
Dengue serotyping with a label-free DNA sensor†
 *ac
*ac
Abstract
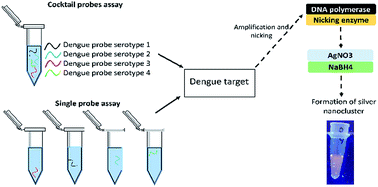
Dengue virus (DENV) is one of the most important mosquito-borne viruses in tropical and subtropical regions. Development of severe forms of dengue viral infection such as dengue fever (DF) and dengue hemorrhagic fever (DHF) has claimed many lives. The standard methods for detecting dengue virus are time consuming, laborious, and require skilful personnel. In this study, we propose a method whereby DENV RNA extracted from dengue infected mosquitoes was converted into DNA for probe hybridization to generate silver nanocluster strands that could be visualised under UV light. Label-free silver nanocluster based DNA sensors are able to provide strong fluorescence upon DNA hybridization. Highly specific DNA sequence detection is possible by taking advantage of the specificity of DNA hybridization kinetics. The proposed system is capable of detecting all four dengue DNA serotypes (DENV1–4) without any cross-reactivity. A single tube assay format showed better hybridisation efficiency with higher fluorescence intensity generated and a lower detection limit compared to a cocktail probe assay format. The method was able to detect as low as 100 nM of amplified double stranded dengue DNA targets using both single and cocktail probe assays. This provides an interesting alternative approach for multiplex DNA sensing utilizing DNA silver nanoclusters as a reporter system.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

