
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntra-individual variance of the human plasma oxylipin pattern: low inter-day variability in fasting blood samples versus high variability during the day†
Annika I.
Ostermann‡
ab,
Theresa
Greupner‡
 c,
Laura
Kutzner
ab,
Nicole M.
Hartung
ab,
Andreas
Hahn
c,
Jan Philipp
Schuchardt
c and
Nils Helge
Schebb
*ab
c,
Laura
Kutzner
ab,
Nicole M.
Hartung
ab,
Andreas
Hahn
c,
Jan Philipp
Schuchardt
c and
Nils Helge
Schebb
*ab
aInstitute for Food Toxicology, University of Veterinary Medicine Hannover, Germany
bChair of Food Chemistry, Faculty of Mathematics and Natural Sciences, University of Wuppertal, Gaußstr. 20, 42119 Wuppertal, Germany. E-mail: schebb@uni-wuppertal.de; Tel: +49-202-439-3457
cInstitute of Food Science and Human Nutrition, Leibniz University Hannover, Germany
First published on 25th September 2018
Abstract
Introduction: Several eicosanoids and other oxylipins are important lipid mediators. Reliable quantification in plasma is important to assess the state of disease, action of drugs and the biology of oxylipins. In order to monitor biological changes, low background variability of oxylipin concentrations in biological samples is essential for proper interpretation of oxylipin biology. However, only little is known about the variation in the oxylipin profile in healthy human subjects. Experimental: Inter-day variation in circulating oxylipins after overnight fasting was investigated in healthy young men on either a standardized or non-standardized diet during a (24 to) 48 h time interval. Intra-day variance was investigated during an 8 h time interval (covering breakfast and lunch meals) in men on a standardized diet with blood sampling at 0, 2, 4, 6 and 8 hours. Free oxylipins in plasma were analyzed using a targeted metabolomics platform for the quantification of 160 oxylipins from different precursors. Analytical variation was evaluated based on quality control plasma samples. Results: Free oxylipins in quality control plasma samples showed low variations (<20% for most analytes). Inter-day variations in fasting blood were in the same range, while significant differences were observed within the day (intra-day variance). Conclusion: Based on the low intra-individual inter-day variance in concentrations of free oxylipins, our results demonstrate the suitability of fasting plasma for the investigation of oxylipin biology. In non-fasting plasma samples, the variations were high during the day. Thus, non-fasting plasma samples appear to be unsuitable to evaluate biologically relevant changes, for instance, those caused by disease or drugs. However, it remains to be determined if the same standardized meal results in reproducible modulations of the oxylipin profile allowing evaluation of the oxylipin pattern during the postprandial state.
1 Introduction
Eicosanoids and other oxylipins are oxygenated metabolites of polyunsaturated fatty acids (PUFAs) and many of them have high biological activity in different physiological processes. They are formed endogenously within the arachidonic acid (C20:4n6, ARA) cascade in a variety of enzymatic and non-enzymatic reactions. Conversion of PUFAs by cyclooxygenases (COXs) may lead to the formation of prostanoids like PGE2, which is involved in the regulation of pain and inflammation or thromboxane A2, a potent mediator in the regulation of platelet aggregation.1 Action of lipoxygenases (LOXs) may give rise to hydroperoxy-PUFAs, which can either be reduced to hydroxy-PUFAs or further metabolized, e.g. by LOXs, to multiple hydroxylated PUFAs.2 In particular, multiple hydroxylated metabolites from the omega-3 (n3)-PUFAs eicosapentaenoic acid (C20:5n3, EPA) or docosahexaenoic acid (C22:6n3, DHA) have been shown to be potent mediators in the resolution of inflammation.2 Cytochrome P450 (CYP) monooxygenase enzymes can act as epoxygenases or ω-hydroxylases resulting dominantly in epoxy-PUFAs or terminally (ω and ω-n) hydroxylated PUFAs.3,4 Epoxy-PUFAs are highly active, e.g. the terminally epoxygenated EPA (17(18)-EpETE) shows strong antiarrhythmic potency3,5 and epoxy-PUFAs from ARA, EPA and DHA show anti-inflammatory properties.6 Moreover, ARA derived epoxy-PUFAs display angiogenic activity,6 while the terminally epoxygenated DHA (19(20)-EpDPE) shows anti-angiogenic properties.7 These highly potent epoxy-PUFAs are converted by the soluble epoxide hydrolase (sEH) to dihydroxy-PUFAs8 with so far poorly characterized biological activity. Additionally, autoxidation of PUFAs may give rise, for instance, to hydro(pero)xy-PUFAs or prostaglandin-like structures, such as isoprostanes.9,10Reliable quantification of oxylipins in biological samples is essential for the understanding of their biological role. However, different endogenous and exogenous factors have been shown to have an impact on the oxylipin profile, which hampers the interpretation of biological data. For instance, genetic variants in PUFA metabolizing enzymes11–13 or enzyme preferences for specific PUFAs might influence oxylipin production.3 Other factors, including age,14–16 sex,17–19 physical exercise20 or health status21–23 may also impact oxylipin concentrations, either directly or indirectly via an influence on PUFA metabolism. Dietary factors, such as fatty acid intake, also influence the oxylipin profile, for example, as recently shown for the amount of alpha-linolenic acid (C18:3n3, ALA) in the diet24 or for supplementation with n3-PUFAs.25 Of note, supplementation with n3-PUFAs resulted in high variations in the individual response to n3-PUFAs25–29 and only part of the effect could be accounted for by the basal status of n3-PUFAs.25,27 This means that even when aiming at modulating the profile of oxylipins, interpretation of results might be complicated due to high inter-individual variations. Apart from these biological variations, (pre-)analytical procedures, such as the time between blood collection and plasma preparation, comprise a factor that is known to influence oxylipin concentrations in the sample.30
In most studies analyzing oxylipin concentrations, blood from the fasting state is used for analysis.24,26,27,31 However, only little is known about the modulation of free oxylipin concentrations in plasma following food ingestion or whether the oxylipin profile underlies circadian variations30,32 and which background variations (inter-day variation of the oxylipin profile in fasting plasma) may have to be expected. Therefore, this study aims to (i) investigate the inter-day variation in the oxylipin profile in samples collected 48 hours apart (with and without standardized nutrition) and (ii) to investigate the intra-day variation of free oxylipins in plasma.
2 Experimental
The data published here are derived from two different studies. The investigator initiated studies were conducted according to the guidelines laid down in the Declaration of Helsinki and all procedures involving human subjects were approved by the ethics committee of the Medical Chamber of Lower Saxony (Hannover, Germany). Written informed consent was obtained from all subjects. Both studies are registered in the German Clinical Trial Register (DRKS00006765 and DRKS00012257) and were conducted at the Institute of Food Science and Human Nutrition, Leibniz University Hannover, Germany.Study design
To investigate the inter- and intra-day variation in the oxylipin profile in a population of healthy volunteers, the samples of two separate studies were used:Proband examination, blood sampling and pre-analytical procedures
At baseline examination (t0), blood pressure, body height, body weight and pulse were measured and the subjects completed a questionnaire to obtain information about changes in medication, diet and lifestyle habits (e.g. physical activity) compared to the screening questionnaire. Additionally, fasting blood samples were taken at baseline (t0), after 24 (t24), 48 (t48) and 72 hours (t72). Non-fasting blood samples were taken after 2 (t2), 4 (t4), 6 (t6) and 8 (t8) hours. Blood samples were obtained by venipuncture of an arm vein using Multiflyneedles (Sarstedt, Nümbrecht, Germany) into serum and EDTA monovettes (Sarstedt). For analysis of plasma oxylipins and triglycerides (TGs), EDTA blood monovettes were centrifuged for 10 min at 1500 × g and 4 °C. Plasma was transferred into 1.5 mL Eppendorf tubes (Sarstedt) and immediately frozen and stored at −80 °C until analysis. All transfer steps were carried out on ice. Serum lipid levels, liver enzymes and small blood picture at baseline (t0) and triglycerides in plasma at t0, t2, t4, t6 and t8 were determined in the LADR laboratory (Laborärztliche Arbeitsgemeinschaft für Diagnostik und Rationalisierung e.V.), Hannover, Germany.Fatty acid analyses in food samples
The total fat content of food samples from Study B was determined by gravimetry after lipid extraction according to Weibull–Stoldt performed as rapid microextraction.33 Concentrations of fatty acids in the lipid extracts were determined by means of gas chromatography with flame ionization detection (GC-FID) following (trans-)esterification to fatty acid methyl esters (FAMEs) as described34 using methyl pentacosanoate (C25:0 methyl ester) as an internal standard (IS). Fatty acid concentrations in food samples were calculated as mg fatty acid/100 g meal.Oxylipin analysis
The concentrations of oxylipins in the plasma of Study A were determined by means of liquid chromatography-mass spectrometry (LC-MS) following solid phase extraction as described.24 Oxylipins in the plasma of Study B were analyzed accordingly with slight modifications described in ref. 35. In brief, after addition of internal standards, antioxidant solution and methanol, plasma samples were frozen at −80 °C overnight. Oxylipins were extracted from the supernatant using Bond Elut Certify II cartridges (200 mg, Agilent, Waldbronn, Germany) and ethyl acetate/n-hexane (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, v/v) with 1% acetic acid as the eluent. Before elution, the samples were dried under vacuum (−200 mbar, 30 sec). LC-MS analysis was carried out using a 1290 Infinity LC System (Agilent, Waldbronn, Germany) with a 6500 QTrap (Sciex, Darmstadt, Germany) operated in scheduled selected reaction monitoring mode following negative electrospray ionization as described.35
:25, v/v) with 1% acetic acid as the eluent. Before elution, the samples were dried under vacuum (−200 mbar, 30 sec). LC-MS analysis was carried out using a 1290 Infinity LC System (Agilent, Waldbronn, Germany) with a 6500 QTrap (Sciex, Darmstadt, Germany) operated in scheduled selected reaction monitoring mode following negative electrospray ionization as described.35
Within the sample batch of Study B, human quality control (QC) plasma samples (n = 15) and randomly assigned duplicate samples of the study population (n = 12) were prepared and analyzed to characterize intra-batch variation of the analytical method (including sample preparation, LC-MS analysis and peak integration).
Calculations and statistics
Oxylipin concentrations in plasma and their relative change [%] are stated as mean ± standard error of the mean (SEM). If the concentration in a sample was below the lower limit of quantification (LLOQ) the 1/2 LLOQ was used to calculate the mean and SEM. The concentration was set to LLOQ if the analyte could not be quantified in more than 50% of the samples. Relative changes were calculated individually for each subject at each time point (x) using the following formula: rel. change [%] = 100 × (conctx/conct0). In QC samples, only those analytes were evaluated which were >LLOQ in ≥80% of the samples. Means were calculated by filling in the LLOQ for analytes <LLOQ and the 95% interval of the standard deviation (95% SD = SD × 1.96) was calculated.The distributions of the sample sets (Study A and B) were analyzed by a Kolmogorov–Smirnov test. Statistical differences between the time points were tested for parametric data with a t-test (Study A) or ANOVA with repeated measurements followed by post hoc t-tests for paired samples with Holm–Bonferroni-adjusted levels of significance (Study B) and for non-parametric data with Wilcoxon-tests (Study A) or the Friedman Test followed by the Dunn–Bonferroni post hoc test (Study B). Statistical tests were only performed for analytes which were quantified in the study population at all time points of the respective studies. Statistical significance was set at p ≤ 0.05 for all analyses. All statistical analyses were carried out with SPSS software (Version 24, SPSS Inc., Chicago, IL, USA).
3 Results
Study population
The study population of Study A is described in detail elsewhere.24 18 male, healthy subjects were included in the study with a mean age of 26.2 ± 4.5 years and a BMI of 24.9 ± 2.0 kg m−2. In Study B, 13 male subjects met the criteria and were included in the study. All participants (mean age 24.6 ± 2.5 years) were healthy and had a normal BMI (24.6 ± 2.0 kg m−2). Liver enzymes and serum lipid profiles were in the normal range (ESI Table S2A†).Quality control plasma
The results of selected oxylipins in the QC samples can be found in Fig. 1. Absolute concentrations of all analytes quantified in the QC samples along with the variation are presented in ESI Table S4.† 73 (out of 160) analytes were quantified in QC plasma in the range of 50 ± 5 pM (11,12-DiHETE) to 11 ± 1 nM (15,16-DiHODE). Based on the relative 95% SD, i.e. the deviation covering 95% of all values measured, 61 of the quantified analytes showed a variation of <20% and only 12 analytes fluctuated >20%. In general, the relative 95% SD decreased with higher concentrations, or more specifically with increasing ratio of determined concentration to LLOQ. However, the variation did not decrease below 4.8%.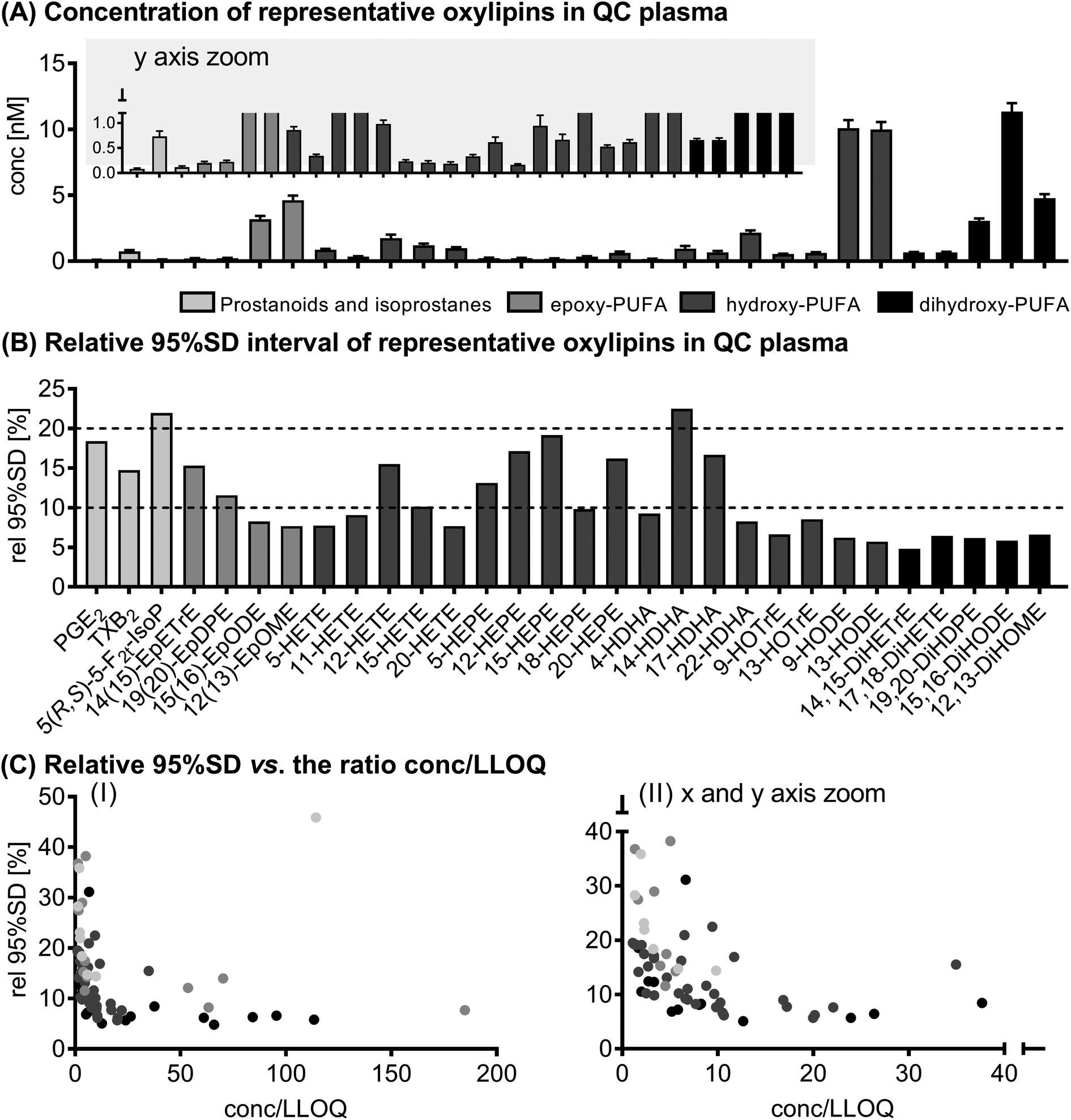 | ||
| Fig. 1 Variation of the analytical method. Shown are concentrations ± 95% interval of the SD (A) and the relative 95% interval of the SD (B) of selected oxylipins in quality control plasma samples (n = 15). In (C) the relative 95% SD is plotted against the ratio of concentration to the lower limit of quantification (conc./LLOQ). Shown are all analytes quantified in quality control plasma (73 analytes, see ESI Table S4†). | ||
The deviation between the first and second analysis of the randomly assigned duplicate samples from the study population was within the range of the deviation of the analytes in the QC samples.
Inter-day variation of free oxylipins in plasma
The results for the inter-day variation of plasma oxylipin concentrations in a non-standardized diet can be found for representative oxylipins in Fig. 2 (all data in ESI Table S5†). Differences between fasting plasma oxylipin concentrations collected at baseline (t0) and after 48 hours (t48) for a non-standardized diet were not significant. However, for some analytes the inter-day variance exceeded the analytical fluctuation, i.e. PGE2, 9(10)-Ep-stearic acid, 11,12-DiHETE and various linoleic acid (C18:2n6, LA) and ALA metabolites.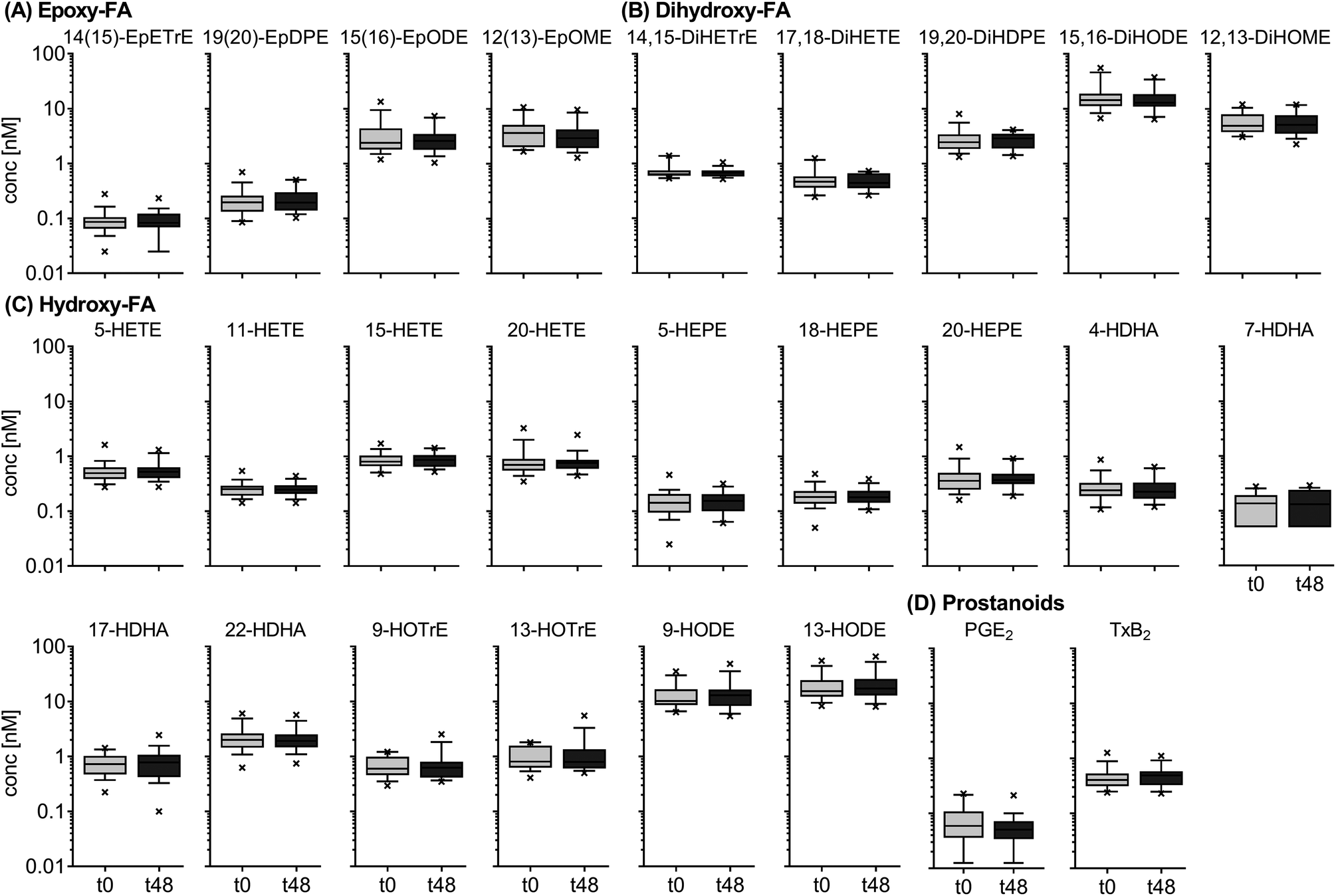 | ||
| Fig. 2 Inter-day variation of circulating oxylipins in subjects on a non-standardized diet. Shown are concentrations ± SEM of selected epoxy-FA (A), dihydroxy-FA (B), hydroxy-FA (C) and prostanoids (D) (n = 18). Plasma was collected at baseline (t0) and after 48 h (t48). No intervention was carried out. | ||
The results for the inter-day variation of oxylipins in fasting plasma samples of subjects on a standardized diet can be found for representative oxylipins in Fig. 3 (all data in ESI Table S6†). Variation of most analytes was within the analytical fluctuation and plasma concentrations remained constant during the observation period (ESI Table S6†). However, the concentration of few hydroxy-PUFAs significantly decreased during the observation period for a standardized diet, i.e. 5-HETE, 8-HETE, 5-HEPE, 4-HDHA, 8-HDHA, 10-HDHA and 17-HDHA. The concentration of 5(R,S)-5-F2t-IsoP, 15-HETE, 20-HDHA and 8,9-DiHETrE also decreased significantly; however, the changes were within the analytical fluctuation. Although not statistically significant, differences for some analytes were higher than expected from the analytical fluctuation, e.g. 12-HHTrE, 10(11)-EpDPE, 11-HETE, 20-HETE, 18-HEPE and 8,9-DiHETE.
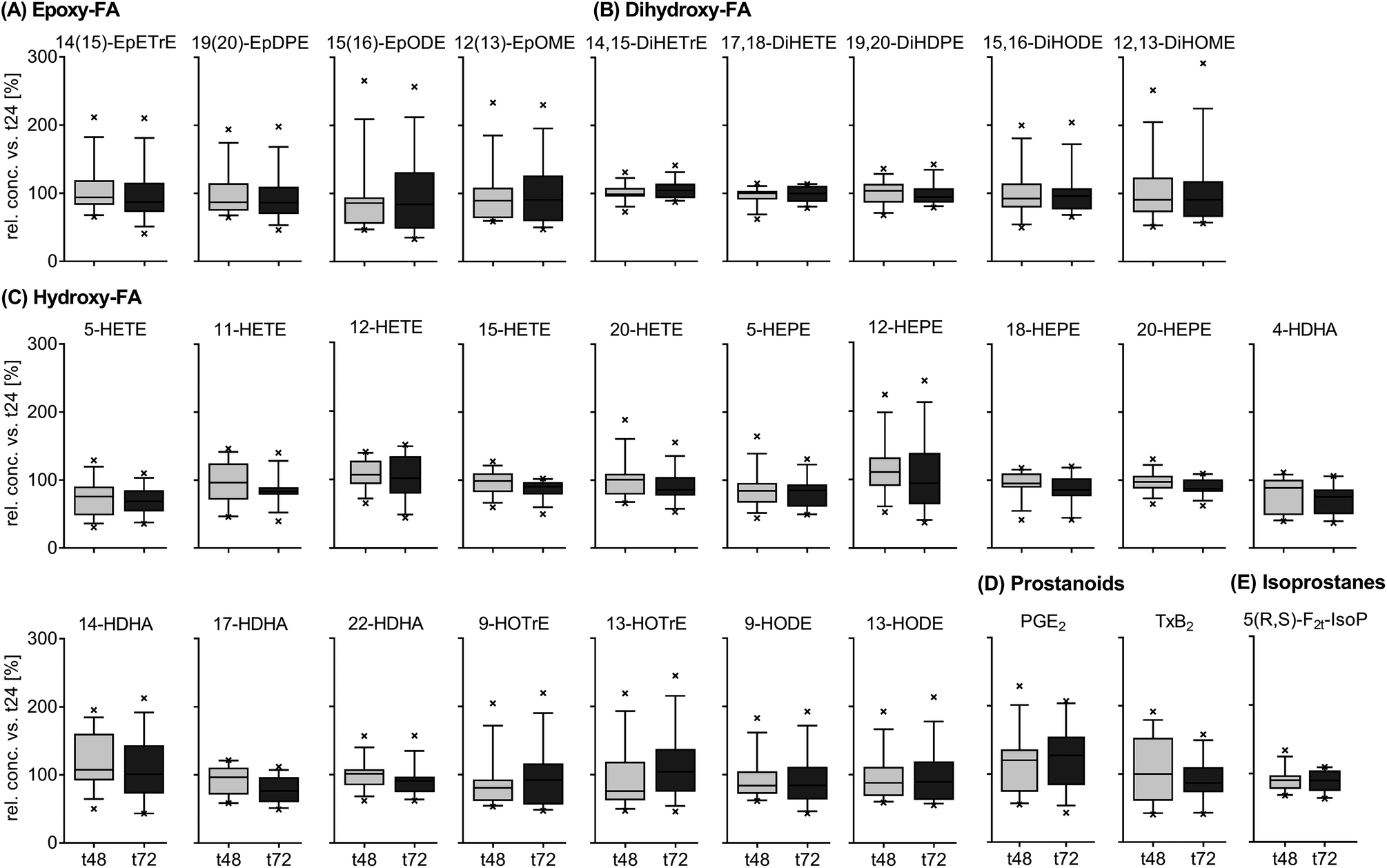 | ||
| Fig. 3 Inter-day variation of circulating oxylipins in subjects on a standardized diet. Shown are means of the relative inter-day change within each subject ± SEM of selected epoxy-FA (A), dihydroxy-FA (B), hydroxy-FA (C), prostanoids (D) and isoprostanes (E) (n = 13). Plasma was collected from study participants on a standardized diet at t24, t48 and t72 and the relative concentrations of oxylipins at t48 and t72 were calculated against t24. | ||
The relative concentrations of representative oxylipins within a 48 hour time interval in both studies (t0 vs. t48 for Study A and t24 vs. t72 for study B) are shown in ESI Fig. S3.† This direct comparison revealed similar inter-day differences in oxylipin concentration in both studies (non-standardized vs. standardized diet). However, the variation in relative concentrations of some analytes was higher in Study A, e.g. 12(13)-EpOME, 5-HEPE or PGE2.
Intra-day variation of free oxylipins in (non-fasting) plasma samples
The results for the intra-day variation can be found for selected oxylipins in Fig. 4 (means, all data in ESI Table S7†) as well as Fig. S4† (individual data values). Plasma oxylipin concentrations were subject to large fluctuations throughout the day (t0 (following overnight fasting) to t8) with significant differences for almost all analytes (ESI Table S7†). Most metabolites from the individual PUFAs showed similar trends during the observation period. Moreover, trends for ARA, EPA and DHA derived metabolites were comparable (except ARA prostanoids increasing from t0 to t8): starting from the highest concentration at t0, concentrations were decreased to a minimum at t2 and increased again at t4. At t6 and t8, concentrations were decreased compared to t4, for some analytes to a similar level compared to t2 (e.g. dihydroxy-PUFAs, 5-HETE, 5-HEPE and 4-HDHA, Fig. 4). In an analogous manner, trends for LA and ALA derived analytes were similar: an increase in analyte concentrations up to t4 with a following decrease. The lowest concentrations were observed at t8 (e.g. epoxy- and hydroxy-PUFAs, Fig. 4). The largest intra-day variations were observed for different LA and ALA metabolites (up to 400% at t4 compared to t0).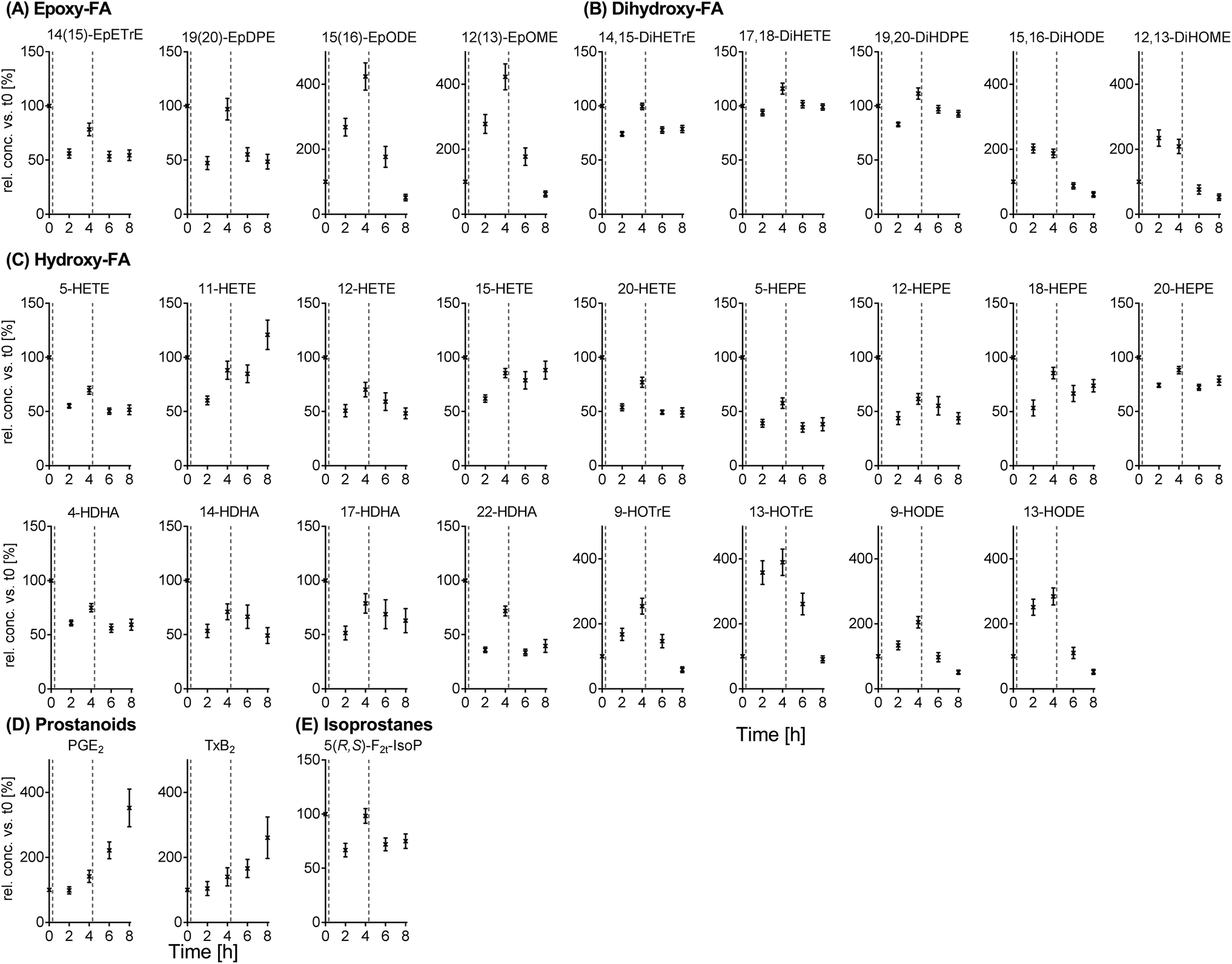 | ||
| Fig. 4 Intra-day, intra-person variation of circulating oxylipins in subjects on a standardized diet. Shown are means of the relative intra-day change within each subject ± SEM of epoxy-FA (A), dihydroxy-FA (B), hydroxy-FA (C), prostanoids (D) and isoprostanes (E) (n = 13). Plasma was collected from study participants on a standardized diet at baseline (t0) and t2, t4, t6 and t8. The relative concentrations of oxylipins were calculated against the baseline. The dotted lines in the diagrams indicate food intake (20 min past sample collection at t0 and t4). | ||
4 Discussion
The physiological effects of oxylipins are diverse and range, for example, from important roles in the regulation of fever and inflammation (e.g. PGE2) to anti-arrhythmic (e.g. 17(18)-EpETE) and anti-angiogenic effects (e.g. 19(20)-EpDPE). However, due to crosstalk between the different pathways of the ARA cascade, physiological effects result more from changes in the whole product pattern rather than from changes in individual mediators.36 In order to investigate and understand oxylipin biology, exact and reliable quantification of a comprehensive pattern of oxylipins is crucial. The pattern of oxylipins can reflect the pathophysiology of diseases,21–23 which may allow using oxylipins as biomarkers of the disease state. Moreover, drugs37 as well as the diet24,25,38 modulate the oxylipin pattern, which is an important factor for their effect on physiology and health. However, biological, time-dependent variations in the oxylipin pattern in healthy individuals have to be kept in mind when interpreting biological data. Therefore, the aim of the present study was to characterize inter- and intra-day variations of the oxylipin profile as an important basis for the interpretation of biological effects.The precision of the analytical method was assessed in QC plasma samples (different aliquots of the same sample). Oxylipin concentrations in the QC samples were in the same range as in the plasma samples of both studies. Most oxylipins (about 84% of 73 analytes) showed a fluctuation of less than 20% within the batch; only 12 analytes showed a higher fluctuation. The variations observed here were comparable to or lower than those previously described for other LC-MS based analytical approaches for the quantification of oxylipins.39,40 It is not surprising that the degree of variation decreased with increasing ratio of concentration to LLOQ, i.e. in general with higher levels in plasma. The minimum variation was ∼5%, which is consistent with the expected (random) relative error of LC(-MS) methods.41 For the interpretation of biological data it is important to keep this analytical variation in mind since high variations might mask biological effects. Therefore, it is crucial to have a well-characterized quantification method to reliably determine endogenous concentrations.
In most studies investigating the biology of oxylipins, fasting blood is used and is usually collected in the morning following overnight fasting.24,26,27,32 Therefore, another crucial parameter for the interpretation of biological effects is the inter-day variability (i.e. from one morning to the next) in the oxylipin profile of a healthy individual. This background variation should be reduced as much as possible in order to allow a better evaluation of biologically relevant effects, e.g. in the course of diseases or during pharmacological intervention. Our results support the suitability of fasting plasma for the investigation of biological effects since inter-day variations were low for oxylipins from all chemical groups in both studies (non-standardized and standardized diet). Except for a few analytes (mainly LA and ALA metabolites), the variation of all oxylipins in the study population on a non-standardized diet was comparable to the analytical variance, indicating stable levels of oxylipins in a healthy person as expected for a homeostasis of lipid mediators. For a standardized diet, the variations observed in the oxylipin profile were slightly more pronounced and often higher than expected from the analytical method. Comparing absolute concentrations between t24, t48 and t72, a slight negative trend could be observed which was more pronounced when baseline concentrations (t0) were taken into account. This decrease might be a result of changes in dietary PUFA intake with the standardized nutrition (which started at lunch the day before baseline blood sampling). For a western diet the consumption of (n6-)PUFA – especially LA – is higher compared to the standardized diet.42 Moreover, it has been previously shown for increases of LA in plasma lipids of subjects changing from a low- to a high-PUFA diet43 that – depending on the lipid class – most changes occurred during the first five days for the high-PUFA diet.
Nevertheless, it can be summarized that inter-day variations in the oxylipin profile of fasting plasma in healthy human subjects are small, which is a prerequisite for the investigation of oxylipin biology in intervention studies. However, the study population of male subjects has been well-characterized and further studies have to show to what extent different lifestyles, physical activity, or the menstrual cycle might influence the oxylipin profile.
The postprandial state differs substantially from the fasting state regarding blood – and thus plasma – lipid composition and fatty acid metabolism. Taking into account that the postprandial state is more representative for the plasma composition during the day for individuals of western countries compared to the fasting state, the question arises whether postprandial plasma might also be a suitable matrix for the analysis of the oxylipin profile. Following a meal, fatty acids are absorbed and reach the blood via lipoproteins, i.e. chylomicrons, resulting in a peak of plasma TGs, which is accompanied by elevated lipolysis.44 Thus, it is not surprising that the intake of fat (within a meal) resulted in remarkable changes in the oxylipin profile; LA and ALA metabolites showed the highest changes throughout the day with a maximum at 4 h post breakfast, which was also reflected in mean plasma TGs (individual maxima at 2–6 h). In a previous study with a (breakfast) meal containing more fat compared to our study (1 g fat per kg body weight (BW) vs. ∼0.44 g kg−1 BW) plasma TGs reached a maximum after 4 h,45 while in another study with a similar fat content compared to our breakfast (∼0.56 g kg−1 BW) most individuals showed two maxima in plasma TGs: the first between 1 and 3 h and the second between 4 and 7 h post ingestion.46 In contrast to breakfast, the lunch meal did not show a marked effect on the profile of LA and ALA derived oxylipins, which might be explained by the higher fat content of the breakfast (36.6 g total fat, 17.8 g saturated fatty acids (SFAs), and 8.68 g monounsaturated fatty acids (MUFAs)) in comparison to the lunch meal (6.8 g total fat, 2.71 g SFAs, and 1.24 g MUFAs). Moreover, the gastric emptying of the more complex lunch meal may occur later and slower because of the higher content in vegetables (fiber) and proteins.
Kardinaal et al. found an increased level of different eicosanoids, i.e. 11- and 12-HETE and 19,20-DiHDPE as well as 8,9- and 11,12-DiHETrE following a high fat challenge (milk shake (∼500 mL) containing 16 g fat/100 g milk shake).47 Strassburg et al. found that a high fat shake (95 g fat) with high SFA content (51 g; and 6 g PUFA (no ALA, EPA or DHA)) increased LA derived oxylipins involved in the LOX pathway while high MUFA (79 g; and 8 g PUFA (no ALA, EPA or DHA)) content led to increased LA derived oxylipins derived from the CYP450 pathway.38 Although a similar differentiation was not possible in our study because of the fat composition of the breakfast, our observations are still in line with those from Strassburg et al.38 since LA oxylipin concentrations from both the LOX as well as the CYP pathway were elevated. However, although increased in the present study, the concentrations of ALA derived oxylipins were not affected by the high fat/high SFA or high fat/high MUFA shakes, which might be a result of the higher ALA content of the meals in comparison to the shakes.
In contrast to LA and ALA metabolites, the concentration of oxylipins derived from ARA, EPA and DHA – with only a few exceptions – decreased 2 h post ingestion, which was accompanied by an increase at t4. The initial decrease – which is in contrast to the observed increase in LA and ALA metabolites – might be a result of the small amount of these PUFAs in the meals and has been observed previously following a meal challenge with low PUFA content.38,39 Moreover, this time course in the concentrations may be explained by changing insulin concentrations as previously discussed by Strassburg et al.:38 a postprandial increase of insulin concentrations is associated with reduced lipolysis and therefore decreased oxylipin concentrations at t2. Decreasing insulin concentrations following t2 may lead to a release of fatty acids and corresponding oxylipin formation at t4.48 A similar decrease – as observed after breakfast – was found following the lunch meal; however, oxylipin concentrations did not increase 4 h post lunch.
Interestingly, the prostanoids PGE2 and TxB2 showed a different course compared to other oxylipins since both increased (almost linearly) over all non-fasting blood samplings from the baseline to t8. PGE2 and TxA2 (the biologically active precursor of TxB2) are important metabolites derived from ARA and are involved in inflammatory responses by regulation of pain and fever (PGE2) or platelet aggregation (TxA2).1 It has been discussed before that a high fat meal might induce postprandial inflammation.49 Regarding changes in pro-inflammatory oxylipins, previous results have been ambiguous38,39,50 while our results clearly show increased concentrations of PGE2 and TxB2 during the day. This increase, however, could not be associated with the time of meal ingestion (as observed for other oxylipin classes). Moreover, at the fasting time points following t8, i.e. t24, t48 and t72, both analytes were found in plasma with similar concentrations as compared to t8. Thus, instead of being associated with the postprandial state, the observed increase in prostanoids PGE2 and TxB2 could be a result of increasing stress levels in the participants due to the number of blood samplings during the study.
In line with previous results,38 our results indicate that intake of dietary fat and its fatty acid composition influence the oxylipin profile. However, it has to be determined if they act as direct precursors for the synthesis of oxylipins or if they indirectly activate intermediate pathways that may lead to oxylipin formation or their release.51 Moreover, further research is needed to investigate whether postprandial plasma might be useful for the investigation of oxylipin patterns, for example, for the identification of biomarkers of diseases or the efficacy of pharmaceutical drugs. Although the pattern of oxylipins is subject to changes induced by ingestion of fat, this diet induced modulation of the oxylipin profile might be reproducible as previously shown for the individual intra-day response of different plasma fatty acids in lipid classes to the same standardized meal.52
5 Conclusion
Overall, our data demonstrate the suitability of fasting plasma for the investigation of the biological role of the oxylipin pattern. Background variations are low, for most analytes within the variation of the analytical method and independent of a standardization of the diet. This makes fasting plasma an ideal matrix for the investigation of oxylipins in pathophysiological states and may allow the identification of biomarkers as well as evaluating the modulation of oxylipin formation by pharmaceuticals, food ingredients and diet.In non-fasting plasma, oxylipin concentrations fluctuated strongly over the day and ingestion of food was followed by changes in the oxylipin profile. Since in western countries individuals are in a postprandial state during most of the day it would be highly interesting to investigate whether a standardized meal might result in similar modifications of the oxylipin profile and if postprandial plasma might be suitable for the investigation of oxylipin biology.
Funding
This study was supported by grants from the German Research Foundation (Grant SCHE 1801 and SCHU 2516) to NHS and JPS.Conflicts of interest
The authors declare that they have no conflicts of interest.Abbreviations
| ALA | Alpha-linolenic acid (C18:3n3) |
| ARA | Arachidonic acid (C20:4n6) |
| BW | Body weight |
| COX | Cyclooxygenase |
| CYP | Cytochrome P450 |
| DHA | Docosahexaenoic acid (C22:6n3) |
| DiHETE | Dihydroxy eicosatetraenoic acid |
| DiHETrE | Dihydroxy eicosatrienoic acid |
| DiHODE | Dihydroxy octadecadienoic acid |
| EPA | Eicosapentaenoic acid (C20:5n3) |
| EpDPE | Epoxy docosapentaenoic acid |
| EpETE | Epoxy eicosatetraenoic acid |
| EpETrE | Epoxy eicosatrienoic acid |
| FAMEs | Fatty acid methyl esters |
| GC-FID | Gas chromatography with flame ionization detection |
| HDHA | Hydroxy docosahexaenoic acid |
| HEPE | Hydroxy eicosapentaenoic acid |
| HETE | Hydroxy eicosatetraenoic acid |
| HODE | Hydroxy octadecadienoic acid |
| IS | Internal standard |
| IsoP | Isoprostanes |
| LA | Linoleic acid (C18:2n6) |
| LC-MS | Liquid chromatography-mass spectrometry |
| LLOQ | Lower limit of quantification |
| LOX | Lipoxygenase |
| MUFA | Monounsaturated fatty acid |
| n3/6 | Omega 3/6 |
| PG | Prostaglandin |
| PUFA | Polyunsaturated fatty acid |
| QC | Quality control |
| SD | Standard deviation |
| sEH | Soluble epoxide hydrolase |
| SFA | Saturated fatty acid |
| TG | Triglyceride |
| Tx | Thromboxane |
Acknowledgements
We would like to thank Vanessa Christa for helping with the fatty acid analyses in food samples and the participants who contributed their time to this project.References
- C. D. Funk, Science, 2001, 294, 1871–1875 CrossRef CAS PubMed.
- C. N. Serhan, Nature, 2014, 510, 92–101 CrossRef CAS PubMed.
- C. Arnold, M. Markovic, K. Blossey, G. Wallukat, R. Fischer, R. Dechend, A. Konkel, C. von Schacky, F. C. Luft, D. N. Muller, M. Rothe and W.-H. Schunck, J. Biol. Chem., 2010, 285, 32720–32733 CrossRef CAS PubMed.
- M. Gabbs, S. Leng, J. G. Devassy, M. Monirujjaman and H. M. Aukema, Adv. Nutr., 2015, 6, 513–540 CrossRef CAS PubMed.
- W.-H. Schunck, A. Konkel, R. Fischer and K.-H. Weylandt, Pharmacol. Ther., 2018, 183, 177–204 CrossRef CAS PubMed.
- W. Wang, J. Zhu, F. Lyu, D. Panigrahy, K. W. Ferrara, B. Hammock and G. Zhang, Prostaglandins Other Lipid Mediators, 2014, 113–115, 13–20 CrossRef CAS PubMed.
- G. Zhang, D. Panigrahy, L. M. Mahakian, J. Yang, J.-Y. Liu, K. S. Stephen Lee, H. I. Wettersten, A. Ulu, X. Hu, S. Tam, S. H. Hwang, E. S. Ingham, M. W. Kieran, R. H. Weiss, K. W. Ferrara and B. D. Hammock, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6530–6535 CrossRef CAS PubMed.
- C. Morisseau and B. D. Hammock, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 37–58 CrossRef CAS PubMed.
- G. L. Milne, Q. Dai and L. J. Roberts, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2015, 1851, 433–445 CrossRef CAS PubMed.
- H. Yin, L. Xu and N. A. Porter, Chem. Rev., 2011, 111, 5944–5972 CrossRef CAS PubMed.
- C. C. Berthelot, S. G. Kamita, R. Sacchi, J. Yang, M. L. Nording, K. Georgi, C. Hegedus Karbowski, J. B. German, R. H. Weiss, R. J. Hogg, B. D. Hammock and A. M. Zivkovic, PLoS One, 2015, 10, e0144996 CrossRef PubMed.
- C. B. Stephensen, P. Armstrong, J. W. Newman, T. L. Pedersen, J. Legault, G. U. Schuster, D. Kelley, S. Vikman, J. Hartiala, R. Nassir, M. F. Seldin and H. Allayee, J. Lipid Res., 2011, 52, 991–1003 CrossRef CAS PubMed.
- B. N. M. Zordoky and A. O. S. El-Kadi, Pharmacol. Ther., 2010, 125, 446–463 CrossRef CAS PubMed.
- S. P. B. Caligiuri, H. M. Aukema, A. Ravandi and G. N. Pierce, Exp. Gerontol., 2014, 59, 51–57 CrossRef CAS PubMed.
- M. Plourde, R. Chouinard-Watkins, M. Vandal, Y. Zhang, P. Lawrence, J. T. Brenna and S. C. Cunnane, Nutr. Metab. (Lond), 2011, 8, 5 CrossRef CAS PubMed.
- M. A. Zulyniak, K. Roke, C. Gerling, S. L. Logan, L. L. Spriet and D. M. Mutch, Mol. Nutr. Food Res., 2016, 60, 631–641 CrossRef CAS PubMed.
- E. J. Giltay, L. J. Gooren, A. W. Toorians, M. B. Katan and P. L. Zock, Am. J. Clin. Nutr., 2004, 80, 1167–1174 CrossRef CAS PubMed.
- A. H. Metherel, J. M. Armstrong, A. C. Patterson and K. D. Stark, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2009, 81, 23–29 CrossRef CAS PubMed.
- K. D. Stark and B. J. Holub, Am. J. Clin. Nutr., 2004, 79, 765–773 CrossRef CAS PubMed.
- R. M. Giordano, J. W. Newman, T. L. Pedersen, M. I. Ramos and C. L. Stebbins, Int. J. Sport Nutr. Exercise Metab., 2011, 21, 471 CrossRef CAS.
- S. L. Lundström, B. Levänen, M. Nording, A. Klepczynska-Nyström, M. Sköld, J. Z. Haeggström, J. Grunewald, M. Svartengren, B. D. Hammock, B.-M. Larsson, A. Eklund, Å. M. Wheelock and C. E. Wheelock, PLoS One, 2011, 6, e23864 CrossRef PubMed.
- M. Monirujjaman, J. G. Devassy, T. Yamaguchi, N. Sidhu, M. Kugita, M. Gabbs, S. Nagao, J. Zhou, A. Ravandi and H. M. Aukema, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2017, 1862, 1562–1574 CrossRef CAS PubMed.
- A. M. Zivkovic, N. Telis, J. B. German and B. D. Hammock, Calif. Agric., 2011, 65, 106–111 CrossRef PubMed.
- T. Greupner, L. Kutzner, F. Nolte, A. Strangmann, H. Kohrs, A. Hahn, N. H. Schebb and J. P. Schuchardt, Food Funct., 2018, 9, 1587–1600 RSC.
- A. I. Ostermann and N. H. Schebb, Food Funct., 2017, 8, 2355–2367 RSC.
- A. H. Keenan, T. L. Pedersen, K. Fillaus, M. K. Larson, G. C. Shearer and J. W. Newman, J. Lipid Res., 2012, 53, 1662–1669 CrossRef CAS PubMed.
- M. L. Nording, J. Yang, K. Georgi, C. Hegedus Karbowski, J. B. German, R. H. Weiss, R. J. Hogg, J. Trygg, B. D. Hammock and A. M. Zivkovic, PLoS One, 2013, 8, e76575 CrossRef CAS PubMed.
- J. P. Schuchardt, I. Schneider, I. Willenberg, J. Yang, B. D. Hammock, A. Hahn and N. H. Schebb, Prostaglandins Other Lipid Mediators, 2014, 109–111, 23–31 CrossRef CAS PubMed.
- A. J. Watkins, S. Sirovica, B. Stokes, M. Isaacs, O. Addison and R. A. Martin, Biochim. Biophys. Acta, Mol. Basis Dis., 2017, 1863, 1371–1381 CrossRef CAS PubMed.
- I. Willenberg, A. I. Ostermann and N. H. Schebb, Anal. Bioanal. Chem., 2015, 407, 2675–2683 CrossRef CAS PubMed.
- J. P. Schuchardt, S. Schmidt, G. Kressel, I. Willenberg, B. D. Hammock, A. Hahn and N. H. Schebb, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2014, 90, 27–37 CrossRef CAS PubMed.
- J. P. Schuchardt, A. I. Ostermann, L. Stork, S. Fritzsch, H. Kohrs, T. Greupner, A. Hahn and N. H. Schebb, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2017, 121, 76–87 CrossRef CAS PubMed.
- E. Schulte, Dtsch. Lebensm.-Rundsch., 2004, 100, 188–189 CAS.
- A. I. Ostermann, M. Müller, I. Willenberg and N. H. Schebb, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2014, 91, 235–241 CrossRef CAS PubMed.
- K. M. Rund, A. I. Ostermann, L. Kutzner, J.-M. Galano, C. Oger, C. Vigor, S. Wecklein, N. Seiwert, T. Durand and N. H. Schebb, Anal. Chim. Acta DOI:10.1016/j.aca.2017.11.002.
- J. Yang, K. Schmelzer, K. Georgi and B. D. Hammock, Anal. Chem., 2009, 81, 8085–8093 CrossRef CAS PubMed.
- H. Gottschall, C. Schmoecker, D. Hartmann, N. Rohwer, K. Rund, L. Kutzner, F. Nolte, A. I. Ostermann, N. H. Schebb and K. H. Weylandt, J. Lipid Res., 2018, 59, 864–871 CrossRef CAS PubMed.
- K. Strassburg, D. Esser, R. J. Vreeken, T. Hankemeier, M. Müller, J. Duynhoven, J. Golde, S. J. Dijk, L. A. Afman and D. M. Jacobs, Mol. Nutr. Food Res., 2014, 58, 591–600 CrossRef CAS PubMed.
- S. Gouveia-Figueira, J. Späth, A. M. Zivkovic and M. L. Nording, PLoS One, 2015, 10, e0132042 CrossRef PubMed.
- K. Strassburg, A. M. L. Huijbrechts, K. A. Kortekaas, J. H. Lindeman, T. L. Pedersen, A. Dane, R. Berger, A. Brenkman, T. Hankemeier, J. van Duynhoven, E. Kalkhoven, J. W. Newman and R. J. Vreeken, Anal. Bioanal. Chem., 2012, 404, 1413–1426 CrossRef CAS PubMed.
- G. D. Christian, Analytical Chemistry, Wiley India Pvt. Limited, 6th edn, 2007 Search PubMed.
- S. Raatz, Z. Conrad, L. Johnson, M. Picklo and L. Jahns, Nutrients, 2017, 9, 438 CrossRef PubMed.
- C. M. Skeaff, L. Hodson and J. E. McKenzie, J. Nutr., 2006, 136, 565–569 CrossRef CAS PubMed.
- R. J. Havel, Proc. Nutr. Soc., 1997, 56, 659–666 CrossRef CAS PubMed.
- T. Tholstrup, B. Sandström, A. Bysted and G. Hølmer, Am. J. Clin. Nutr., 2001, 73, 198–208 CrossRef CAS PubMed.
- J. M. Olefsky, P. Crapo and G. M. Reaven, Am. J. Clin. Nutr., 1976, 29, 535–539 CrossRef CAS PubMed.
- A. F. M. Kardinaal, M. J. van Erk, A. E. Dutman, J. H. M. Stroeve, E. van de Steeg, S. Bijlsma, T. Kooistra, B. van Ommen and S. Wopereis, FASEB J., 2015, 29, 4600–4613 CrossRef CAS PubMed.
- K. Jelic, C. E. Hallgreen and M. Colding-Jørgensen, Ann. Biomed. Eng., 2009, 37, 1897–1909 CrossRef PubMed.
- M. Herieka and C. Erridge, Mol. Nutr. Food Res., 2014, 58, 136–146 CrossRef CAS PubMed.
- A. M. Wolfer, M. Gaudin, S. D. Taylor-Robinson, E. Holmes and J. K. Nicholson, Anal. Chem., 2015, 87, 11721–11731 CrossRef CAS PubMed.
- G. C. Shearer and J. W. Newman, Curr. Atheroscler. Rep., 2009, 11, 403–410 CrossRef CAS PubMed.
- A. M. Zivkovic, M. M. Wiest, U. Nguyen, M. L. Nording, S. M. Watkins and J. B. German, Metabolomics, 2009, 5, 209–218 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ay01753k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |