
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-throughput method for orthophosphate determination of thermostable membrane-bound pyrophosphatase activity
Keni
Vidilaseris
 a,
Juho
Kellosalo
b and
Adrian
Goldman
*ac
a,
Juho
Kellosalo
b and
Adrian
Goldman
*ac
aDepartment of Biosciences, Division of Biochemistry, University of Helsinki, Helsinki, Finland
bInstitute of Biotechnology, University of Helsinki, Finland
cSchool of Biomedical Sciences, Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, UK. E-mail: a.goldman@leeds.ac.uk
First published on 15th January 2018
Abstract
Membrane-bound pyrophosphatases (mPPases) are homodimeric integral membrane proteins that hydrolyse pyrophosphate into orthophosphates coupled to the active transport of protons or sodium ions across membranes. They occur in bacteria, archaea, plants, and protist parasites. As they are essential in protist parasites and there are no homologous proteins in animals and humans, these enzymes represent an excellent drug target for treating protistal diseases. Experimental screening to find drug candidates is an important step to discover new hit compounds. For that, a cheap, simple, and robust assay is needed. Here we report the application of the molybdenum blue reaction method for a medium throughput microplate activity assay of the hyperthermophilic bacterium Thermotoga maritima mPPase and the possible application of the assay to screen inhibitors of membrane-bound pyrophosphatases.
Introduction
Inorganic pyrophosphate (PPi) is the by-product of diverse cellular reactions within the cell, such as polynucleotide synthesis and lipid metabolism. Inside the cell, the level of PPi is controlled by inorganic pyrophosphatases (PPase),1,2 enzymes that cleave the phosphoanhydride bond of pyrophosphate into two orthophosphates. There are two types of PPases, soluble PPases (sPPases) and membrane bound PPases (mPPases). sPPases have been studied from many different sources, and their structures and functions have been studied extensively.2–6 Membrane-bound pyrophosphatases (mPPases) are homodimeric integral membrane proteins that hydrolyse pyrophosphate into orthophosphate and couple this to the active transport of protons or sodium ion across membranes. mPPases were first discovered in photosynthetic bacteria and plants.7 Later it was shown that these enzymes occurred in all kingdoms of life except animalia and fungi.7 In bacteria, mPPases localise in the inner membrane and in acidocalcisome-like organelles.8 In plants, these proteins mainly reside in the vacuolar membranes and are important for plant growth under stress environments,9–12 while in protist parasites, such as Plasmodium spp., Toxoplasma spp., Trypanosoma spp., and Leishmania spp., mPPases localise mostly though not exclusively to the acidocalcisome,13 a vesicle rich in calcium, phosphorus, and magnesium. In these parasites, mPPases are important for maintaining the acidity of the acidocalcisome and its functions for osmoregulation upon passage of the parasite from the insect vector to the mammalian host.14 It has been shown that knocking out mPPase expression in T. gondii produced parasite cells more sensitive to extracellular environments that make them avirulent in mice.15As they are essential in protist parasites and because they have no homologous proteins in multicellular organisms, mPPases may be an excellent potential drug target for protistal diseases.16 For that, a simple, cheap, and sensitive method to determine the reaction product of mPPases is needed to screen drug candidates. Several methods have been reported for determining the phosphate released from the enzymatic reaction of pyrophosphatase, including colourimetric,17–21 fluorometric,22,23 enzymatic,24–26 and radiolabeling27 methods. Each method has its drawbacks and advantages. The colourimetric method based on the molybdenum blue reaction, first developed by Fiske and Subbarow,17 is the most commonly used. This method involves the formation of 12-phosphomolybdic acid (PMA) from orthophosphate and molybdate under acid conditions, which is then reduced to a dark blue colour of phosphomolybdenum species.28 Here we explore the use of the colourimetric molybdenum blue method for medium to high throughput analysis of the activity of the hyperthermophilic bacterium T. maritima mPPase (TmPPase) as a model system.
TmPPase is a sodium pumping mPPase and has maximum activity in the presence of millimolar concentrations of sodium and potassium ions at 71 °C.29,30 The structures of TmPPase and the H+-pump mPPase from mung bean (Vigna radiata) (VrPPase)31,32 are very similar and show complete conservation of the position and identity of all the catalytic residues. Protist mPPases are H+-pumps, like VrPPase, and therefore have conserved residues in the catalytic region too. The available structures can be used for structure-based drug design experiments and for experimental screening of compounds.
Experimental
Chemicals
Hydrochloric acid (HCl), soybean lecithin, ascorbic acid, sodium chloride, imidodiphosphate (IDP), calcium chloride, sodium fluoride, sodium phosphate dibasic (Pi) and sodium pyrophosphate (Na2PPi) were purchased from Sigma. Ammonium heptamolybdate tetrahydrate, sodium meta-arsenite, acetic acid, potassium chloride, dimethyl sulfoxide (DMSO), ethanol and magnesium chloride were purchased from Merck. Trisodium citrate dehydrate was purchased from Fluka and dodecyl maltoside (DDM) was purchased from Melford.Reagent preparation
Assay
The protocol is illustrated in Fig. 1. 40 μL reaction mixture per well containing 75 mM Tris-HCl pH 8.0, 3 mM MgCl2, 125 mM KCl, 25 mM NaCl, and 0.2 μL of reactivated TmPPase30 were added to PCR tube strips (8 tubes for each strip) using a multichannel pipette and sealed with an adhesive sealing sheet (Thermo Scientific). Then, the sealing sheet was cut to separate each strip. The samples were then preincubated for 5 minutes at 71 °C on a 96-well heating block with a 20 second interval between each strip for ease of performing the subsequent steps. After incubation, 10 μL of 2 mM sodium pyrophosphate was added to each tube, mixed, and incubated for 1–10 minutes. The reaction was stopped by putting each strip on the cooling apparatus for 10 minutes. The cooling apparatus was made by placing a 96-well PCR plate (Bio-Rad) on a polystyrene Petri dish (size 150 mm × 15 mm) (Sigma) filled with water and then frozen for 1 h. After 5 minutes of cooling, strips were centrifuged briefly to collect the condensed moisture below the sealing sheet, put back onto the cooling apparatus, and the sealing sheets were removed. After 10 minutes of cooling, 60 μL of solution A + B was added to each tube and they were then incubated for 10 minutes. Finally, 90 μL of sodium arsenite solution was added, incubated at room temperature for at least 30 minutes to reach stable colour production, and then transferred into the clear PS microplate 96 wells (Thermo Scientific) with a multichannel pipette. The absorbance was measured at 860 nm using MultiSkan Go (Thermo Scientific). For standard calibration, 50 μL of reaction mixtures containing 0–800 μM of Pi were tested. A reaction mixture with no Pi added was used as a blank. All measurements were done in triplicate. Analysis and regression were performed with Prism 6 software (GraphPad Software, San Diego, CA).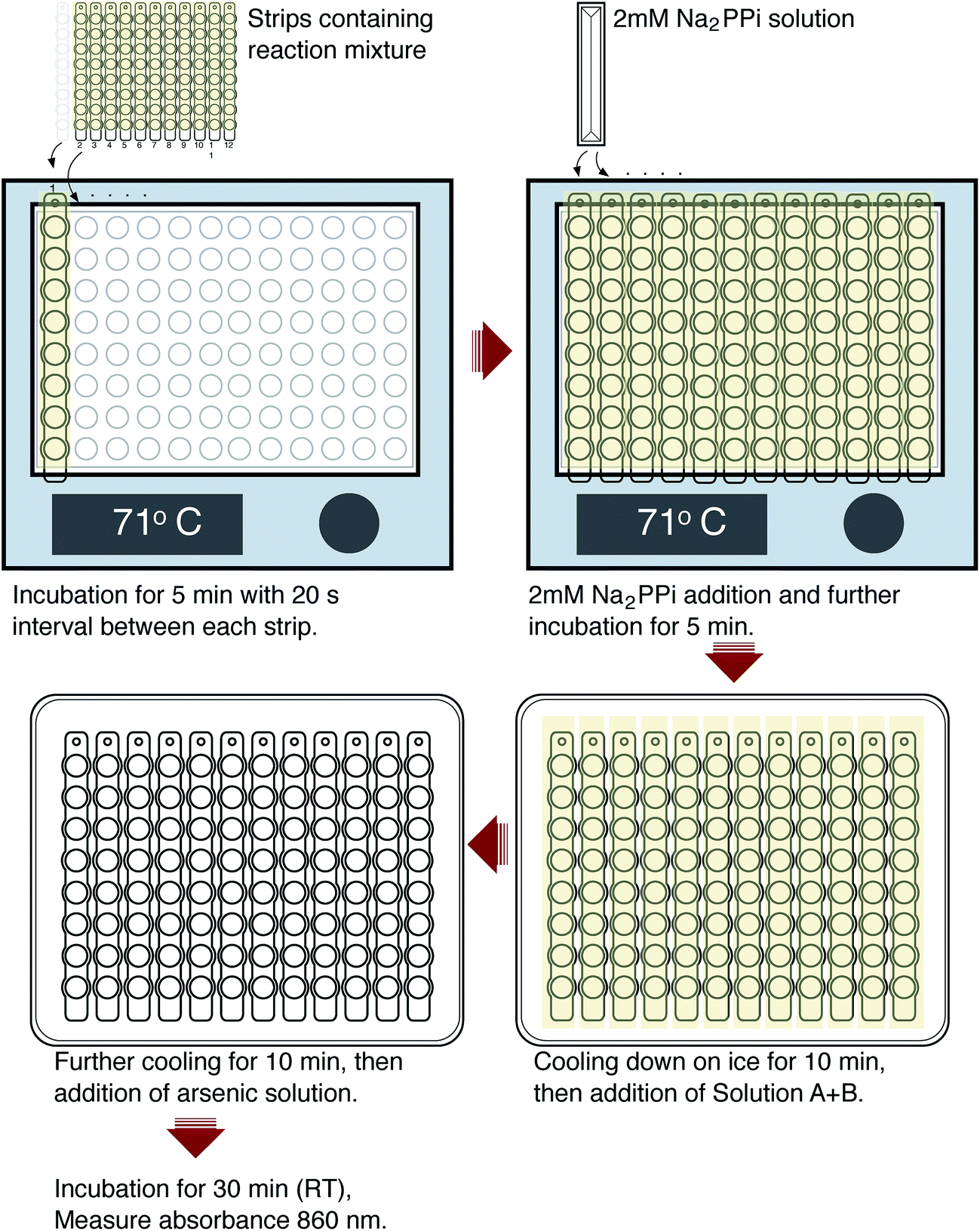 | ||
| Fig. 1 Step by step illustration of the assay. | ||
Results and discussion
Method development
In drug development projects, experimental screening is an important step to discover new hit compounds. For that, a cheap, simple, and robust assay is needed. Here we explore the viability of a colourimetric method based on the molybdenum blue reaction for a medium throughput assay to determine the activity of TmPPase. We adapted the existing protocol30,33 for mPPase activity assays to a 96 well format using twelve PCR tubes strips (Fig. 1). We chose the molybdenum blue assay of Baginski et al.34 over a similar assay from Heinonen and Lahti20 and the malachite-green assay21 because it is 2-fold more sensitive than the former and, unlike the latter, it does not show interference in the presence of high concentrations of phospholipid (data not shown), which we need for the activity of our enzyme (see below). Thus, even though the malachite green assay would be 10-fold more sensitive21 than the Baginski assay34 it is impractical for our purposes.The reaction volume for each tube is 50 μL, which consists of 40 μL of reaction buffer containing enzyme and 10 μL of substrate (PPi). We used the optimal temperature of TmPPase activity (71 °C)30 for the reaction condition in a 96 well heating block. During initial testing, loss of solution because of evaporation led to poorly reproducible results. Sealing the tubes with capping strips did not help due to the difficulty in the closing and opening the caps during the assay, which leads to delays in the timing. We solved this problem by using an adhesive sealing sheet which we cut to fit the tube strips. With this sealing, we can rapidly open and close the tube strips during the assay. In the assay, 40 μL of reaction mixtures (reactivated enzyme in the reaction buffer) were incubated in the heating block for 5 minutes to raise the temperature of the reaction mixture to 71 °C. After the addition of 10 μL of PPi, the reaction mixture was incubated at 71 °C for 1–10 minutes for the enzymatic reaction. The reaction was stopped by putting the strip tubes onto the 96-well cooling apparatus for 10 minutes. During this time, strips were spun down briefly to collect the any water vapour that had condensed on the sealing sheet, thus minimising the differences in the reaction volumes in each tube.
After enzyme inactivation, 60 μL of solution A + B (see above) was added to the reaction mixture to determine the amount of phosphate produced according to the method of Baginski and coworkers.34 The addition of solution A + B generates a dark blue colour that appears immediately and develops over a 10 minute time on the cooling apparatus (0 °C). Incubation at 0 °C prevents acid-catalysed hydrolysis compared to incubation at room temperature.35 Solution A and B cannot be added individually to the reaction mixture because it produces colour inconsistency and lowers the intensity of the final product.36
Subsequent addition of 90 μL of the arsenic–citrate solution to the reaction mixture at room temperature was used to stabilise the dark blue solution, which has absorption maxima at 709 nm and 860 nm (Fig. 2). Arsenite appears to enhance the colour formation while citrate complexes excess molybdate in the solution,34 thus inhibiting further colour development due to any phosphate liberated by non-enzymatic PPi cleavage.35 For Pi determination, we measured at 860 nm based on the observation of He & Honeycutt37 that measurement at longer wavelengths increased the detection limit and sensitivity. We observed that, at 860 nm, the blue colour developed within 30 minutes of incubation at room temperature and was stable for at least five hours (Fig. 3).
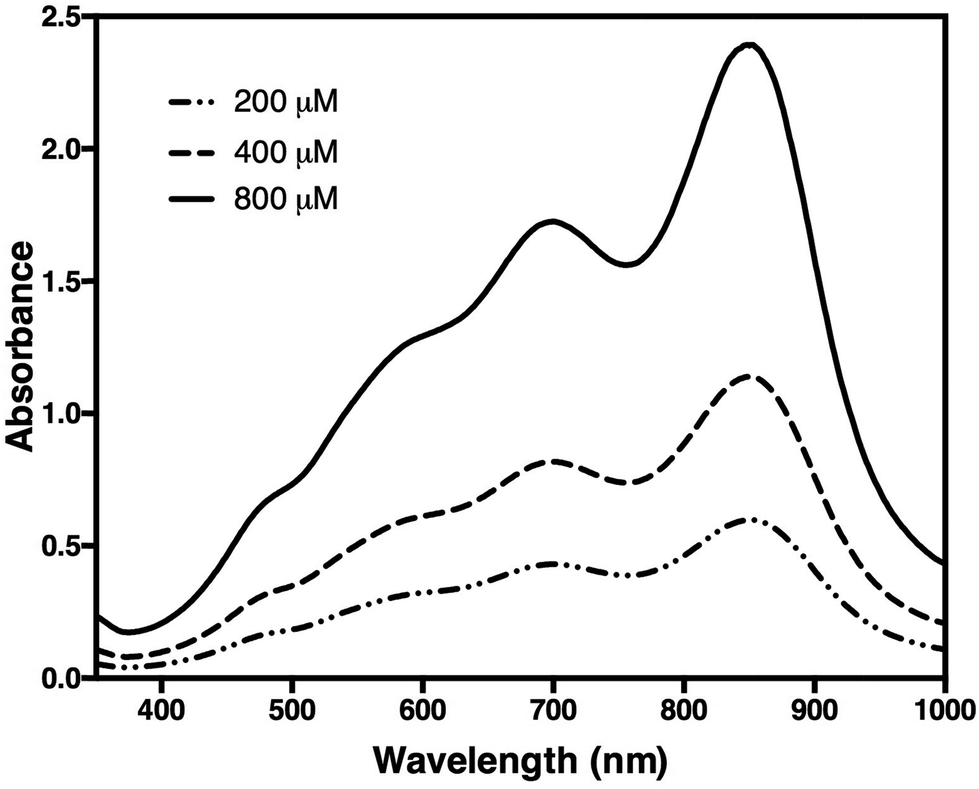 | ||
| Fig. 2 Spectra of a sample containing 200, 400, and 800 μM of orthophosphate (Pi). Measurement was carried out in 10 mm cuvettes against control buffer. Two peaks were observed at 709 nm and 860 nm. | ||
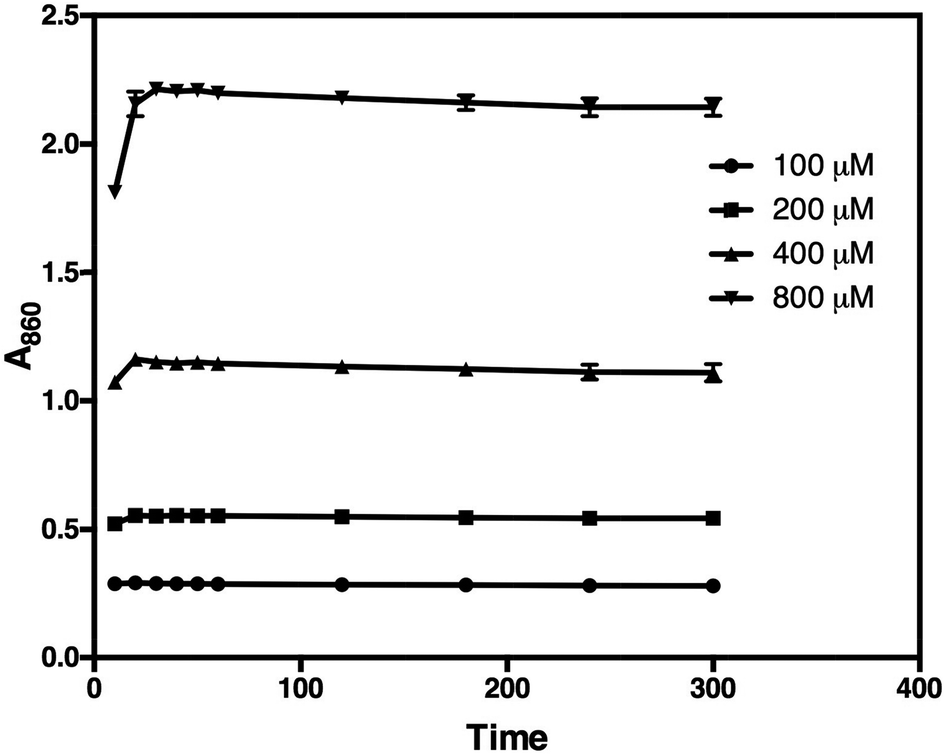 | ||
| Fig. 3 The blue colour development of 100, 200, 400, and 800 μM Pi (50 μL reaction mixture) and its stability over time at the wavelength of 860 nm. The absorbance reaches a maximum after 30 minutes of incubation at room temperature. | ||
The molybdenum blue assay we use is sensitive to interference from ≥1 mM MgCl2, ≥5 mM EDTA and ≥20% glycerol38 and high concentrations of phosphoryl compounds (2–10 mM PPi, ATP, β-glycerophosphate and p-nitrophenylphosphate).35 As the assay conditions that we employ do not require interfering concentrations of these reagents, the assay is well suited for our purposes.
The activity of Thermotoga maritima mPPase
To test the method, we determined the activity of TmPPase, a thermophilic mPPase from T. maritima and its inhibition by known inhibitors. We used this enzyme because it can be easily produced and purified, has good stability, high specific activity, and also has a conserved catalytic region with other mPPases including the parasite homologs. TmPPase expression and purification have been previously described.30 Before use, the pure protein was reactivated with 4.52% DDM and 12 mg mL−1 soybean lecithin. Based on the calibration curve (Fig. 4), the lower and the upper limit of the assay were 10 μM and 800 μM of Pi, respectively. After 24 hours, the absorbance maxima of the calibration solutions decreased by 10%, but were still linear over the range tested (data not shown). Based on this method, TmPPase has higher specific activity (140.7 ± 7.01 μmol Pi per mg per min) compared to our previously reported values (20 μmol Pi per mg per min), which used 0.015% DDM and 0.12 mg mL−1 soybean lecithin for reactivation.30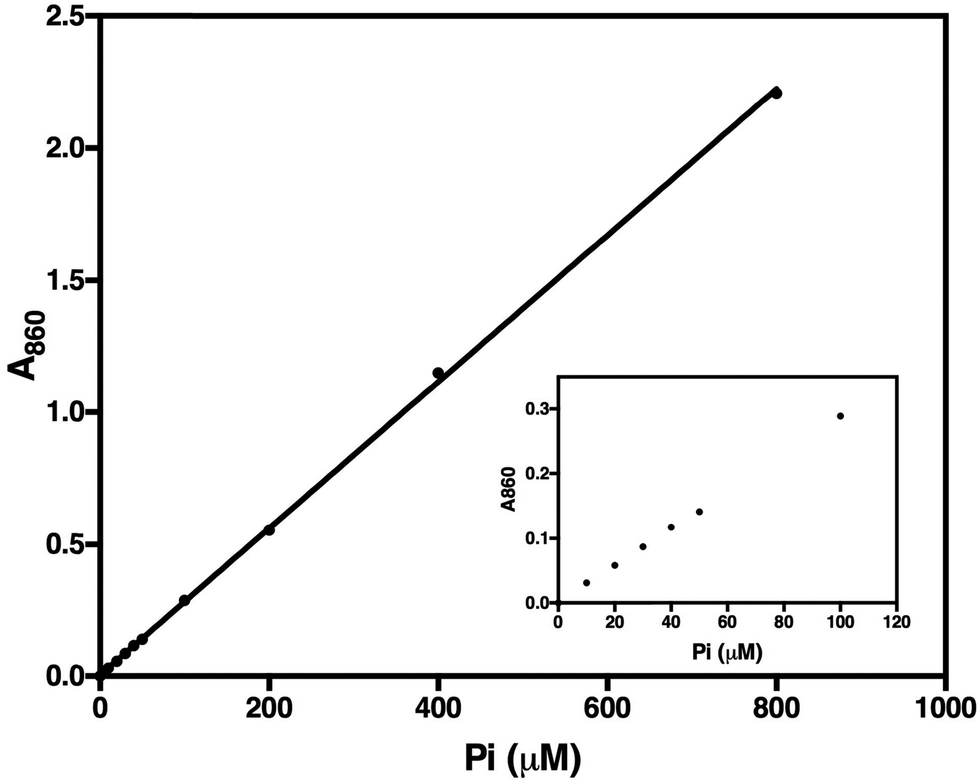 | ||
| Fig. 4 Calibration curve of orthophosphate (Pi) determination in 50 μL reaction mixture after 30 minutes incubation at room temperature. Inset shows the absorbance linearity of 0 to 100 μM Pi. The line fit is linear over the concentration range tested (y = 0.0555x + 0.0059; r2 = 1.000). | ||
In the initial testing, we tested reaction times from 1–10 minutes (Fig. 5). As the phosphate release as a function of time was linearly correlated in the tested time range, we chose 5 minutes as a suitable reaction time for high-throughput screening. This was because the absorbance of the phosphate released was in this case in the mid-range of the calibration standard (Fig. 4), which also makes the concentration of the phosphate suitable for the assay. To test the accuracy of this assay we carried out measurements on five different plates on three different days and calculated a Z′-factor39 of 0.736 for the assay, which indicates that the 5 minute incubation is sufficient for reliable screening.
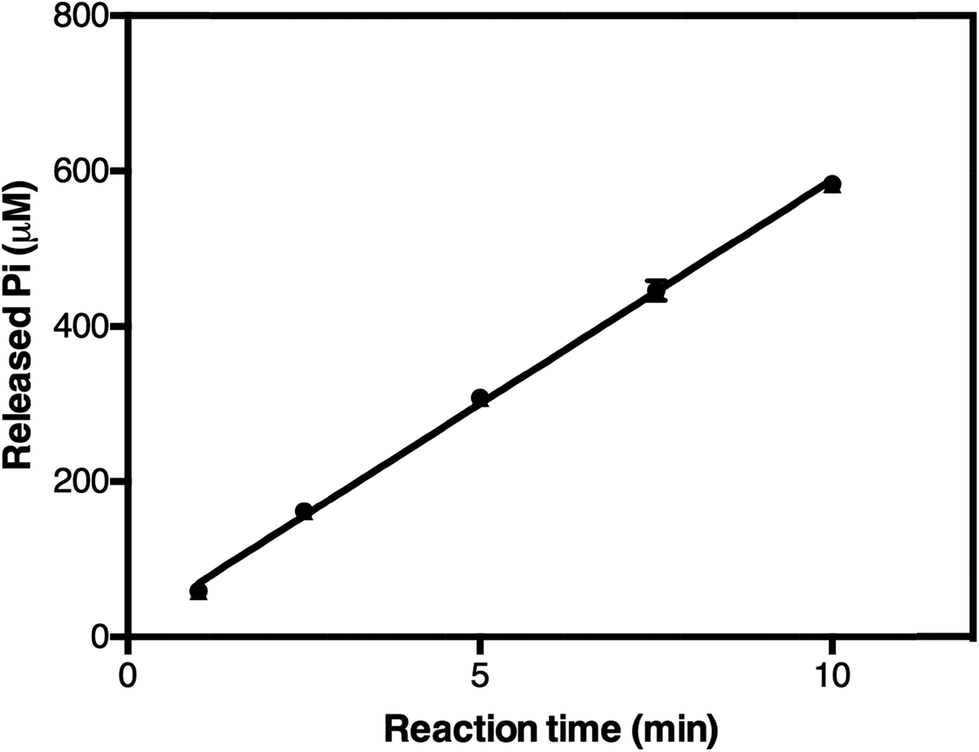 | ||
| Fig. 5 The release of orthophosphate in TmPPase-containing 50 μL reaction mixtures after different incubation times at 71 °C. The line fit is linear over the time range tested (y = 57.76x + 11.36; r2 = 0.9972). | ||
We tested two mPPase inhibitors, imidodiphosphate (IDP) and Ca2+, and one inhibitor, fluoride, of soluble PPases. Structures of TmPPase in complex with IDP40 and Ca2+ (ref. 31) has been solved. IDP binds to the hydrolytic centre of the enzyme, and Ca2+ to the conserved aspartic residues (D688, D692, and D669) in the hydrolytic centre. Both IDP and CaCl2 inhibited the activity of TmPPase with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50 of 1.90 ± 0.02 (IC50 = 80.0 μM) for IDP and 2.42 ± 0.009 (IC50 = 264.0 μM) for total Ca2+ (Fig. 6). Even though fluoride did not inhibit the TmPPase activity at low concentration, at higher concentration it inhibited with logIC50 of 3.66 ± 0.008 (IC50 = 4.6 mM) (Fig. 6). This is probably due the sequestering of Mg2+ ion by F− from the reaction mixture and enzyme active site to form MgF2. This result is similar with the fluoride inhibition of mPPase from R. rubrum and V. radiata with the Ki values of 4.8 and 3.4 mM, respectively.41
IC50 of 1.90 ± 0.02 (IC50 = 80.0 μM) for IDP and 2.42 ± 0.009 (IC50 = 264.0 μM) for total Ca2+ (Fig. 6). Even though fluoride did not inhibit the TmPPase activity at low concentration, at higher concentration it inhibited with logIC50 of 3.66 ± 0.008 (IC50 = 4.6 mM) (Fig. 6). This is probably due the sequestering of Mg2+ ion by F− from the reaction mixture and enzyme active site to form MgF2. This result is similar with the fluoride inhibition of mPPase from R. rubrum and V. radiata with the Ki values of 4.8 and 3.4 mM, respectively.41
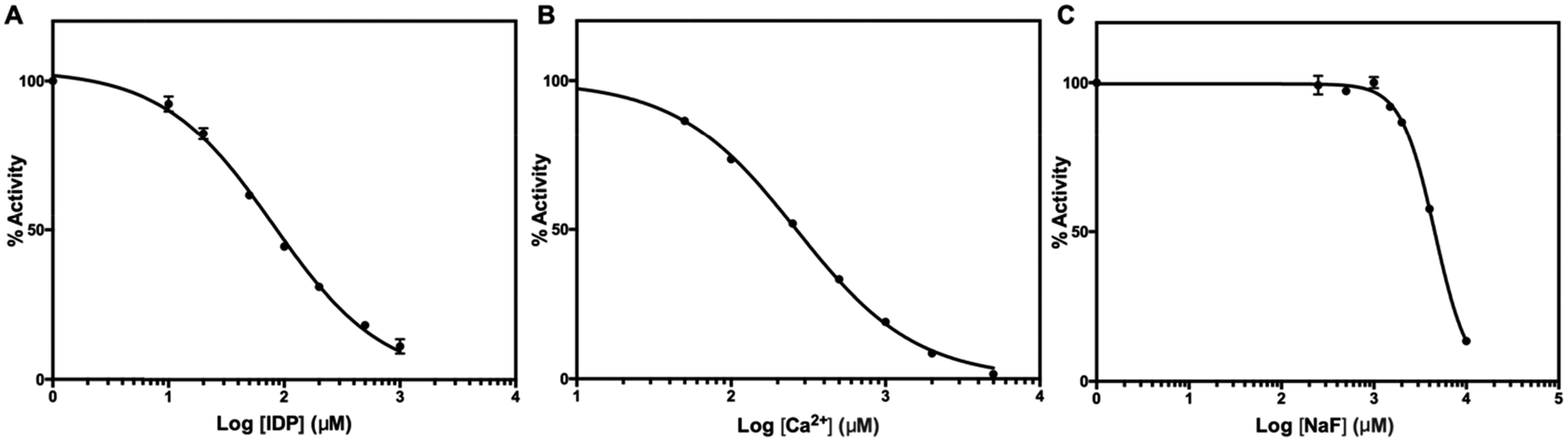 | ||
| Fig. 6 Inhibition of TmPPase by (A) IDP, (B) CaCl2, and (C) NaF. IDP, CaCl2, and NaF inhibited TmPPase with the logIC50 of 1.90 ± 0.022 (IC50 = 80.0 μM), 2.42 ± 0.009 (IC50 = 264.0 μM), and 3.66 ± 0.008 (IC50 = 4.6 mM), respectively. | ||
For experimental screening of mPPase inhibitors, target compounds are usually dissolved in organic solvents such as DMSO or ethanol. To check whether either organic solvent inhibits TmPPase activity, we performed the same assay as before. DMSO and ethanol inhibited the TmPPase activity at very high concentration with logIC50 of 2.85 ± 0.014 (IC50 = 709 mM) and 2.88 ± 0.015 (IC50 = 766 mM), respectively (Fig. 7). Even though both solvents have similar IC50, DMSO did not affect the TmPPase activity at 100 mM concentration, while ethanol decreased the activity to about 92%. Therefore, DMSO is a better solvent for target compounds when screening mPPase inhibitors.
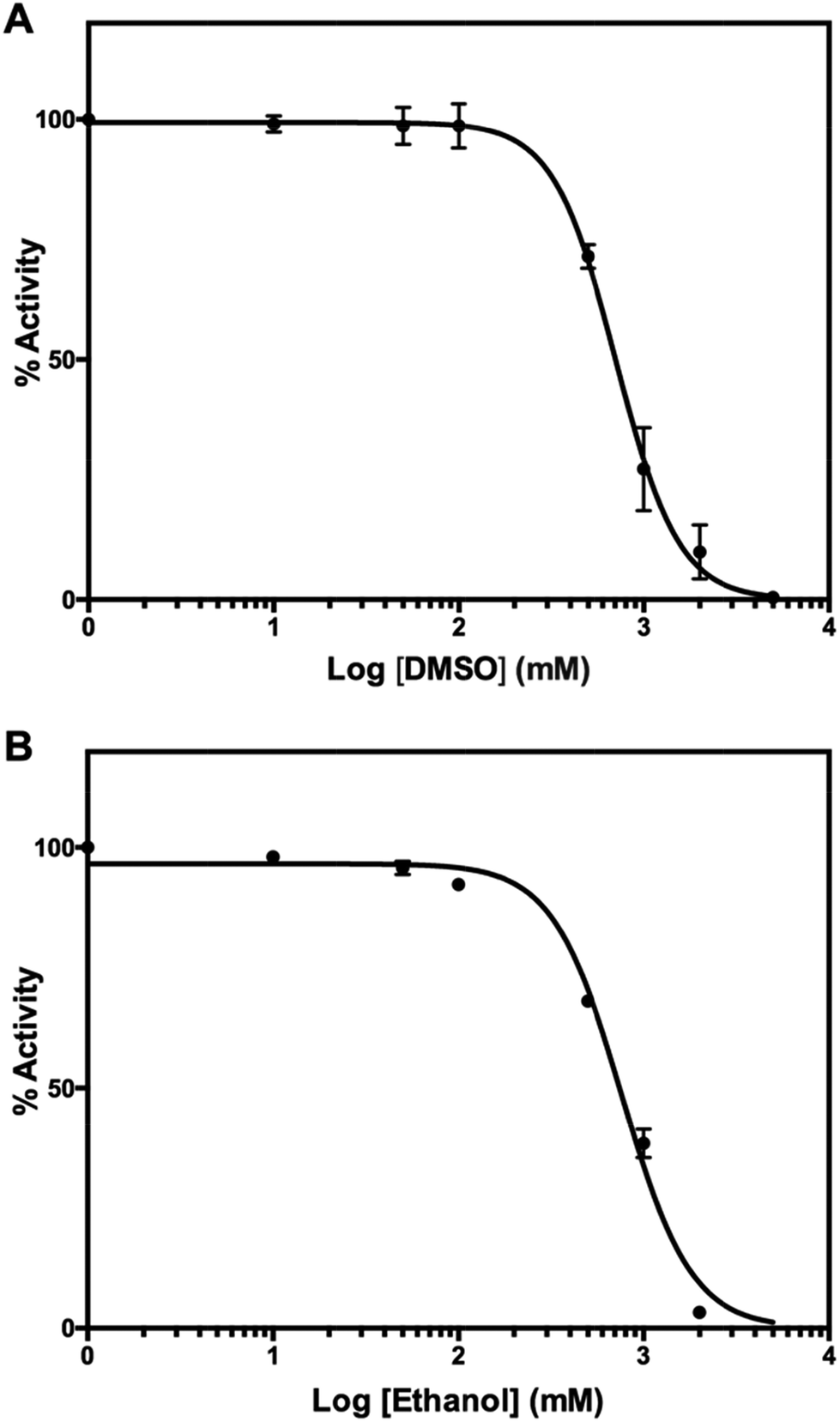 | ||
| Fig. 7 Effect of organic solvent, (A) DMSO and (B) ethanol, on the activity of TmPPase. | ||
Conclusions
In conclusion, we have shown that the molybdenum blue method can be applied for medium-throughput (96 well plate) assay of TmPPase activity at high temperature. The assay is a simple, cheap, and sensitive way to determine the amount of phosphate released from the reaction. The colour developed by this method is also stable for at least 12 hours, so there is no need to measure the absorbance at the same time as the assay is being performed. The method can be further miniaturised for a 384-well plate assay, especially for testing mesophilic mPPases, which have optimal activity at ambient temperature. In this case sealing during the reaction to prevent evaporation should not be necessary. We are currently using this method to screen libraries to find drug candidates against various parasitic diseases.Conflicts of interest
All authors declare no conflict of interest in this paper.Acknowledgements
This work is supported by the grants from Erkon Säätiö and the BBSRC (BB/M021610/1) to Adrian Goldman and the Academy of Finland (No. 308105) to Keni Vidilaseris and (No. 299762) to Juho Kellosalo. The authors thank Violeta Manole for her technical help.References
- T. Kajander, J. Kellosalo and A. Goldman, FEBS Lett., 2013, 587, 1863–1869 CrossRef CAS PubMed.
- T. C. Chao, H. Huang, J. Y. Tsai, C. Y. Huang and Y. J. Sun, Proteins: Struct., Funct., Bioinf., 2006, 65, 670–680 CrossRef CAS PubMed.
- T. Salminen, J. Käpylä, P. Heikinheimo, J. Kankare, A. Goldman, J. Heinonen, A. A. Baykov, B. S. Cooperman and R. Lahti, Biochemistry, 1995, 34, 782–791 CrossRef CAS PubMed.
- V. R. Samygina, A. N. Popov, E. V. Rodina, N. N. Vorobyeva, V. S. Lamzin, K. M. Polyakov, S. A. Kurilova, T. I. Nazarova and S. M. Avaeva, J. Mol. Biol., 2001, 314, 633–645 CrossRef CAS PubMed.
- P. Heikinheimo, J. Lehtonen, A. Baykov, R. Lahti, B. S. Cooperman and A. Goldman, Structure, 1996, 4, 1491–1508 CrossRef CAS PubMed.
- M. C. Merckel, I. P. Fabrichniy, A. Salminen, N. Kalkkinen, A. A. Baykov, R. Lahti and A. Goldman, Structure, 2001, 9, 289–297 CrossRef CAS PubMed.
- A. Serrano, J. R. Pérez-Castiñeira, M. Baltscheffsky and H. Baltscheffsky, IUBMB Life, 2007, 59, 76–83 CrossRef CAS PubMed.
- M. Seufferheld, M. C. F. Vieira, F. A. Ruiz, C. O. Rodrigues, S. N. J. Moreno and R. Docampo, J. Biol. Chem., 2003, 278, 29971–29978 CrossRef CAS PubMed.
- J. Li, H. Yang, W. A. Peer, G. Richter, J. Blakeslee, A. Bandyopadhyay, B. Titapiwantakun, S. Undurraga, M. Khodakovskaya, E. L. Richards, B. Krizek, A. S. Murphy, S. Gilroy and R. Gaxiola, Science, 2005, 310, 121–125 CrossRef CAS PubMed.
- E. V. D'yakova, A. L. Rakitin, A. M. Kamionskaya, A. A. Baikov, R. Lahti, N. V. Ravin and K. G. Skryabin, Dokl. Biol. Sci., 2006, 409, 346–348 CrossRef.
- F. Gao, Q. Gao, X. Duan, G. Yue, A. Yang and J. Zhang, J. Exp. Bot., 2006, 57, 3259–3270 CrossRef CAS PubMed.
- F. Brini, M. Hanin, I. Mezghani, G. A. Berkowitz and K. Masmoudi, J. Exp. Bot., 2007, 58, 301–308 CrossRef CAS PubMed.
- M. T. McIntosh, Y. M. Drozdowicz, K. Laroiya, P. A. Rea and A. B. Vaidya, Mol. Biochem. Parasitol., 2001, 114, 183–195 CrossRef CAS PubMed.
- R. Docampo, W. de Souza, K. Miranda, P. Rohloff and S. N. J. Moreno, Nat. Rev. Microbiol., 2005, 3, 251–261 CrossRef CAS PubMed.
- J. Liu, D. Pace, Z. Dou, T. P. King, D. Guidot, Z.-H. Li, V. B. Carruthers and S. N. J. Moreno, Mol. Microbiol., 2014, 93, 698–712 CrossRef CAS PubMed.
- N. R. Shah, K. Vidilaseris, H. Xhaard and A. Goldman, AIMS Biophys., 2016, 3, 171–194 CrossRef.
- C. H. Fiske and Y. Subbarow, J. Biol. Chem., 1925, 66, 375–400 CAS.
- A. A. Baykov, O. A. Evtushenko and S. M. Avaeva, Anal. Biochem., 1988, 171, 266–270 CrossRef CAS PubMed.
- J. Deng, Q. Jiang, Y. Wang, L. Yang, P. Yu and L. Mao, Anal. Chem., 2013, 85, 9409–9415 CrossRef CAS PubMed.
- J. K. Heinonen and R. J. Lahti, Anal. Biochem., 1981, 113, 313–317 CrossRef CAS PubMed.
- B. Martin, C. J. Pallen, J. H. Wang and D. J. Graves, J. Biol. Chem., 1985, 260, 14932–14937 CAS.
- D. H. Vance and A. W. Czarnik, J. Am. Chem. Soc., 1994, 116, 9397–9398 CrossRef CAS.
- K. Xu, Z. Chen, L. Zhou, O. Zheng, X. Wu, L. Guo, B. Qiu, Z. Lin and G. Chen, Anal. Chem., 2015, 87, 816–820 CrossRef CAS PubMed.
- M. Hill and B. Arrio, Anal. Biochem., 1997, 254, 249–253 CrossRef CAS PubMed.
- J. Eriksson, S. Karamohamed and P. Nyrén, Anal. Biochem., 2001, 293, 67–70 CrossRef CAS PubMed.
- R. H. Upson, R. P. Haugland, M. N. Malekzadeh and R. P. Haugland, Anal. Biochem., 1996, 243, 41–45 CrossRef CAS PubMed.
- H. P. Bloemers, M. L. Stephenson and P. C. Zamecnik, Anal. Biochem., 1970, 34, 66–70 CrossRef CAS PubMed.
- E. A. Nagul, I. D. McKelvie, P. Worsfold and S. D. Kolev, Anal. Chim. Acta, 2015, 890, 60–82 CrossRef CAS PubMed.
- G. A. Belogurov, A. M. Malinen, M. V. Turkina, U. Jalonen, K. Rytkönen, A. A. Baykov and R. Lahti, Biochemistry, 2005, 44, 2088–2096 CrossRef CAS PubMed.
- J. Kellosalo, T. Kajander, M. G. Palmgren, R. L. Lopéz-Marqués and A. Goldman, Protein Expression Purif., 2011, 79, 25–34 CrossRef CAS PubMed.
- J. Kellosalo, T. Kajander, K. Kogan, K. Pokharel and A. Goldman, Science, 2012, 337, 473–476 CrossRef CAS PubMed.
- S. M. Lin, J. Y. Tsai, C. D. Hsiao, Y. T. Huang, C.-L. Chiu, M. H. Liu, J. Y. Tung, T. H. Liu, R. L. Pan and Y. J. Sun, Nature, 2012, 484, 399–403 CrossRef CAS PubMed.
- R. L. López-Marqués, J. R. Pérez-Castiñeira, M. J. Buch-Pedersen, S. Marco, J.-L. Rigaud, M. G. Palmgren and A. Serrano, Biochim. Biophys. Acta, Biomembr., 2005, 1716, 69–76 CrossRef PubMed.
- E. S. Baginski, P. P. Foa and B. Zak, Clin. Chim. Acta, 1967, 15, 155–158 CrossRef CAS.
- M. Hørder, Anal. Biochem., 1972, 49, 37–47 CrossRef.
- M. J. Black and M. E. Jones, Anal. Biochem., 1983, 135, 233–238 CrossRef CAS PubMed.
- Z. He and C. W. Honeycutt, Commun. Soil Sci. Plant Anal., 2005, 36, 1373–1383 CrossRef CAS.
- S. Chifflet, A. Torriglia, R. Chiesa and S. Tolosa, Anal. Biochem., 1988, 168, 1–4 CrossRef CAS PubMed.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- K. M. Li, C. Wilkinson, J. Kellosalo, J. Y. Tsai, T. Kajander, L. J. C. Jeuken, Y. J. Sun and A. Goldman, Nat. Commun., 2016, 7, 13596 CrossRef PubMed.
- A. A. Baykov, E. B. Dubnova, N. P. Bakuleva, O. A. Evtushenko, R. G. Zhen and P. A. Rea, FEBS Lett., 1993, 327, 199–202 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |