
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple lateral flow assays for microbial detection in stool
Wendy A.
Henderson†
 *a,
Lichen
Xiang†
ab,
Nicolaas H.
Fourie
a,
Sarah K.
Abey
a,
Eric G.
Ferguson
a,
Ana F.
Diallo
a,
Natnael D.
Kenea
a and
Chang Hee
Kim
*b
*a,
Lichen
Xiang†
ab,
Nicolaas H.
Fourie
a,
Sarah K.
Abey
a,
Eric G.
Ferguson
a,
Ana F.
Diallo
a,
Natnael D.
Kenea
a and
Chang Hee
Kim
*b
aDivision of Intramural Research, National Institute of Nursing Research, National Institutes of Health, Department of Health and Human Services, Bethesda, MD 20892, USA. E-mail: hendersw@mail.nih.gov
bGoDx, 510 Charmany Drive, Suite 257, Madison, WI 53719, USA. E-mail: chang@godiagnostic.com
First published on 20th September 2018
Abstract
Diarrheal diseases claim the lives of 1300 children daily, mostly in the developing world. We have developed a simple lateral flow assay capable of detecting E. coli and EPEC DNA and RNA rapidly (<15 minutes) at the point-of-need, directly from stool without nucleic acid extraction or molecular amplification. The limit of detection of the method is 1 nM using synthetic DNA target substrates spiked into stool. However, due to the endogenous amplification of the 23S rRNA targets, we were able to detect the endogenous EPEC in pea-sized (5 mg) stool without labor-intensive and time-consuming nucleic acid purification or target amplification using enzymes. The significance of this method is that it is rapid (<15 minutes) and simple (without nucleic acid purification or molecular amplification) and does not require instrumentation, or access to a laboratory, cold chain or electric power. Thus, it is well-suited for point-of-need use in remote and/or resource-limited settings in the developing world where the mortality due to diarrheal diseases is especially high. The rapid testing of stool pathogens in real time at the point-of-need will decrease the loss of patients to follow-up, and enable patients to be treated earlier with the appropriate therapeutics in both the developed and developing world settings.
Introduction
Diarrheal diseases are one of the leading causes of mortality and morbidity worldwide. Approximately 1300 diarrhea related deaths occur every day, in children under the age of 5.1 A majority of the burden is on populations where pathogen transmission occurs mainly through overcrowding, poor sanitation, limited access to clean water and inadequate food hygiene.2 South Asia and Sub-Saharan Africa account for 90% of the total childhood mortality rate due to diarrhea.3 Identification of the pathogens that cause diarrheal disease is one of the proven ways to save lives.4 However, the diagnosis and treatment of diarrheal diseases in resource-limited settings are mainly based on patient or caregiver reported clinical symptoms, which makes it often inaccurate and causes preventable medical complications and unnecessary use of broad spectrum antibiotics.5In the United States (US), the burden of diarrheal diseases remains significant, mainly due to mass production and distribution of food, long-term care, child daycare and municipal water systems.6 Approximately 179 million diarrhea related diseases are reported each year in the US resulting in 473![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 832 hospitalizations and 5072 deaths.7 Underserved US populations (e.g., economically depressed communities), and vulnerable populations such as the immunocompromised, the very young, the elderly, and those in palliative care are particularly at risk of diarrheal diseases, and can benefit from improved, rapid and affordable pathogen detection.
832 hospitalizations and 5072 deaths.7 Underserved US populations (e.g., economically depressed communities), and vulnerable populations such as the immunocompromised, the very young, the elderly, and those in palliative care are particularly at risk of diarrheal diseases, and can benefit from improved, rapid and affordable pathogen detection.
Accurate point-of-need detection of pathogens responsible for diarrheal diseases (and other GI symptoms), leading to early detection and early intervention, is imperative in mitigating the burden of GI symptoms and infections on morbidity and mortality. Empowering healthcare providers with less costly, simple tools is key to reaching this goal. The current gold standard in enteropathogen testing involves stool enteric pathogen culture, immunological identification, and/or multiplexed PCR laboratory tests to identify the presence of an array of bacterial and viral enteropathogens.8 The Department of Laboratory Medicine at the National Institutes of Health (NIH) Clinical Center has recently switched from culture-based methods to multiplexed PCR based methods as their standard of care in identifying enteric pathogens from stool. PCR based simultaneous testing for multiple pathogens is both sensitive and accurate.8,9 However, these methods are expensive and require highly skilled technicians and dedicated laboratory resources with overhead costs.10 Even in the developed world, the multiplexed PCR tests are laboratory tests that require the shipping of samples from outpatient primary or tertiary healthcare providers to laboratories where instrumentation and technicians are available. This causes delays in diagnosis, infection control, and treatment and loss of patients to follow-up. In addition, the conventional methods for identification of gastrointestinal pathogens may not be readily available to healthcare providers in limited resource environments and may constitute an unnecessary financial burden for testing of GI complaints, specifically diarrhea.
Nanoparticle based lateral flow assays (LFAs) offer a unique solution for point-of-need pathogen detection tests. LFA is a paper-based technology offering a low-cost, simple, rapid and user-friendly detection test device that requires no electricity or instrumentation. Antibody-based LFAs have been developed for the detection of a variety of microorganisms (e.g., viruses – HIV-1, bacteria – Mycobacterium tuberculosis, parasites – Plasmodium falciparum, Listeria) in blood,11 urine,12 food,13–17 water18 and other substrates.19,20 However, they exhibit low sensitivity (50–75%).21
Nucleic acid based LFAs (NALFAs) use DNA conjugated nanoparticles (often gold, e.g., DNA–AuNPs) as a detection probe, which hybridizes to a species or strain specific complementary target sequence.22,23 The hybridization produces a colorimetric signal indicating the detection of the target DNA sequence. The intense red color of DNA–AuNPs provides a platform for colorimetric detection using AuNPs as a signal label to trace hybridization of the target DNA.24 Recently, CRISPR-Cas has been used in conjunction with isothermal amplification of DNA to detect DNA on paper strip lateral flow assays. This technology may simplify sample preparation and increase the sensitivity of DNA detection on lateral flow strips by sequence-specific cleavage of reporter groups from amplified DNA. However, these methods require enzymatic processing and added time (over 1.5 hours).25–28
In this study, we report the development of a simple paper strip lateral flow assay for the detection of Escherichia coli (E. coli) nucleic acids in human stool, using 23S rDNA–AuNP conjugated probes, which requires minimal sample preparation and no nucleic acid amplification. The visual identification of colorimetric signals from the E. coli DNA in clinical fecal samples was possible without the use of PCR or any laboratory equipment such as microscopes. The point-of-need test that we have developed combines the ease of use of the rapid immunoassays with the high sensitivity of molecular assays.
Experimental
Reagents
The biotinylated DNA capture and control probes, streptavidin, TCEP and colloidal Au-NPs were purchased from IDT (Coralville, IA, USA), Fisher Scientific (Hanover Park, IL, USA), Thermo Fisher (Rockford, IL, USA), and Millipore-Sigma (St. Louis, MO, USA), respectively. The average diameter of the Au-NPs used in this study is 15 nm. The buffers and stock solution were prepared using RNAse/DNAse free water, purchased from Qiagen (Valencia, CA, USA). Detergent Tween 20 was purchased from Millipore-Sigma (St. Louis, MO, USA). The nitrocellulose membrane, glass fiber and reagent absorption pad were purchased from Millipore (Billerica, MA, USA).The 23S rDNA sequences were obtained from the Microbes database of the National Center for Biotechnology Information (NCBI). Detection and capture probes were designed from unique target sequences 55–60 nucleotides in length within NCBI accession number NC_018658.1 for E. coli O104:H4. Unique target sequences were selected based on the absence of matching alignments in them against other species within the same family of microbes29 and other known diarrhea-causing microbes in humans from an NCBI Microbes Blast search. For capture probes, a biotin moiety (5′Biosg, Integrated DNA Technologies, IDT) followed by eight adenosine residue (A8) spacers were added to the 5′ end. For the detection probes, eight adenosine residue (A8) spacers followed by dithiol phosphoramidites (DTPA, IDT) were added to the 3′ end. A positive control for detection probes was designed by adding eight adenosine residue (A8) spacers followed by a biotin moiety (3′Bio, IDT) to a sequence probe which aligns with detection probes in the opposite direction. A 7-nucleotide spacer was included between the detection and capture probes. The sequences of the probes are listed in Table 1.
| Oligo name | 5′ | Sequence | 3′ |
|---|---|---|---|
| a TS = target sequence; C = capture; probe D = detection probe; DC = positive control capture probe. | |||
| DC | GAG CGT TCT GTA AGC CTG CGA AAA AAA AA | Biotin | |
| C | Biotin | AA AAA AAA TAC CTC CAG CAT GCC TCA CAG | |
| D | TCG CAG GCT TAC AGA ACG CTC AAA AAA AA | Dithiol | |
| TS | GGG TAG GGG AGC GTT CTG TAA GCC TGC GAA GGT GTG CTG TGA GGC ATG CTG GAG GTA | ||
Preparation of thiol–DNA conjugated gold nanoparticles
Conjugation of the target DNA to the surface of the AuNPs was performed using the following procedure. Briefly, 1 μl of immobilized tris(2-carboxyethyl)phosphine (TCEP, Thermo Fisher Scientific, Rockford, IL USA) was added to 50 μl of the thiol-modified oligonucleotide for breakage of the disulfide bonds. The solution was lightly vortexed and incubated for 30 minutes at room temperature. The supernatant was then added to 1 ml of colloidal gold measuring 15 nm in diameter. The colloid solution was incubated for 16 hours at 4 °C before gradually adding phosphate buffered saline (PBS). The solution was centrifuged at 20000g for 15 minutes, the supernatant was removed and the DNA–AuNP conjugates were redispersed in 1 ml PBS. After a second washing procedure, DNA–AuNP conjugates were resuspended in 100 μl buffer containing 20 mM NaPO4, 5% BSA, 0.25% Tween® 20, and 10% sucrose and then stored at 4 °C.
Preparation of the lateral flow strips
A schematic description of the NALFA is presented in Fig. 1. The strip is about 6.0 cm long; its development involved a backing card (used to hold all the pads), a glass fiber conjugate pad (1.0 × 30 cm, GFCP203000, Millipore), a cellulose absorbent pad (CFSP223000), a nitrocellulose membrane (Millipore HFB 24004) containing a capture and a control region and a cellulose absorbent pad facilitating liquid flow. To prepare the capture and control probes, 10 μl of 1 nM biotinylated primers was added onto 38.3 μl of streptavidin–PBS solution and 41.7 μl of PBS. The mixture was incubated for 1 hour at room temperature and then 10 μl of ethanol was added for a final concentration of 100 μM for both the capture and control probes. Both capture and control probe solutions were manually applied to the nitrocellulose membrane in the capture and control regions, respectively. Using a micropipette, the AuNP conjugates were manually added to the conjugate pad and then the strips were incubated at −20 °C for 15 min before being stored in a dry place at room temperature.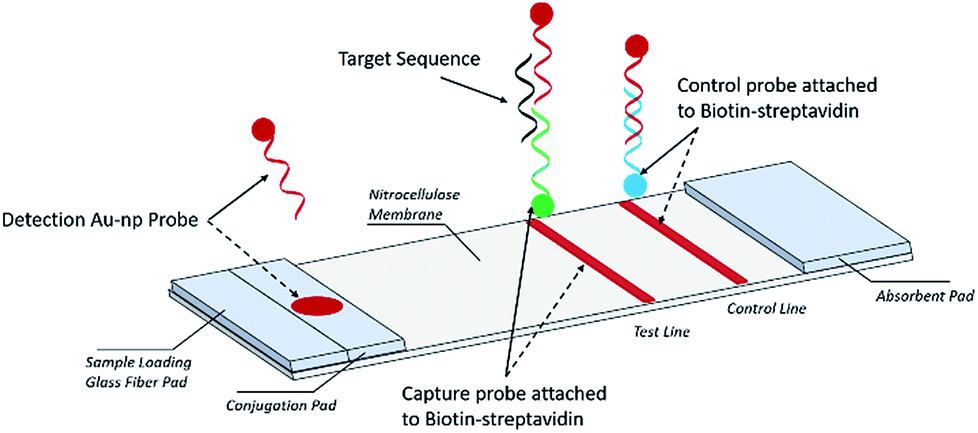 | ||
| Fig. 1 Schematic depicting the lateral flow assay. | ||
Stool samples
Stool samples were collected from participants who enrolled and consented to a natural history protocol at the National Institutes of Health Clinical Center (NCT00824941). After collection, a part of the sample was delivered to the research study lab stocked into a −80 °C freezer within 24 hours of sample collection. A sample confirmed to test positive via PCR with the standard clinical diagnostic laboratory methods for Enteropathogenic E. coli (EPEC) (National Institutes of Health, Department of Laboratory Medicine) was used. The limit of detection for the PCR standard clinical diagnostic laboratory method was 103 CFU per ml. A control sample, rigorously screened for the absence of reported infectious pathogens by Openbiome, a public stool bank, was used to test the LFA. For each sample, 60 mg of stool was diluted in 1 ml SSC buffer and manually mixed to give a homogeneous sample solution. In the control stool sample solution, the commercially purchased E. coli (2011C-3911) target sequence oligonucleotide was added at different concentrations (0, 1, 25, 50, 75 and 100 nM). All experiments utilizing human samples were approved for use by the Office of Human Subjects Research or the Institutional Review Board of the National Institutes of Health.Assay design
The nucleic acid lateral flow assay was designed to detect the target DNA or RNA sequences (e.g., E. coli) as shown in Fig. 1. A stool sample is mixed with buffer and added to the sample pad composed of glass fiber, with minimal sample preparation. If the target is present, it will hybridize to the detection probes conjugated with the AuNP label at the conjugate pad (Fig. 1). The target–detection probe hybrid would then migrate along the nitrocellulose pad to the test zone where it is captured by hybridization to a target capture probe and generates a red signal, detectable by the naked eye or a smartphone scanner. Detection probes that are not bound to the target sequence migrate further on the nitrocellulose pad and hybridize to the control capture probe immobilized on the control line, generating a red color at the control line.Assay procedure
A total of 85 μl of sample solution containing a desired concentration of target bacterial DNA was manually applied to the sample pad. Target DNA first hybridized with AuNP conjugated detection probes (AuNP–DP) to form a AuNP–DP–target DNA complex at the conjugate pad. The solution containing the AuNP–DP–target DNA complex migrated by capillary action, passing the glass fiber part of the strip into the capture or test region. At this point, a second hybridization took place between the AuNP–DP–target DNA complex and the capture probes immobilized on the test region, producing a red signal in the test region. In the absence of target DNA in the sample solution, no colored signal is detectable in the test region. The excess AuNP conjugated detection probes (AuNP–DP) then reached the control region where hybridization with the control probe occurred, producing a red signal. The red signal at the control zone indicates that the tool is working (Fig. 1). For the endogenous stool test, 50 mg of stool sample were weighed and then diluted with 1 ml 2% Tween 20 solution, following by vortexing to homogenize the stool. The EPEC detection probe, control probe, and capture probe were prepared via the same procedure as that described earlier. The DNA sequences of the EPEC probes are listed in Table 2. After the stool dilution and preparation of lateral flow strips, 80 μl nuclease free water, 1 mM target sequence oligonucleotides, and 50 mg ml−1 diluted stool were loaded onto the negative control, spike-in oligonucleotide, and endogenous stool strips, respectively. The lateral flow strip results were scanned with a Canon LiDE120 Color Image Scanner.| Oligo name | 3′ | Sequence | 5′ |
|---|---|---|---|
| a TS = target sequence; C = capture; probe D = detection probe; DC = positive control capture probe. | |||
| DC | CAG CCC GGA GGG CTG CAT TAC AAA AAA AA | Biotin | |
| C | Biotin | AA AAA AAA GCT CGG CTT TCA GCC CTC TTG | |
| D | GTA ATG CAG CCC TCC GGG CTG AAA AAA AA | S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S S |
|
| TS | GGA TCC CT CAG CCC GGA GGG CTG CAT TAC C CAA TAT CAA GAG GGC TGA AAG CCG AGC | ||
Quantitative analysis
The images of reacted strips were scanned with a Canon LiDE120 Color Image Scanner for further analysis of optical densities. The quantitative analysis of each test was performed using ImageJ open-source software. Analysis routines were used to obtain region-of-interest (ROI) outlines for each test area. The intensities within the ROIs were measured on the original 8-bit grayscale image to obtain the optical density of each test. The collected optical density data were graphed in Microsoft Excel and fit to a linear rise-to-maximum standard line with the equation f(x) = y − (ax + b). The R square values were calculated using the Excel program to represent the precision of the regression line.Results and discussion
Initial experiments with target sequence DNA oligonucleotides allowed for the estimation of the lower detection limits of the assay and the performance of the assay across a spectrum of target sequence concentrations (Fig. 2). As shown by our preliminary data, using different concentrations of synthetic DNA oligonucleotide E. coli targets (23S rDNA) spiked into PBS buffer, the signal from gold nanoparticles was linear (R2 = 0.969) in the range of 1 nM to 100 nM, and the limit of detection was 1 nM in 50 microliters of sample input (Fig. 2).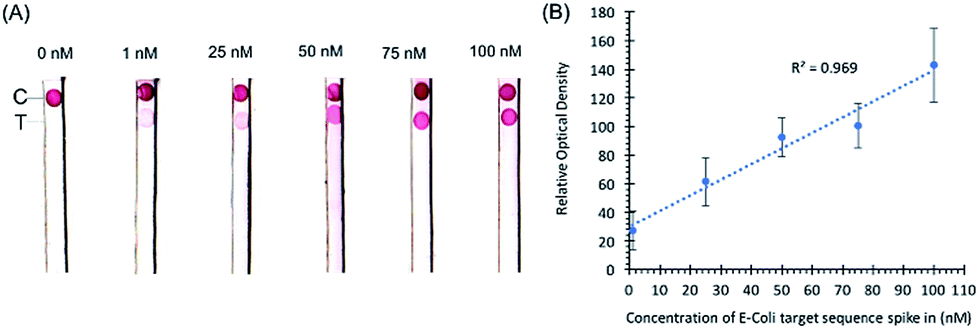 | ||
| Fig. 2 (A) Images of nitrocellulose diagnostic strips with different concentrations of E. coli target sequence oligonucleotides spiked into PBS samples. The concentrations of the spiked oligonucleotides are 1, 25, 50, 75, and 100 nM. (B) Statistical curve of the optical density of signals as a function of target sequence concentration. The error bars represent optical density standard deviations from three replicates of samples. The linear regression line follows the function f(x) = y − ax + b; the R square value of the regression equals 0.969. | ||
In a separate experiment, when a pea-sized stool sample (1 g) diluted in 1× PBS buffer and spiked-in with E. coli sequence-containing oligonucleotides was added to the sample loading glass fiber pad, the sample loading pad absorbed most of the impurities as shown by the clear nitrocellulose membrane in our preliminary data in Fig. 3. Any remaining impurities did not interfere with the hybridization of the probe conjugated with the AuNPs even with the capture probe at the control line. The spike-in experiment shows that stool as a substrate does not interfere with the control hybridization reaction at the control line and the ability of the assay to detect the target sequence at the test line. The gold nanoparticle signal is linear (R2 = 0.905) in the range of 1 nM to 100 nM. No sample preparation was required for this assay other than the dilution of stool samples.
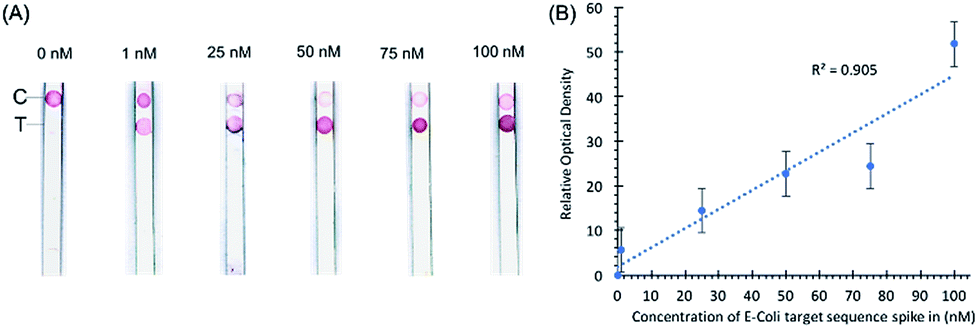 | ||
| Fig. 3 (A) Images of nitrocellulose diagnostic strips with different concentrations of target sequence oligonucleotides spiked into stool samples. The concentrations of the spike-in oligonucleotides are 1, 25, 50, 75, and 100 nM. (B) Statistical curve of the optical density of signals as a function of target sequence concentration. The error bars represent optical density standard deviations from three replicates of samples. The linear regression line follows the function f(x) = y − ax + b; the R square value of the regression equals 0.905. | ||
Next, a human stool sample confirmed to test positive via PCR by the standard clinical diagnostic laboratory methods (National Institutes of Health, Department of Laboratory Medicine) for the selected strain of EPEC was tested. EPEC (Enteropathogenic E. coli) is a pathogenic E. coli. Thus, our method is comparable in sensitivity to the clinical diagnostic standard (103 CFU per ml). Fig. 4 shows the positive detection of endogenous EPEC 23S rRNA in a stool sample resuspended in a PBS solution. Note that most of the stool impurities are absorbed by the sample pad.
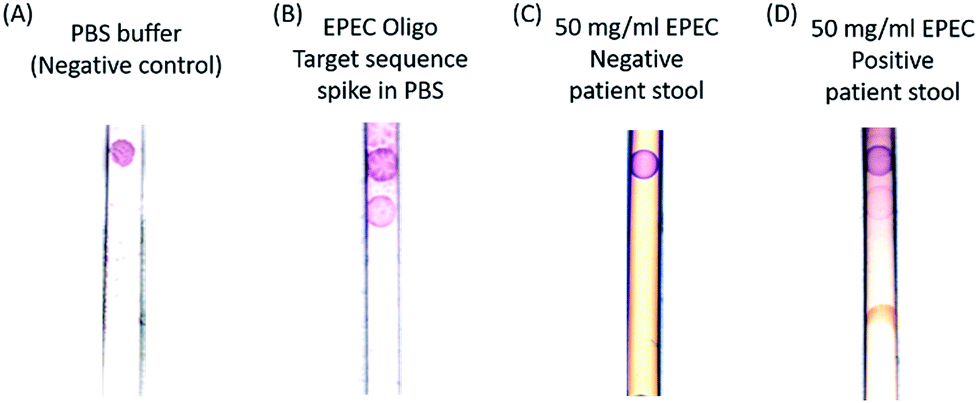 | ||
| Fig. 4 Diluted stool test results. From left to right: (A) the negative control sample with only PBS buffer loaded on the strip; (B) EPEC target sequence oligonucleotides spiked into PBS buffer, and then tested on strips; (C) 50 mg EPEC negative but C. diff positive patient stool diluted with 1 ml 2% Tween 20 solution tested on a lateral flow strip; and (D) 50 mg EPEC positive patient stool diluted with 1 ml 2% Tween 20 solution at room temperature and then tested on a lateral flow strip. | ||
In the negative control sample, only the control band binds to the gold nanoparticles and turns to red color, and the positive band is clearly absent. In contrast, the target sequence oligonucleotide spike-in sample has two bands, which are the control and pathogen DNA capture bands from top to bottom. To test the specificity of the probes, an EPEC negative but Clostridium difficile (C. diff) positive patient stool sample and an EPEC positive stool sample were tested, and the results are presented in Fig. 4C and D. Only the 50 mg ml−1 EPEC positive endogenous patient stool dilution sample has both control and capture bands, and the EPEC negative stool has the control band only. The principle of the method relies on the presence of lysed bacterial material in stool and the design of the capture probe to bind to both the complementary rDNA sequences, of which each bacterial cell may only have one copy, and complementary rRNA sequences of which each bacterial cell has many copies. Bacterial populations in stool encompass cells in all stages of the life cycle, including dead cells with compromised cell membranes releasing molecular material. There are approximately 107 cells in one CFU (colony forming unit) and 103 ribosomal RNAs per cell; therefore there are approximately 1010 equivalents of target sequence oligonucleotides in 1 CFU of EPEC.30 This is equivalent to the 1 nM limit of detection that we found experimentally using our target sequence oligonucleotides. By designing probes in such a way as to be complementary to both rDNA and rRNA sequences, the amount of available molecular material for detection is dramatically increased and the sensitivity of the assay is increased without requiring nucleic acid purification and amplification.
The paper strip test is robust and rapid. Together with the minimal sample processing, it is ideal for application in the field at the point-of-need, especially in resource limited settings as minimal technical skills and no specialized equipment or materials are required to run the assay or assess the results. At present the assay is designed to be qualitative only and efforts to develop it to facilitate simple, low-tech quantitative assessment are underway.
Conclusions
We have developed a simple and rapid lateral flow assay for the detection of DNA and RNA of E. coli and EPEC in stool without sample preparation or nucleic acid amplification. The detection of E. coli and EPEC only requires the use of a mild detergent, and the sample pad absorbs most of the impurities. The use of ribosomal RNA and DNA targets obviates the need for time-consuming nucleic acid amplifications using PCR or isothermal amplification methods that use enzymes that require a cold chain for storage and transport. The test is semi-quantitative in the range of 1 nM to 100 nM of DNA analyte.This test has the potential to be developed into a simple and rapid test for the point-of-need detection in real time of enteric pathogens that cause diarrheal diseases. Such a test can enable earlier diagnosis, infection control and treatment of patients with the appropriate therapeutics, faster infection control to limit the spread of infectious diseases and reduced loss of patients to follow-up. The test is particularly well-suited to resource-limited and remote settings in developing countries where mortality and morbidity due to diarrheal diseases are the highest.
Conflicts of interest
WAH, LX, NHF, SKA, EGF, and CHK are named inventors on related patent applications. CHK has ownership interest in GoDx. The work described herein is under the terms of a Clinical Cooperative Research and Development Agreement (CRADA) between NIH and GoDx.Acknowledgements
The research reported in this publication was supported by the U.S. Department of Health and Human Services, National Institutes of Health, National Institute of Nursing Research, Division of Intramural Research to Wendy A. Henderson, 1Z1ANR000018-01-7; the National Center For Advancing Translational Sciences under Award Number R43TR001912 to GoDx Inc; and the U.S. Department of Health and Human Services, National Institutes of Health, Intramural Research Training Awards to Nicolaas H. Fourie, Sarah K. Abey, Eric G. Ferguson, Ana F. Diallo, and Natnael D. Kenea. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Notes and references
- United Nations Childrens Fund, https://data.unicef.org/topic/child-health/diarrhoeal-disease/.
- D. T. Leung, M. J. Chisti and A. T. Pavia, Pediatr. Clin. North Am., 2016, 63, 67–79 CrossRef PubMed.
- L. Liu, S. Oza, D. Hogan, J. Perin, I. Rudan, J. E. Lawn, S. Cousens, C. Mathers and R. E. Black, Lancet, 2015, 385, 430–440 CrossRef.
- Centers for Disease Control, https://www.cdc.gov/healthywater/global/diarrhea-burden.html#two.
- A. F. Diallo, X. Cong, W. A. Henderson and J. McGrath, Int. J. Nurs. Stud., 2017, 66, 82–92 CrossRef PubMed.
- A. L. Shane, R. K. Mody, J. A. Crump, P. I. Tarr, T. S. Steiner, K. Kotloff, J. M. Langley, C. Wanke, C. A. Warren, A. C. Cheng, J. Cantey and L. K. Pickering, Clin. Infect. Dis., 2017, 65, e45–e80 CrossRef PubMed.
- E. Scallan, P. M. Griffin, F. J. Angulo, R. V. Tauxe and R. M. Hoekstra, Emerging Infect. Dis., 2011, 17, 16–22 CrossRef PubMed.
- J. A. Platts-Mills, J. Liu and E. R. Houpt, Mucosal Immunol., 2013, 6, 876–885 CrossRef PubMed.
- K. H. Rand, E. E. Tremblay, M. Hoidal, L. B. Fisher, K. R. Grau and S. M. Karst, Diagn. Microbiol. Infect. Dis., 2015, 82, 154–157 CrossRef PubMed.
- S. G. Beal, E. E. Tremblay, S. Toffel, L. Velez and K. H. Rand, J. Clin. Microbiol., 2018, 56, e02012-17 CrossRef PubMed.
- A. J. Baeumner, N. A. Schlesinger, N. S. Slutzki, J. Romano, E. M. Lee and R. A. Montagna, Anal. Chem., 2002, 74, 1442–1448 CrossRef PubMed.
- D. A. Williams, T. Kiiza, R. Kwizera, R. Kiggundu, S. Velamakanni, D. B. Meya, J. Rhein and D. R. Boulware, Clin. Infect. Dis., 2015, 61, 464–467 CrossRef PubMed.
- D. Tang, J. C. Sauceda, Z. Lin, S. Ott, E. Basova, I. Goryacheva, S. Biselli, J. Lin, R. Niessner and D. Knopp, Biosens. Bioelectron., 2009, 25, 514–518 CrossRef PubMed.
- W. Wang, L. Liu, S. Song, L. Xu, J. Zhu and H. Kuang, Food Agric. Immunol., 2017, 28, 274–287 CrossRef.
- W. Wang, L. Liu, L. Xu, H. Kuang, J. Zhu and C. Xu, Part. Part. Syst. Charact., 2016, 33, 388–395 CrossRef.
- W. Wang, W. Wang, L. Liu, L. Xu, H. Kuang, J. Zhu and C. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 15591–15597 CrossRef PubMed.
- M. Feng, Q. Yong, W. Wang, H. Kuang, L. Wang and C. Xu, Food Agric. Immunol., 2013, 24, 481–487 CrossRef.
- A. J. Baeumner, R. N. Cohen, V. Miksic and J. Min, Biosens. Bioelectron., 2003, 18, 405–413 CrossRef PubMed.
- A. Carrio, C. Sampedro, J. Sanchez-Lopez, M. Pimienta and P. Campoy, Sensors, 2015, 15, 29569 CrossRef PubMed.
- P. Damborsky, K. M. Koczula, A. Gallotta and J. Katrlik, Analyst, 2016, 141, 6444–6448 RSC.
- J. Platts-Mills, J. Liu and E. Houpt, Mucosal Immunol., 2013, 6, 876–885 CrossRef PubMed.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef PubMed.
- X. Mao, Y. Ma, A. Zhang, L. Zhang, L. Zeng and G. Liu, Anal. Chem., 2009, 81, 1660–1668 CrossRef PubMed.
- W. Zhao, M. M. Ali, S. D. Aguirre, M. A. Brook and Y. Li, Anal. Chem., 2008, 80, 8431–8437 CrossRef PubMed.
- S. Chen, X. Yu and D. Guo, Viruses, 2018, 10, 40 CrossRef PubMed.
- D. S. Chertow, Science, 2018, 360, 381–382 CrossRef PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, M. J. Kellner, J. Joung, J. J. Collins and F. Zhang, Science, 2018, 360, 439–444 CrossRef PubMed.
- C. Myhrvold, C. A. Freije, J. S. Gootenberg, O. O. Abudayyeh, H. C. Metsky, A. F. Durbin, M. J. Kellner, A. L. Tan, L. M. Paul, L. A. Parham, K. F. Garcia, K. G. Barnes, B. Chak, A. Mondini, M. L. Nogueira, S. Isern, S. F. Michael, I. Lorenzana, N. L. Yozwiak, B. L. MacInnis, I. Bosch, L. Gehrke, F. Zhang and P. C. Sabeti, Science, 2018, 360, 444–448 CrossRef PubMed.
- K. H. Wilson, R. Blitchington, B. Hindenach and R. C. Greene, J. Clin. Microbiol., 1988, 26, 2484–2488 Search PubMed.
- C. Pohlmann, I. Dieser and M. Sprinzl, Analyst, 2014, 139, 1063–1071 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |