higher (p < 0.05) protein content was recorded for the undifferentiated ASCs as compared to the differentiated ASCs, whereas once again, no significant difference was observed between osteo- and adipo-differentiated ASCs. This result suggests that some protein content was lost during the decellularization process for the differentiated ASCs. During differentiation, the shape of the ASCs changes due to changes in the actin cytoskeleton structure,27,28 which is the cellular component that interacts with the ECM via the integrins.13,29–33 Hence, we postulate that upon differentiation, the interaction between the cells and the ECM resulted in higher amounts of ECM proteins being directly attached to the cell surface. Therefore, as a result of the decellularization process, these cell-associated ECM proteins were lost to a greater extent in the differentiated cultures as compared to undifferentiated cultures.
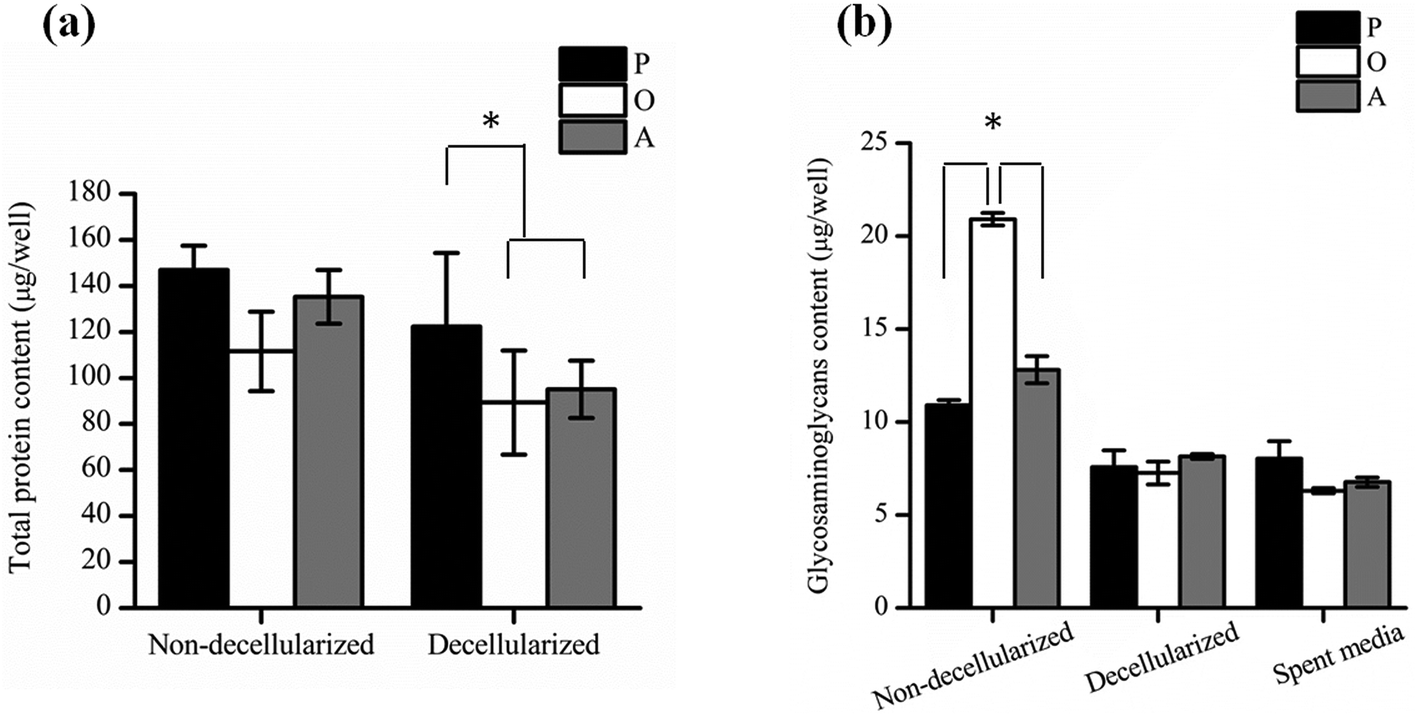 | ||
| Fig. 4 Quantification of the amount of (a) total protein and (b) glycosaminoglycans (GAG) secreted by the ASCs as measured by the BCA assay and GAG assay respectively, before and after decellularization of cells cultured in proliferation (P), osteogenic (O) and adipogenic (A) media (*p < 0.05). | ||
In addition, the amount of GAGs (Fig. 4b) secreted by the ASCs was found to be significantly higher (p < 0.05) in osteo-differentiated ASCs as compared to the undifferentiated and the adipo-differentiated ASCs before decellularization. However, after decellularization, the amount of GAG was found to be similar for all the cells. Hence, even though the osteo-differentiated ASCs produced the highest amount of GAG, the O-ECM that was subsequently used for cellular studies had a similar GAG composition to the P-ECM and A-ECM. Since GAGs are known to also be secreted into the media by the cells of monolayer cultures in vitro,34,35 the amount of GAG in the spent media of undifferentiated, osteo-differentiated and adipo-differentiated cells was measured. However, the results (Fig. 4b) showed that there was no significant difference in the amount of GAG secreted by the cells in the three media (i.e. spent media). Hence, the high amount of GAG secreted originally by the osteo-differentiated ASCs could have been lost during the decellularization process itself. As this phenomenon was only specific to osteo-differentiated ASCs, this suggests that the GAGs were loosely bound to the cell surface after osteogenic differentiation36,37 and were therefore easily removed upon decellularization.
Further analysis of the ECM composition was also carried out by quantifying the amount of collagen secreted by the ASCs in the three media. As seen in Fig. 5a, the amount of collagen secreted by the ASCs upon osteo-differentiation was significantly higher (p < 0.05) than the undifferentiated and adipo-differentiated ASCs. Hence, the overall total protein secretion by the ASCs was similar in quantity (Fig. 4a) but not in composition, since there was a significant difference in the collagen content. This observation of tissue-specific differences in the collagen/protein content was also reported by others,38–41 thus highlighting the importance of the ECM composition in controlling tissue function, development and homeostasis.
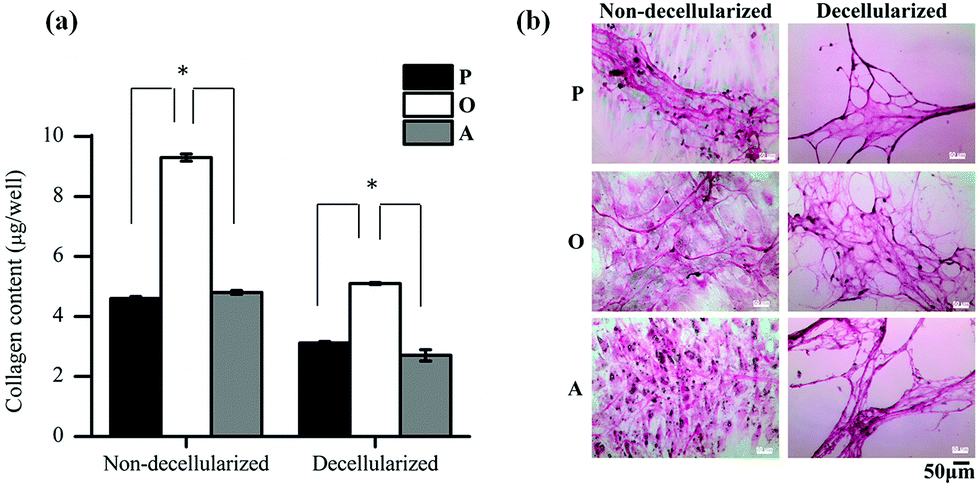 | ||
| Fig. 5 Comparison of collagen secreted by the ASCs under different in vitro conditions. (a) Amount of collagen and (b) morphology of the fibrous collagen stained with Sirius Red, before and after decellularization of cells cultured in proliferation (P), osteogenic (O) and adipogenic (A) media (*p < 0.05). | ||
In addition, previous studies also showed that collagen is secreted by the cells as a result of differentiation.42–45 In our present work, when the collagen fibers present in the deposited ECM were stained with Sirius Red before and after decellularization (Fig. 5b), a mesh-like collagen structure was visible for the P-ECM and O-ECM, with the O-ECM consisting of a more intricate network of collagen fibers. On the other hand, collagen fibers observed for the A-ECM were denser and more closely packed after the decellularization process. As the adipo-differentiated ASCs contained lipid-filled cells (Fig. 1b(ii)), it is possible that after the removal of lipids, the collagen fibers surrounding the cells were not able to maintain their original structure, and hence were found bundled together in a denser structure. It has previously been shown that ECM deposited by cells at different time points in culture and under different environmental conditions such as hypoxia and shear have different compositions.46,47 To our knowledge, the present work is the first study to show evidence that the differentiation of ASCs into different lineages will affect the unique composition and structure of the ECM secreted by the differentiated cells.
Overall, the reduction in the amount of protein and GAGs in the decellularized matrices appears to be the result of the decellularization process itself, although this observation was more evident in differentiated cells than in undifferentiated cells. Hence, further optimization of the decellularization method is still necessary, especially for quantifying the loosely bound GAG in the osteo-differentiated cells. However, differentiation of ASCs did indeed lead to changes in the amount of collagen (Fig. 5a), as well as reorganisation of the ECM structure (Fig. 5b).
Proliferation and morphology of ASCs cultured on cell-secreted ECM-coated TCP substrates
Undifferentiated ASCs were seeded onto TCP coated with the ECM from ASCs previously cultured in proliferation (P), osteogenic (O) and adipogenic (A) media. The proliferation of these cells was measured over 7 days and compared with the cells grown on uncoated TCP substrates. It was found that there was no significant difference in the proliferation of cells on the different substrates up to day 5 (Fig. 6). However, on day 7, a significantly higher number of cells were observed on the uncoated TCP as compared to ECM-coated TCP substrates. This suggests that the ECM coatings did not result in the enhancement of proliferation of the ASCs.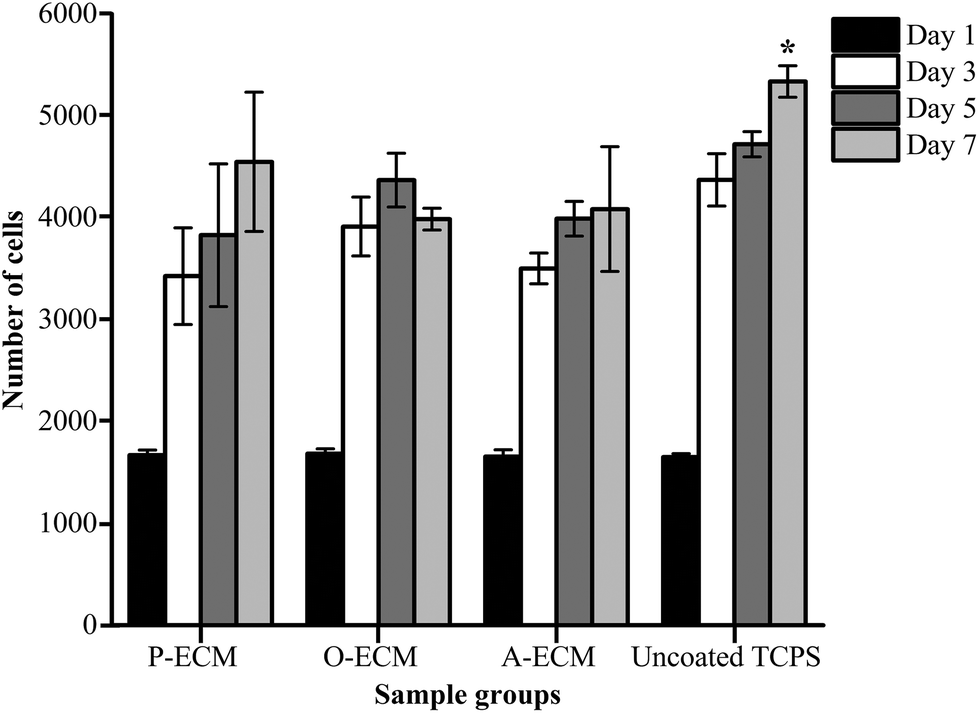 | ||
| Fig. 6 Proliferation of cells (ASCs) cultured on ECM-coated and uncoated TCPs substrates. | ||
To further analyse the cell spreading and shape of the ASCs seeded onto the ECM-coated substrates, staining of actin filaments (red) and the nucleus (blue) was carried out. As shown in Fig. 7a, it can be observed that the ASCs displayed a typical fibroblast-like morphology when grown on top of the different types of ECM-coated wells. Hence, the ECM coating material itself did not affect the cellular morphology and shape of the ASCs cultured on them. In general, a change in the cell shape and morphology is generally representative of a change in cell behaviour.31 As no visible differences were observed, it appears that the different cell-secreted ECM coatings did not bring about any major changes to the cellular properties of the ASCs that were growing on them.
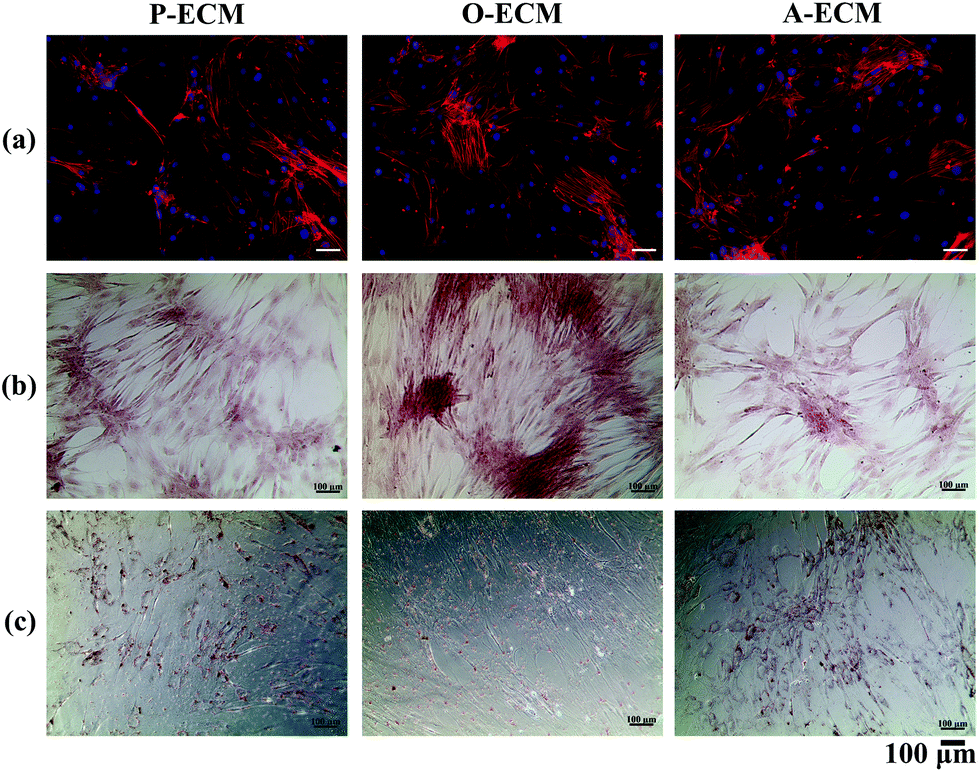 | ||
| Fig. 7 Characterization of ASCs cultured on plates coated with the ECM secreted by cells in proliferation (P-ECM), osteogenic (O-ECM) and adipogenic (A-ECM) media and stained with (a) cytoskeleton stain (actin: red, nucleus: blue), (b) Alizarin Red and (c) Oil Red O for cell spreading, mineralization and lipid accumulation respectively. | ||
Differentiation of ASCs cultured on cell-secreted ECM-coated TCP
In order to assess the efficacy of the cell-secreted ECM proteins for triggering ASC differentiation, Alizarin Red and Oil Red O staining was carried out to stain for the presence of mineralization and lipid accumulation, respectively. As seen in Fig. 7b and c, no positive Alizarin Red and Oil Red O staining was visible for ASCs cultured on all three types of ECM-coated TCPs, which is unlike the staining results observed for after the cells were differentiated by biological cues from the different differentiation culture media (Fig. 1). Mineralized ECM secretion occurs when cells undergo terminal osteogenesis and lipid droplets accumulate in cells that are terminally differentiated into adipocytes.22,23 As both these phenomena were not observed (Fig. 7), it can be concluded that the ECM secreted by ASCs in the differentiated state by itself is not capable of inducing differentiation of ASCs into either osteogenic or adipogenic lineages.Nevertheless, further probing into the gene expression analysis of ASCs cultured on the cell-secreted ECM-coated TCP was carried out for early (peroxisome proliferator associated receptor gamma (PPARγ)) and late (adiponectin) adipogenic markers (Fig. 8). PPARγ expression was found to be significantly higher (p < 0.05) in ASCs cultured on A-ECM, as compared to those cultured on P-ECM and O-ECM. The PPARγ gene is expressed early in the process of adipogenesis and further induces a cascade of events that support adipogenesis of ASCs.48 Hence, a significantly higher expression level of PPARγ (p < 0.05) in ASCs cultured on A-ECM (Fig. 8) implies that very early events of adipogenesis have indeed started to take place in these cells as a result of the adipo-differentiated ECM. On the other hand, the reduction in the expression of PPARγ in cells cultured on O-ECM reflects the down regulation of adipogenic signaling pathways in ASCs cultured on O-ECM. This phenomenon further supports the opposing relationship between the signaling pathways of adipogenesis and osteogenesis of ASCs.49 Furthermore, no significant difference in expression levels between the ASCs cultured on the different types of ECM-coated TCPs was observed for adiponectin. Adiponectin is a late marker of adipogenesis as it is a protein secreted by mature adipocytes.50 The absence of any increase in the expression of this gene concurs with the observation of the absence of Oil Red O staining (Fig. 7c), which is a marker for late stage adipogenesis.51 Taken together, this suggests that the ECM secreted by adipo-differentiated ASCs by itself is unable to trigger terminal adipogenesis of ASCs. We postulate that a combination of other microenvironmental factors along with the cell-secreted ECM would provide a better substrate for terminal adipogenesis of ASCs. In fact, even when the ECM from adipose tissue itself was used to coat TCP, no significant difference between the expression levels of PPARγ and adiponectin was observed (Fig. S1†), despite the fact that both adipose tissue itself and cell-secreted A-ECM had different ECM compositions (ESI Table 2†).
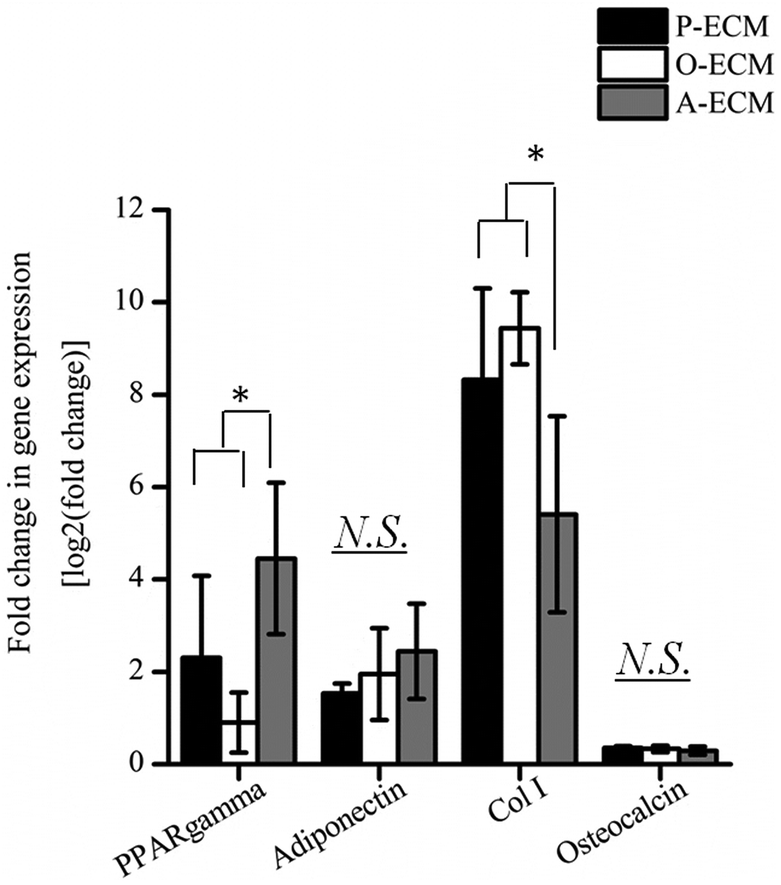 | ||
| Fig. 8 The fold change in gene expression of adipogenic markers (PPARγ and adiponectin) and osteogenic markers (Col I and osteocalcin) for ASCs cultured on the ECM secreted by cells in proliferation (P-ECM), osteogenic (O-ECM) and adipogenic (A-ECM) media (*p < 0.05). | ||
Gene expression levels of early (Col I) and late osteogenic markers (osteocalcin) were also measured for ASCs cultured on the three types of ECM-coated TCP (Fig. 8). Overall, the expression of Col I was found to be significantly higher (p < 0.05) as compared to osteocalcin. Between the different ECM-coated surfaces, ASCs cultured on the A-ECM had significantly lower levels of expression of Col as compared to those cultured on P-ECM and O-ECM, whereas no significant differences were observed in the osteocalcin expression levels. Together with the absence of Alizarin Red staining for calcium deposits (Fig. 7b), it appears that on its own, the ECM of the osteo-differentiated ASCs was also unable to induce terminal osteogenesis of ASCs. Overall the gene expression analysis suggests that the ECM secreted by ASCs after osteo- and adipo differentiation when coated onto TCP, can induce the differentiation of ASCs into the respective lineages, but only to a small extent, since only the genes involved in the initial phases of the differentiation signaling pathways are upregulated. However, terminal differentiation could not be triggered by the ECM secreted by the differentiating ASCs alone.
Cell-integrin interaction between the ASCs and ECM
The interaction between ASCs and ECM was analysed by measuring the amount of integrins present on the cell surface of ASCs cultured on ECM-coated and uncoated substrates (Fig. 9). Integrin alpha (α) 6 and integrin beta (β) 1 are known to play important roles in cell–ECM interactions.52 Specifically with regard to ASCs, these integrins have been previously shown to partake in events that affect the preservation of a stable stem cell population and also those that control the differentiation of stem cells.10,18,53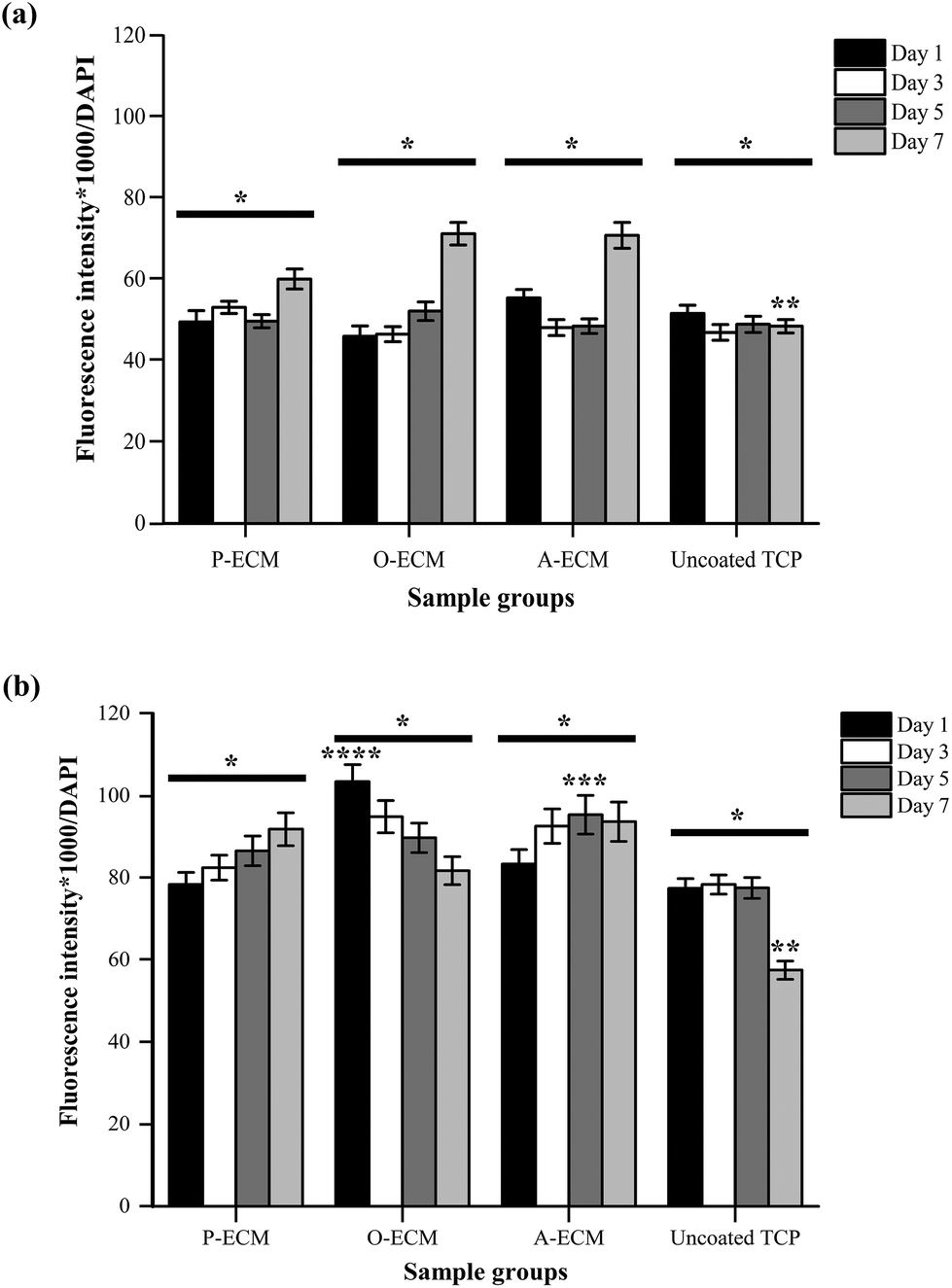 | ||
| Fig. 9 Relative quantification of integrins (a) alpha 6 and (b) beta 1 produced by the ASCs when cultured on ECM-coated and uncoated TCP substrates on days 1, 3, 5 and 7 (*p < 0.05) (* significant difference between the different substrates, ** significant difference between the integrin production on day 7, *** significant difference between the integrin beta 1 production on day 5, **** significant difference between the integrin beta 1 production on day 1). | ||
We found that a higher amount of surface integrin α6 was expressed by ASCs cultured on ECM-coated substrates (P-ECM, O-ECM and A-ECM) than those cultured on the uncoated TCP substrates (Fig. 9a). The surface integrin β1 on time points from day 1 through 7 was dependent on the type of substrate used for culturing the cells. The overall trend of higher production of surface integrin β1 over time was observed when the cells were cultured on P-ECM and A-ECM, whereas lower production of surface integrin β1 was observed on day 7 for O-ECM and uncoated TCP (Fig. 9b). This suggested that the presence of cell-secreted ECM did indeed play a role in triggering the presentation of integrins α6 and β1 on the cell surface, which in turn affected subsequent cell–material interactions.
Overall, a higher expression of integrin α6 and integrin β1 was found in ASCs cultured on ECM-coated substrates as compared to those cultured on uncoated TCP substrates after 7 days of culture (Fig. 9). Integrin α6 has previously been shown to function as a receptor for laminin in the basement membrane and therefore plays an important role in facilitating cell–ECM interaction.54 Hence, the significantly higher production of surface integrin α6 after 7 days of culture on ECM-coated substrates as compared to uncoated TCP (Fig. 9a) substrates indicated that the ECM material was able to improve cell attachment.
In earlier studies, the role of the ECM material in promoting cell attachment to otherwise inert materials was also demonstrated using an adipose tissue-derived ECM material.20,21 Integrin β1 plays a critical role in processes including cell adhesion, tissue repair and immune response through the cell–ECM interaction52,55 and its secretion is modulated by the presence of cell-secreted ECM on the substrates. Integrin α6 has also been known to be expressed in MSCs undergoing adipogenesis and in adult human subcutaneous adipose tissue.53 However, a significantly high expression of integrin α6 was not observed when cultured on A-ECM substrates as compared to other substrates. Once again, this is an indication that the A-ECM substrate alone was unable to trigger terminal differentiation of the ASCs in concurrence with the gene expression results (Fig. 8). This further reinforced the two-way relationship between cellular processes and the ECM.7
The expression of surface integrin β1 increased with time when ASCs were cultured on P-ECM and A-ECM, with a significantly higher production on day 5 on A-ECM as compared to other substrates at the same time point (Fig. 9b). Integrin β1 has been previously extensively researched and reviewed for its multiple roles in regulating stem cell fate, tissue morphogenesis and many other cellular processes.10 In an important recent study, integrin β1 mediated signalling was shown to preserve undifferentiated progenitor properties in ASCs during in vitro expansion.56 ASCs cultured on P-ECM were in the presence of proliferative cues from both the ECM and the media as compared to the uncoated TCP, where the proliferative cues came only from the media alone. As such, a higher production of surface integrin β1 in cells cultured on P-ECM was observed as compared to those cultured on the uncoated TCP, which also further confirmed the role of integrin β1 in preserving the undifferentiated state of the ASCs. As a similar trend was also observed in cells cultured on A-ECM, it appears that the presence of A-ECM alone was unable to bring about terminal adipogenic differentiation of the ASCs. This is further confirmed by the gene expression results (Fig. 8), where no significant increase in the expression of adiponectin was observed. Taken together, the ASCs when cultured on A-ECM in proliferation media, did not differentiate into adipocytes.
Integrin β1 also plays an important role in early osteogenic matrix formation during osteogenesis.52,57 A significantly high production of surface integrin β1 in ASCs cultured on substrates coated with the ECM from osteogenic differentiated ASCs (i.e. O-ECM) on the first day of culture itself (Fig. 9b) suggests that the ECM proteins from O-ECM facilitated rapid cell attachment and interaction with the ASCs present. However, over time in culture, the cells interacted with components of both the O-ECM and the proliferation media. In this microenvironment milieu of multiple cues, it was observed that the quantity of integrin β1 decreased with increasing time in culture (Fig. 9b). This decrease in integrin β1 from the initial high quantities was accompanied by no significant difference in the expression of osteogenic genes (i.e. Col I and osteocalcin) (Fig. 8), suggesting that the cells remained in their undifferentiated state over time in culture even though the initial cell attachment and interaction with O-ECM suggest otherwise. Overall, O-ECM was unable to trigger terminal differentiation of ASCs into osteocytes when they were cultured in proliferative media.
Experimental section
Isolation and in vitro culture of ASCs
All cell culture reagents were procured from Life Technologies, USA unless otherwise mentioned. ASCs were isolated from the human adipose tissue using previously established protocols.8,20,21,58 Adipose tissue samples were obtained from elective surgery carried out at Tan Tock Seng Hospital, Singapore (DSRB ref.: 2012/00071) and National University Hospital, Singapore (DSRB ref.: 2011/01721). Cells were cultured in vitro according to previously established protocols2,7,8,59 involving the use of proliferation media that constitutes Dulbecco's modified Eagle's medium/Ham's F12 (DMEM-F12) (Sigma) supplemented with 10% fetal bovine serum (FBS) (Hyclone), 10 mM MEM-non-essential amino acids (MEM-NEAA) and 1% Pen/Strep. Media were changed every other day and cells were passaged at ∼80% confluence. Cells from passages 2 (P2) to 4 (P4) were used for experiments with a seeding density of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2. The cells were routinely visually inspected for their morphology using a phase contrast microscope (Primo Vert, Zeiss) and images were captured using a Zeiss camera and Axio vision software (Zeiss).
000 cells per cm2. The cells were routinely visually inspected for their morphology using a phase contrast microscope (Primo Vert, Zeiss) and images were captured using a Zeiss camera and Axio vision software (Zeiss).
Differentiation of ASCs
ASCs were differentiated into the osteogenic and adipogenic lineages by replacing the proliferation media with specific differentiation media on day 7 of culture as per previously established protocols.2,5,8,21 Fresh media were changed twice a week until day 28. Differentiation supplements were purchased from Sigma unless mentioned otherwise and are as follows: the osteogenic differentiation media constituted the addition of 0.1 μM dexamethasone, 50 μM ascorbic acid phosphate and 10 mM β-glycerol phosphate to the proliferation media. For adipogenic differentiation, 1 μM dexamethasone, 10 μM insulin, 200 μM indomethacin, and 0.5 mM IBMX were added to the proliferation media. The successful differentiation of ASCs in the two lineages was qualitatively assessed by staining the calcium deposits and lipid droplets present in osteoblasts and adipocytes with Alizarin Red and Oil Red O, respectively, using previously established protocols.2,5,8,21 Briefly, the differentiated cells were fixed with 4% paraformaldehyde for 30 min at 4 °C and incubated with the respective stain for 30 min at room temperature. After 30 min, the cells were washed with distilled water and viewed under a bright field microscope (Primo Vert, Carl Zeiss) and images were captured using the Zeiss camera and Axio vision software (Carl Zeiss).Decellularization of 2D cultured ASCs
All decellularization chemicals and reagents were purchased from Sigma, unless otherwise stated. Decellularization of the cultured cells was carried out by treating the cells with 0.5% Triton X-100 and 20 mM ammonium hydroxide (NH4OH) at 37 °C for 5 min. Next, the wells were treated with DNAse, RNAse (Promega) and lipase for 1 h at 37 °C and subsequently subjected to three freeze thaw cycles. After that, the wells were treated with 0.1% glutaraldehyde for 6 h at 4 °C. They were then treated with 0.1 M glycine in 1× phosphate buffered saline (PBS). The ECM-coated TCP plates were then used immediately for ECM characterization and cellular studies or stored at −80 °C for later use. TCP wells containing the ECM of cells exposed to proliferation, osteogenic and adipogenic differentiation media were referred to as P-ECM, O-ECM and A-ECM respectively.Confirmation of decellularization
Confirmation of decellularization was determined by the absence of nuclear and cellular materials.20,21 Visual assessment of the presence of cells was carried out by staining with a nuclear stain 4′,6-diamidino-2-phenylindole (DAPI) and viewing under a fluorescence microscope.60 Briefly, plates were washed with 1× PBS and incubated with DAPI (1:1000) in 1× PBS for 10 min at 4 °C. After incubation, the plates were washed thrice with 1× PBS and visualized under the Axio Observer Fluorescence Microscope (Carl Zeiss) and images were acquired using ZEN 2012 (Carl Zeiss) software.
The DNA content before and after decellularization was quantified using the Quant-iT™ PicoGreen® dsDNA Assay Kit (Life Technologies, USA) following previously established protocols.20,21 Briefly, samples were prepared by adding 1× TE buffer (from the kit) + 0.1% Triton X-100 and freeze–thawed three times (−80 °C to 37 °C). Briefly, 1 mL of sample was added to 1 mL of Quant-iT™ PicoGreen® reagent working solution and incubated for 5–10 min at room temperature in dark conditions. Fluorescence was measured at 480 and 520 nm and the amount of DNA was calculated from a standard curve.
Retention of ECM proteins by immunostaining
The various extracellular matrix (ECM) proteins present before and after decellularization were stained by immunostaining and visualized under a fluorescence microscope following previously established protocols.21,61 In brief, the non-decellularized and decellularized wells of tissue culture plates were first fixed with 4% paraformaldehyde for 20 min, followed by treatment with 0.1% Triton X-100 for 5 min and then treated with a blocking solution made up of 1% bovine serum albumin (BSA) in 1× PBS for 30 min at 4 °C. After 3 washes with 1× PBS, primary antibodies specific to ECM proteins such as fibronectin (FN) (ab23750, Abcam), laminin (LM) (ab11575, Abcam), Collagen I (Col I) (ab292, Abcam) and Collagen IV (Col IV) (ab6586, Abcam) were diluted in 1× PBS (1:500) to stain individual wells for 1 h at 4 °C. After 3 washes with 1× PBS, secondary antibodies that were goat-anti rabbit were used for 1 h at 4 °C in dark conditions. After 3 washes with 1× PBS, counterstaining with DAPI was carried out. Fluorescence was then visualized under the Axio Observer Fluorescence Microscope (Carl Zeiss) and images were acquired using ZEN 2012 (Carl Zeiss) software.
Quantification of total ECM protein content by BCA assay
Total protein content in each well was measured using the bicinchoninic acid (BCA) assay.7 Cell lysates were prepared by washing the wells with the cultured cells briefly with chilled 1× PBS. 1 mL radioimmunoprecipitation assay buffer (RIPA) buffer (1% Nonidet P-40, 0.5% deoxycholate, 5 M NaCl and 1 M Tris, pH 7.4)62 with a Halt-cocktail protease inhibitor (Thermo Scientific) was added and stored on ice for 30 min. The well plates were then sonicated (Elma Sonic) for 2–5 min in 10 s sonication–10 s rest cycles. After sonication, the cell lysates were collected in microtubes and centrifuged at 10000g for 20 min at 4 °C. The supernatant was used for determining the protein concentration by the BCA assay. The Pierce™ BCA protein assay kit (Thermo scientific) was used to measure the concentration of proteins in the samples, according to the manufacturer's instructions. Briefly, the BCA working reagent was prepared and 200 μL was added to 25 μL of sample in a 96-well microplate and mixed thoroughly on a plate shaker for 30 s. The plate was covered and incubated at 37 °C for 30 min. Absorbance was read at 562 nm using a plate reader (SpectraMax® Gemini, Molecular Devices) after cooling the plate at room temperature. The protein content in the samples was calculated against a standard curve using different concentrations of albumin protein provided in the kit.
Quantification of collagen content in the ECM by Sirius Red assay
The amount of collagenous protein was analysed using the Sirius Red/Fast Green collagen staining kit (Chondrex, Inc.) as per the manufacturer's instructions. Briefly, the non-decellularized and decellularized well plates were washed with 1× PBS and fixed with 4% paraformaldehyde for 10 min at 4 °C. After washing with 1× PBS, 200 μL of dye solution was added and incubated for 30 min at room temperature. Next, the dye solution was removed and plates were washed with distilled water until all the dye was removed. At this stage, the non-decellularized and decellularized plates stained for collagen were visualized under a bright light microscope (Primo Vert, Carl Zeiss) and images were captured using the Zeiss camera and Axio vision software (Carl Zeiss). 1 mL of dye extraction buffer was added to each well and gently mixed by pipetting until all the colour was eluted, this was then used to read the OD values at 540 nm and 605 nm using a plate reader (Molecular Devices). The amount of collagen was calculated using eqn (1):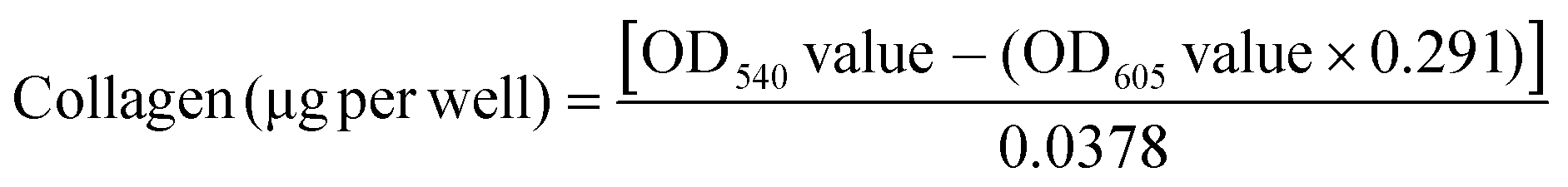 | (1) |
Quantification of glycosaminoglycan (GAG) content
The amount of glycosaminoglycans (GAG) secreted by the cells was analyzed by the Blyscan™ GAG assay (Biocolor) according to the manufacturer's instructions.7 Briefly, each well was treated with 1 mL of papain extraction reagent. The plates were incubated in 65 °C for 3 h and the content was transferred to microtubes and centrifuged at 10000g for 10 min. The supernatant was then used for analyzing the GAG content using the assay. 1 mL of Blyscan dye reagent was added to 100 μL of test sample. The tubes were shaken for 30 min at room temperature, and centrifuged at 12000 rpm for 10 min. The supernatant was discarded and the coloured pellet was dissociated with 0.5 mL of dissociation reagent. The tubes were then vortexed and centrifuged at 12000 rpm for 5 min. 200 μL of the sample was transferred to each well of the 96-well plate and absorbance was measured at 656 nm using a plate reader (SpectraMax® Gemini, Molecular Devices). The GAG content of the sample was calculated against a standard curve using different concentrations of the standard GAG provided in the kit.
Cell culture on decellularized cell-secreted ECM-coated TCP
ASCs grown in proliferation, osteogenic and adipogenic media in 24-well tissue culture plates were decellularized using already described methods under sterile conditions. Undifferentiated ASCs were then seeded at a density of 15000 cells per cm2 and cultured for 7 days in proliferation media at 37 °C in 5% CO2 with media changes every other day.
Cell proliferation and morphology
The proliferation of cells cultured on decellularized matrices was measured using previously established protocols.2,63,64 Briefly, at various time points through 7 days, the cells were incubated with the 10% Presto Blue® cell viability reagent for 30 min. After that, the solution was transferred to wells of a 96-well plate and fluorescence was measured at 560 nm/590 nm wavelengths using the SpectraMax M2 microplate reader (Molecular Devices). Cell numbers were calculated against a standard curve plotted with a known number of cells. On day 7, the cells were stained for the actin and nucleus to assess the spreading and morphology of the cells on the ECM-coated plates. The staining was carried out using the FAK100 kit (Merck Millipore) as per the manufacturer's instructions. In brief, the cells were fixed with 4% paraformaldehyde for 30 min at 4 °C. After washing with 1× PBS thrice, the cells were treated with 0.1% Triton X-100 for 5 min at room temperature. Subsequently, the cells were blocked with a 1% BSA solution for 30 min at 4 °C. The cells were then incubated with the TRITC-conjugated antibody (1:500) (FAK100 kit) for actin for 30 min at 4 °C in the dark. The cells were then counterstained with DAPI (1:1000) for 30 min at 4 °C, washed with 1× PBS and viewed under the Zeiss Axio Observer Z1 inverted fluorescence microscope fitted with a camera (Carl Zeiss) and images were acquired using Zen software (Carl Zeiss). The differentiation of the cells cultured on the cell-secreted ECM-coated TCP into the osteogenic and adipogenic lineages was analysed by staining with Alizarin Red and Oil Red O as previously mentioned.
Gene expression measured by quantitative polymerase chain reaction (qPCR)
Gene expression was measured by a quantitative polymerase chain reaction (qPCR) carried out using previously established protocols.2,7,8 The total RNA of the cells was extracted from the ASCs using RNA-Solv (Omega bio-tek) and RNAeasy Mini Kit (Qiagen). The cDNA was synthesized from the total RNA using iScript™ cDNA Synthesis Kit (Biorad), which was used as the template in a 20 μl reaction mixture containing the specific primer pair (Sigma) of each gene and qPCR was carried out using the SYBR® Green Supermix (Biorad). The list of the primers used in this study is presented in ESI Table 1.†Cell–ECM interaction
The cell–ECM interaction was analysed by studying the presence of integrin α6 and integrin β1. Cells cultured on ECM-coated and uncoated substrates were stained with antibodies for each of the integrins. In brief, the cells were fixed with 4% paraformaldehyde for 30 min at 4 °C. After washing with 1× PBS thrice, the cells were treated with 0.1% Triton X-100 for 5 min at room temperature. Subsequently, the cells were blocked with a 1% BSA solution for 30 min at 4 °C. The cells were then incubated with primary rabbit antibodies for integrin α6 and integrin β1 for 30 min at 4 °C. Next, the cells were treated with the secondary goat anti-rabbit antibody FITC for 30 min at 4 °C in the dark. The cells were then counterstained with DAPI (1:1000) for 30 min at 4 °C, washed with 1× PBS and analysed. The fluorescence intensity of each well was measured using a SpectraMax M2 plate reader (Research Instruments) with Softmax Pro V5.4 software. A well scan with pattern setting as fill and at a density of 9 was selected to perform the reading, which allows us to record 69 reading points from each well. Quantification of the integrins was calculated by normalizing the fluorescence intensity of the integrin to the corresponding spot fluorescence intensity of DAPI.
Statistics
The Mann–Whitney U test was used for statistical analysis of the comparison between the non-decellularized and decellularized groups. The Kruskal Wallis, non-parametric one-way ANOVA analysis was used for statistical analysis between the undifferentiated, adipo-differentiated and osteo-differentiated groups. Statistically significance was assumed when p < 0.05. For the integrin quantification, the statistical analysis was performed using Graphpad Prism 7 XML. Two-way ANOVA with a Tukey's multiple comparison test was carried out to analyze the effect of the type of substrate on integrin production by the cells.Conclusions
In the present study, undifferentiated and osteo- and adipo-differentiated cultures of ASCs were decellularized, leaving behind the ECM proteins on the TCP substrates. Analysis of the ECM produced in different media showed that the osteo-differentiated ASCs secreted higher amounts of collagen and GAGs as compared to the undifferentiated and adipo-differentiated ASCs. However, the total amount of protein secreted by the cells in all the three media was similar despite the different amounts of collagen and GAGs secreted, which supports earlier findings that tissue-specific differences in the ECM composition exist. In addition, upon culturing undifferentiated ASCs on TCP substrates coated with the ECM secreted by differentiated ASCs, a slight upregulation in genes expressed during the early events of differentiation was observed. However, terminal differentiation into osteogenic and adipogenic lineages of the ASCs could not be triggered by the cell-secreted ECM alone. Furthermore, integrin subunits α6 and β1 were found to be involved in the interaction of the ASCs with the ECM coating and their expression was dependent on the type of ECM (i.e. P-ECM, O-ECM or A-ECM) and the time point in culture. This study clearly demonstrated the important role of the ECM in cell–material interactions for early differentiation of ASCs. A combination of the biochemical cues from the cell-derived ECM and the physical cues from material-related properties such as topography, composition, porosity and stiffness can be used to mimic the native microenvironment of the stem cells at different stages of differentiation. This in turn will lead to the establishment of tissue-specific stem cell differentiation strategies for various tissue engineering applications that do not require the use of supplementary biological agents and media supplements that contain xenogeneic materials. Such platforms will allow for drug testing as well as translation into clinical applications of tissue engineering and regenerative medicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Nanyang Technological University and National Healthcare Group innovation seed grant (NTU-NHG, M4060911.706022).References
- M. Tobita, H. Orbay and H. Mizuno, Discov. Med., 2011, 11, 160–170 Search PubMed.
- V. Guneta, N. S. Tan, S. K. J. Chan, V. Tanavde, T. C. Lim, T. C. M. Wong and C. Choong, Exp. Cell Res., 2016, 348, 155–164 CrossRef CAS PubMed.
- W. Tsuji, J. P. Rubin and K. G. Marra, World J. Stem Cells, 2014, 6, 312 CrossRef PubMed.
- H. Mizuno, M. Tobita and A. C. Uysal, Stem Cells, 2012, 30, 804–810 CrossRef CAS PubMed.
- S. P. Hoo, Q. L. Loh, Z. Yue, J. Fu, T. T. Tan, C. Choong and P. P. Chan, J. Mater. Chem. B, 2013, 1, 3107–3117 RSC.
- B. Luo and C. Choong, J. Biomater. Appl., 2015, 29, 903–911 CrossRef CAS PubMed.
- V. Guneta, Q. L. Loh and C. Choong, J. Biomed. Mater. Res., Part A, 2016, 104, 1090–1101 CrossRef CAS PubMed.
- V. Guneta, N. S. Tan, S. Sugii, T. C. Lim, T. C. M. Wong and C. Choong, Ann. Plast. Surg., 2016, 76, 569–575 CrossRef CAS PubMed.
- D. L. Jones and A. J. Wagers, Nat. Rev. Mol. Cell Biol., 2008, 9, 11–21 CrossRef CAS PubMed.
- M. F. Brizzi, G. Tarone and P. Defilippi, Curr. Opin. Cell Biol., 2012, 24, 645–651 CrossRef CAS PubMed.
- Y. Sun, C. S. Chen and J. Fu, Annu. Rev. Biophys., 2012, 41, 519–542 CrossRef PubMed.
- E. Yim and M. P. Sheetz, Stem Cell Res. Ther., 2012, 3, 41 CrossRef CAS PubMed.
- F. M. Watt and W. T. Huck, Nat. Rev. Mol. Cell Biol., 2013, 14, 467–473 CrossRef CAS PubMed.
- G. Bassi, L. Pacelli, R. Carusone, J. Zanoncello and M. Krampera, Transfus. Apher. Sci., 2012, 47, 193–198 CrossRef PubMed.
- J. M. Gimble and F. Guilak, Cytotherapy, 2003, 5, 362–369 CrossRef PubMed.
- P. C. Baer and H. Geiger, Stem Cells Int., 2012 Search PubMed , 812693.
- P. G. di Summa, D. F. Kalbermatten, W. Raffoul, G. Terenghi and P. J. Kingham, Tissue Eng., Part A, 2012, 19, 368–379 CrossRef PubMed.
- A. S. Zanetti, C. Sabliov, J. M. Gimble and D. J. Hayes, J. Biomed. Mater. Res., Part B, 2013, 101, 187–199 CrossRef PubMed.
- P. G. di Summa, D. F. Kalbermatten, W. Raffoul, G. Terenghi and P. J. Kingham, Tissue Eng., Part A, 2012, 19, 368–379 CrossRef PubMed.
- B. Luo, Q. L. Loh, M. T. C. Wong, N. S. Tan and C. Choong, J. Mater. Chem. B, 2014, 2, 7795–7803 RSC.
- B. Luo, S. Yuan, S. E. M. Foo, M. T. C. Wong, T. C. Lim, N. S. Tan and C. Choong, Adv. Healthcare Mater., 2015, 4, 613–620 CrossRef CAS PubMed.
- J. Fan, C. S. Im, Z.-K. Cui, M. Guo, O. Bezouglaia, A. Fartash, J.-Y. Lee, J. Nguyen, B. M. Wu and T. Aghaloo, Tissue Eng., Part A, 2015, 21, 2053–2065 CrossRef CAS PubMed.
- G. Yu, Z. E. Floyd, X. Wu, T. Hebert, Y.-D. C. Halvorsen, B. M. Buehrer and J. M. Gimble, Adipose-Derived Stem Cells: Methods and Protocols, 2011, vol. 702, pp. 193–200 Search PubMed.
- P. M. Crapo, T. W. Gilbert and S. F. Badylak, Biomaterials, 2011, 32, 3233–3243 CrossRef CAS PubMed.
- H. Lu, T. Hoshiba, N. Kawazoe and G. Chen, Biomaterials, 2011, 32, 2489–2499 CrossRef CAS PubMed.
- T. Hoshiba, N. Kawazoe, T. Tateishi and G. Chen, Adv. Mater., 2010, 22, 3042–3047 CrossRef CAS PubMed.
- P. S. Mathieu and E. G. Loboa, Tissue Eng., Part B, 2012, 18, 436–444 CrossRef CAS PubMed.
- Z. A. Schiller, N. R. Schiele, J. K. Sims, K. Lee and C. K. Kuo, Stem Cell Res. Ther., 2013, 4, 79–89 CrossRef CAS PubMed.
- F. Guilak, D. M. Cohen, B. T. Estes, J. M. Gimble, W. Liedtke and C. S. Chen, Cell Stem Cell, 2009, 5, 17–26 CrossRef CAS PubMed.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS PubMed.
- P. S. Mathieu and E. G. Loboa, Tissue Eng., Part B, 2012, 18, 436–444 CrossRef CAS PubMed.
- B. Trappmann, J. E. Gautrot, J. T. Connelly, D. G. Strange, Y. Li, M. L. Oyen, M. A. C. Stuart, H. Boehm, B. Li and V. Vogel, Nat. Mater., 2012, 11, 642–649 CrossRef CAS PubMed.
- N. Wang and D. E. Ingber, Biophys. J., 1994, 66, 2181–2189 CrossRef CAS PubMed.
- J. T. Gallagher, N. Gasiunas and S. L. Schor, Biochem. J., 1980, 190, 243–254 CrossRef CAS PubMed.
- T. P. Dale, A. de Castro, N. J. Kuiper, E. K. Parkinson and N. R. Forsyth, PLoS One, 2015, 10, e0133745 Search PubMed.
- J. Kerrigan, J. Mansell and J. Sandy, Am. J. Orthod., 2000, 27, 227–233 CrossRef CAS PubMed.
- R. J. Naik, P. Chandra, A. Mann and M. Ganguli, J. Biol. Chem., 2011, 286, 18982–18993 CrossRef CAS PubMed.
- V. Z. Beachley, M. T. Wolf, K. Sadtler, S. S. Manda, H. Jacobs, M. R. Blatchley, J. S. Bader, A. Pandey, D. Pardoll and J. H. Elisseeff, Nat. Methods, 2015, 12, 1197–1204 CrossRef CAS PubMed.
- C. Bonnans, J. Chou and Z. Werb, Nat. Rev. Mol. Cell Biol., 2014, 15, 786–801 CrossRef CAS PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, Molecular Cell Biology, W. H. Freeman, New York, 4th edn, 2000, Available from: https://www.ncbi.nlm.nih.gov/books/NBK21475/ Search PubMed.
- C. Nie, D. Yang, J. Xu, Z. Si, X. Jin and J. Zhang, Cell Transplant., 2011, 20, 205–216 Search PubMed.
- H.-M. Chung, C.-H. Won and J.-H. Sung, Expert Opin. Biol. Ther., 2009, 9, 1499–1508 CrossRef CAS PubMed.
- H. Mizuno, J. Nippon Med. Sch., 2009, 76, 56–66 CrossRef PubMed.
- L. E. Kokai, K. Marra and J. P. Rubin, Transl. Res., 2014, 163, 399–408 CrossRef CAS PubMed.
- M. L. Decaris and J. K. Leach, Ann. Biomed. Eng., 2011, 39, 1174–1185 CrossRef PubMed.
- N. Datta, Q. P. Pham, U. Sharma, V. I. Sikavitsas, J. A. Jansen and A. G. Mikos, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2488–2493 CrossRef CAS PubMed.
- T. C. Otto and M. D. Lane, Crit. Rev. Biochem. Mol. Biol., 2005, 40, 229–242 CrossRef CAS PubMed.
- A. W. James, P. Leucht, B. Levi, A. L. Carre, Y. Xu, J. A. Helms and M. T. Longaker, Tissue Eng., Part A, 2010, 16, 2605–2616 CrossRef CAS PubMed.
- G. Yu, Z. E. Floyd, X. Wu, T. Hebert, Y.-D. C. Halvorsen, B. M. Buehrer and J. M. Gimble, in Adipose-Derived Stem Cells, Springer, 2011, pp. 193–200 Search PubMed.
- A.-M. Rodriguez, C. Elabd, F. Delteil, J. Astier, C. Vernochet, P. Saint-Marc, J. Guesnet, A. Guezennec, E.-Z. Amri and C. Dani, Biochem. Biophys. Res. Commun., 2004, 315, 255–263 CrossRef CAS PubMed.
- S. Gronthos, P. Simmons, S. Graves and P. G. Robey, Bone, 2001, 28, 174–181 CrossRef CAS PubMed.
- A. Noro, T. Sillat, I. Virtanen, S. Ingerpuu, N. Bäck, Y. T. Konttinen and M. Korhonen, J. Histochem. Cytochem., 2013, 61, 719–730 CrossRef PubMed.
- Y. Kikkawa, N. Sanzen, H. Fujiwara, A. Sonnenberg and K. Sekiguchi, J. Cell Sci., 2000, 113, 869–876 CAS.
- Z. Lu, B. Z. Doulabi, C. Huang, R. A. Bank and M. N. Helder, Tissue Eng., Part A, 2009, 16, 81–90 CrossRef PubMed.
- N. Di Maggio, E. Martella, A. Frismantiene, T. J. Resink, S. Schreiner, E. Lucarelli, C. Jaquiery, D. J. Schaefer, I. Martin and A. Scherberich, Sci. Rep., 2017, 7, srep44398 CrossRef PubMed.
- V. Volloch and B. R. Olsen, Matrix Biol., 2013, 32, 365–371 CrossRef CAS PubMed.
- B. A. Bunnell, M. Flaat, C. Gagliardi, B. Patel and C. Ripoll, Methods, 2008, 45, 115–120 CrossRef CAS PubMed.
- V. Guneta, J. K. Wang, S. Maleksaeedi, Z. M. He, M. T. C. Wong, C. Choong and J. Biomimetics, Biomater. Biomed. Eng., 2014, 21, 101–115 CrossRef CAS.
- G. M. Xiong, S. Yuan, C. K. Tan, J. K. Wang, Y. Liu, T. T. Y. Tan, N. S. Tan and C. Choong, J. Mater. Chem. B, 2014, 2, 485–493 RSC.
- J. K. Wang, B. Luo, V. Guneta, L. Li, S. E. M. Foo, Y. Dai, T. T. Y. Tan, N. S. Tan, C. Choong and M. T. C. Wong, Mater. Sci. Eng., C, 2017, 75, 349–358 CrossRef CAS PubMed.
- B. S. Soh, C. M. Song, L. Vallier, P. Li, C. Choong, B. H. Yeo, E. H. Lim, R. A. Pedersen, H. H. Yang and M. Rao, Stem Cells, 2007, 25, 3029–3037 CrossRef CAS PubMed.
- Y. Y. Chun, J. K. Wang, N. S. Tan, P. P. Y. Chan, T. T. Y. Tan and C. Choong, Macromol. Biosci., 2016, 16, 276–287 CrossRef CAS PubMed.
- J. K. Wang, G. M. Xiong, B. Luo, C. C. Choo, S. Yuan, N. S. Tan and C. Choong, J. Mater. Sci. Mater. Med., 2016, 27, 1–15 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7bm00695k |
| This journal is © The Royal Society of Chemistry 2018 |