
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-high throughput functional enrichment of large monoamine oxidase (MAO-N) libraries by fluorescence activated cell sorting†
Joanna C.
Sadler
 abc,
Andrew
Currin
abc and
Douglas B.
Kell
*abc
abc,
Andrew
Currin
abc and
Douglas B.
Kell
*abc
aSchool of Chemistry, The University of Manchester, 131 Princess St, Manchester M1 7DN, UK
bThe Manchester Institute of Biotechnology, The University of Manchester, 131 Princess St, Manchester M1 7DN, UK
cCentre for the Synthetic Biology of Fine and Speciality Chemicals (SYNBIOCHEM), The University of Manchester, 131 Princess St, Manchester M1 7DN, UK. E-mail: joanna.sadler@manchester.ac.uk; andrew.currin@manchester.ac.uk; dbk@manchester.ac.uk; Web: http://dbkgroup.org/@dbkell
First published on 10th September 2018
Abstract
Directed evolution enables the improvement and optimisation of enzymes for particular applications and is a valuable tool for biotechnology and synthetic biology. However, studies are often limited in their scope by the inability to screen very large numbers of variants to identify improved enzymes. One class of enzyme for which a universal, operationally simple ultra-high throughput (>106 variants per day) assay is not available is flavin adenine dinucleotide (FAD) dependent oxidases. The current high throughput assay involves a visual, colourimetric, colony-based screen, however this is not suitable for very large libraries and does not enable quantification of the relative fitness of variants. To address this, we describe an optimised method for the sensitive detection of oxidase activity within single Escherichia coli (E. coli) cells, using the monoamine oxidase from Aspergillus niger, MAO-N, as a model system. In contrast to other methods for the screening of oxidase activity in vivo, this method does not require cell surface expression, emulsion formation or the addition of an extracellular peroxidase. Furthermore, we show that fluorescence activated cell sorting (FACS) of large libraries derived from MAO-N under the assay conditions can enrich the library in functional variants at much higher rates than via the colony-based method. We demonstrate its use for directed evolution by identifying a new mutant of MAO-N with improved activity towards a novel secondary amine substrate. This work demonstrates, for the first time, an ultra-high throughput screening methodology widely applicable for the directed evolution of FAD dependent oxidases in E. coli.
Introduction
Directed evolution (DE) is a powerful tool for the improvement and optimisation of enzymes and has been widely adopted throughout the field of biotechnology.1–5 For example, engineered enzymes have been employed in the synthesis of pharmaceuticals,4,6,7 biofuels,8,9 detergents,10 animal feed supplements11,12 and biosensors.13 A typical DE workflow comprises three main stages: (1) identification of a suitable parent enzyme; (2) creation of genetic diversity; and (3) selection or screening for the variants with improved fitness. This cycle may be performed iteratively until a protein with the required fitness is obtained. While it is accepted that large, diverse variant libraries are required to navigate the vast fitness landscape intelligently,14,15 a lack of robust methods to screen or select from large numbers (>106) of variants efficiently represents a major bottleneck in many DE projects. To address this, ultra-high throughput screening techniques have been developed using fluorescence activated cells sorting (FACS),16–21 fluorescence activated droplet sorting (FADS)22,23 and microchamber arrays coupled with optical or robotic extraction.24,25Despite these advances, there are still many classes of biotechnologically important enzymes for which a generally applicable, ultra-high throughput assay is not available. One such example is that of flavin adenine dinucleotide (FAD) dependent oxidases, representing a large and diverse class of enzymes which catalyse the removal of a hydride equivalent from the substrate and release the oxidised product, producing hydrogen peroxide as a by-product. These enzymes use molecular oxygen to regenerate the cofactor in situ, making them operationally simple and economically viable for use in industrial settings and, as such, a popular target for DE. In particular, the monoamine oxidase from Aspergillus niger (MAO-N)26–30 has been heavily engineered to oxidise a wide range of primary, secondary and even tertiary amines.31–36 When coupled with a chemical reducing agent, a dynamic kinetic resolution can be achieved to yield enantiomerically pure amines, which are valuable building blocks for many fine and speciality chemicals, including many pharmaceuticals.37,38 The most widely used screening assay for MAO-N and variants thereof involves a colony-based screen in which colonies form a red colour when amine-oxidising enzyme activity produces H2O2 in the presence of an enzyme substrate, a chromogenic peroxidase substrate and an added (extracellular) peroxidase.37,39 However, for the analysis of large (>105) variant libraries that explore a wider area of sequence space, a higher throughput assay is required. Furthermore, as in silico approaches to protein engineering become increasingly popular, there is a need to develop methods for the quantification of the relative fitness of variants in a library, which is not catered for by the existing colony-based screen.14,40–42
While there have been no reports of an ultra-high throughput screening and sorting assay for MAO-N variants to date, two FACS assays for the directed evolution of FAD dependent glucose oxidase (GOx) in yeast have been reported.43,44 Both of these assays were based on a yeast surface expression system in a single or double emulsion. The first employs a vanadium bromoperoxidase-coupled fluorescence reporter in a double emulsion to detect hydrogen peroxide production,44 while the second uses horseradish peroxidase (HRP) and a fluorescein precursor in a single emulsion to couple GOx activity to a fluorescent output.43 In both cases, FACS of the droplets resulted in enrichment of the variant libraries in active clones. Despite these advances, the requirement for yeast surface expression, addition of an exogenous peroxidase and the delivery of substrates into the droplets limits the widespread application of these assays for the DE of other oxidases.
Given that a number of fluorogenic dyes for intracellular detection of reactive oxygen species (ROS) are commercially available, we were motivated to develop a method that could be used without the need for yeast cell-surface expression and droplet formation, using E. coli as a host. The present paper describes the development and optimisation of just such an assay and its application to identify improved variants through directed evolution. The overall process is shown in Fig. 1. First, E. coli cells are transformed with DNA encoding the enzyme of interest, or a variant library thereof, and enzyme expression is induced, such that each cell harbours a single variant. Next, the induced cells are treated with a fluorogenic probe which is sensitive to oxidation by H2O2 in the presence of an endogenous, intracellular peroxidase. When the amine substrate is added, cells expressing active variants exhibit higher fluorescence due to the production of more H2O2 and are sorted by FACS. We illustrate the utility of this process by employing the method to identify a mutant of MAO-N D526,37 exhibiting activity towards a substrate for which no activity is observed for the parent enzyme. Although demonstrated for MAO-N, this method could be applied to the analysis of any enzyme that produces H2O2 as a by-product of its reaction.
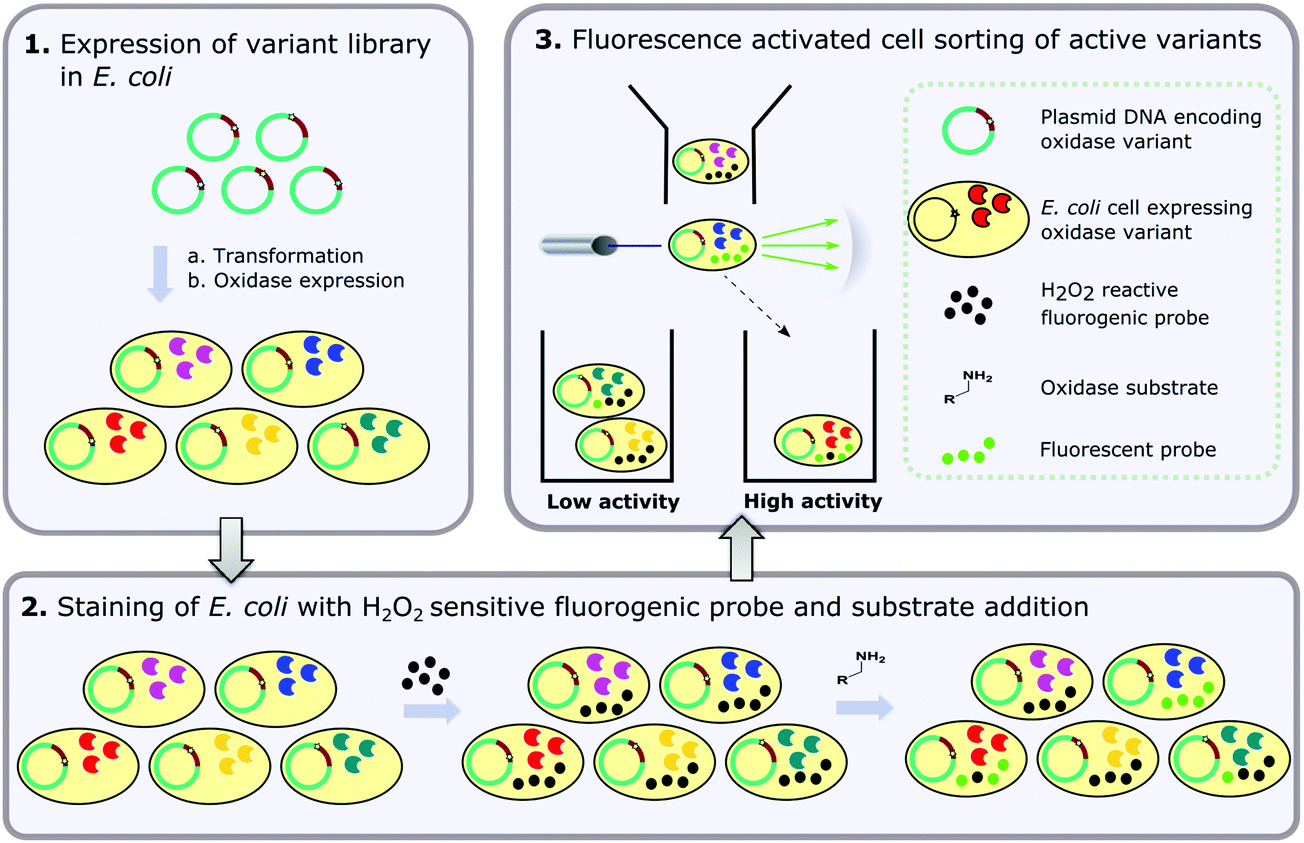 | ||
| Fig. 1 Overview of the flow cytometric assay for detection of amine oxidase activity in E. coli with the three main steps comprising (1) expression of the amine oxidase variant library in E. coli; (2) staining of E. coli cells with a fluorogenic probe which is sensitive to oxidation by H2O2 in the presence of an intracellular, endogenous peroxidase and (3) fluorescence activated cell sorting (FACS) of active variants based on fluorescence of the oxidised probe. | ||
Materials and methods
Preparation of MAO-N D5 gene construct and variant libraries
The codon optimised gene for MAO-N D5 was prepared using GeneGenie and SpeedyGenes as previously described5–7 and cloned into the pBbE2K expression vector,39 such that the encoded protein contains an N-terminal His10-tag. Variants were created using asymmetric PCR mutagenesis as previously described45,46 and cloned into the pBbE2K vector, with the resulting library encoding 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 variants. All cloning reactions were performed using the In-Fusion cloning kit (Takara Clontech) according to the supplier's protocol.
000 variants. All cloning reactions were performed using the In-Fusion cloning kit (Takara Clontech) according to the supplier's protocol.
Preparation of cell samples (single clones)
Chemically competent cells (T7 Express and 5-alpha) were purchased from New England Biolabs. 25 μL E. coli cells were transformed with 1–5 ng plasmid DNA according to the supplier's protocol. 3 mL of Terrific Broth (TB) containing 0.4% (v/v) glycerol and 50 μg mL−1 kanamycin was inoculated with a single colony from the transformation and incubated at 37 °C, 180 rpm (5.1 cm orbit diameter) and grown to an OD600 of 0.6 before inducing protein expression by the addition of anhydrotetracycline (as a 20 μM solution in ethanol) to a final concentration of 200 nM. Cultures were then incubated at 30 °C, 180 rpm (5.1 cm orbit diameter) for 4 hours before harvesting the cells by centrifugation (9500g, 5 minutes at 4 °C). The supernatant was discarded and the cell pellet stored at −80 °C for at least 3 hours prior to analysis by flow cytometry. This freeze–thaw step is crucial for reproducible probe uptake and cell fluorescence.Preparation of cell samples (variant libraries)
25 μL E. coli cells (T7 Express) were transformed with 1–5 ng plasmid DNA or 1 μL cloning reaction according to the supplier's protocol. 3 mL of Lysogeny Broth (LB) containing 50 μg mL−1 kanamycin was inoculated with 400 μL outgrowth (SOC medium) culture and incubated at 37 °C overnight. 3 mL TB containing 0.4% (v/v) glycerol and 50 μg mL−1 kanamycin was inoculated with 1% (v/v) overnight culture and incubated at 37 °C, 180 rpm (5.1 cm orbit diameter) to an OD600 of 0.6. Protein expression was induced and cells collected as described above for single clones.Analysis by flow cytometry
Cell pellets were thawed on ice and resuspended in 100 mM, pH 6.2 potassium phosphate buffer containing 100 mM KCl, 3 mM MgCl2 and 1% (v/v) butan-1-ol, to an OD600 of 1. C-H2DCFDA (as a 10 mM solution in DMSO) was added to a final concentration of 250 μM and the cells were incubated at 37 °C, 180 rpm (5.1 cm orbit diameter) for 1 hour. The cells were harvested by centrifugation (9500g, 5 minutes, 4 °C) and the supernatant discarded. The cell pellet was resuspended in 50 mM potassium phosphate buffer containing 100 mM KCl and 3 mM MgCl2, pH 7.5 and the reaction initiated by the addition of the amine substrate (as a 100 mM solution in 100 mM potassium phosphate buffer containing 100 mM KCl and 3 mM MgCl2) to a final concentration of 10 mM. The substrates used were (S)-(−)-α-methylbenzylamine (AMBA), amylamine (AA), benzylamine (BZA), butylamine (BTA), cyclohexylamine (CHA), or N-(4-methoxybenzyl)ethenamine (MBEA). Dyes and substrates were purchased from Sigma-Aldrich, Thermo Fisher Scientific or Enamine and used without further purification. Catalase from bovine liver (to a final concentration of 0.01 mg mL−1) was added at the time of substrate addition to scavenge H2O2 in the extracellular media, thereby minimising potential ‘cross-talk’ between cells. The reaction was quenched by the addition of ascorbic acid (as a 100 mM solution in Milli-Q water) to a final concentration of 10 mM. The flow cytometric cell sorter used was the Sony SH800S and the fluorescein product was excited by a single laser operating at 488 nm and detecting fluorescence in the FL2 channel (525/50 nm). Forward scatter (FSC) and side scatter (SSC) data were collected with detection thresholds of 2% and 30%, respectively and data output in files obeying the FCS 3.0 standard.41 Cells were gated on FSC-A vs. SSC-A and singlets identified by sub-gating on FSC-A vs. FSC-H. Flow cytometric data were analysed and figures were prepared using the FlowJo v10 software package.Functional enrichment of libraries by FACS
For isolation of functional variants from MAO-N D5 libraries, 1000 cells in the 99th percentile of the FL2-H channel were sorted into an empty collection tube. Amplification of the gene variants was performed by PCR containing 25 μL KAPA HiFi HotStart ReadyMix (Roche Diagnostics), 4 μL sorted cells from FACS, 500 nM forward primer (from 10 μM stock), 500 nM reverse primer (from 10 μM stock) and Milli-Q water to a final volume of 50 μL. The PCR was thermocycled as follows: 95 °C for 5 minutes, then 35 cycles of 98 °C for 20 seconds, 65 °C for 15 seconds and 72 °C for 40 seconds, followed by 72 °C for 2 minutes. The amplified MAO-N genes were purified by gel extraction using a NucleoSpin DNA purification kit (Macherey-Nagel) according to the manufacturer's protocol, eluting in 20 μL elution buffer. The resulting genes were cloned into the pBbE2K expression vector39 using the In-Fusion cloning kit (Takara Clontech) and gene expression performed as described above for variant libraries.Results
Assay development
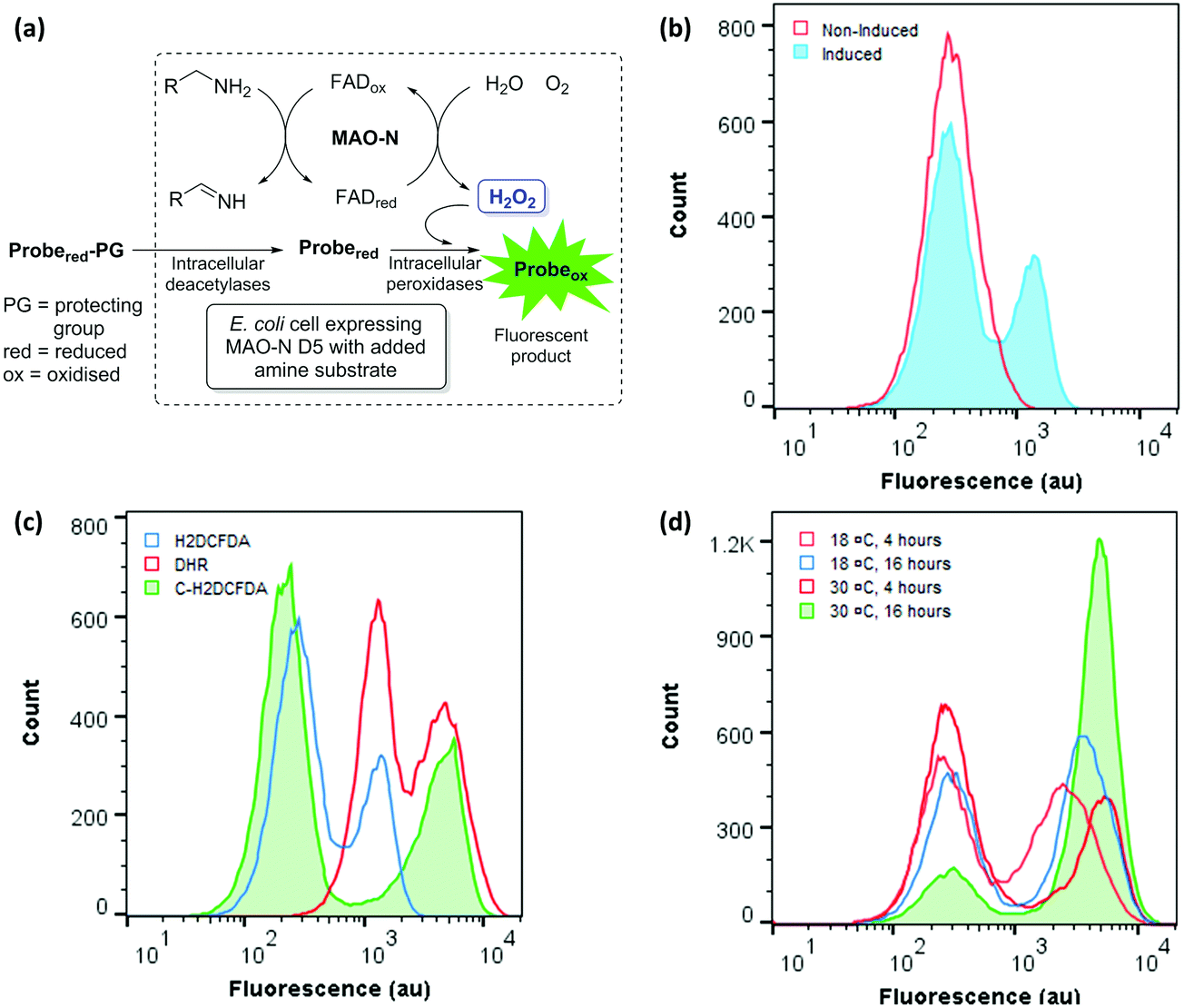 | ||
| Fig. 2 (a) Coupling of MAO-N activity to fluorescence in vivo. (b) FCM analysis of induced and non-induced E. coli cells containing the MAO-N expression construct, treated with H2DCFDA and incubated with AMBA. (c) Comparison of fluorogenic dyes: FCM analysis of E. coli cells expressing MAO-N D5 stained with H2DHDCF, C-H2DHDCF or DHR and incubated with AMBA. (d) Optimisation of conditions for the expression of MAO-N D5 in E. coli: FCM analysis of E. coli cells incubated at 18 °C or 30 °C for 4 or 16 hours after inducing MAO-N D5 expression. | ||
Encouraged by these data, three ‘dihydro’ dyes H2DCFDA, C-H2DCFDA and DHR were tested with induced and non-induced cells (Fig. 2c). While DHR was ineffective at distinguishing between the induced and non-induced samples, both H2DCFDA and C-H2DCFDA successfully reported a distinct second population for the induced sample, which is hereafter referred to as the ‘positive’ population. Cells displaying lower fluorescence (102–103 au) are hereafter referred to as the ‘negative population’. C-H2DCFDA was selected for further development as it displayed a higher fluorescence for the positive population relative to the negative population, which would enhance resolution during sorting experiments. The effect of induction time and temperature on the ratio of the two populations was also investigated, with the aim of maximising the proportion of cells in the positive population relative to the negative population (Fig. 2d). A higher proportion of the cells gave a positive fluorescence reading following a 4-hour induction than those that had been incubated overnight, with a 30 °C post induction temperature resulting in 85% of the total gated population exhibiting a ‘positive’ readout relative to the non-induced control.
Optimisation of assay conditions
Having obtained proof of concept and identified a suitable ROS probe, we sought to determine the linear range of the assay. A time course experiment was performed, in which the fluorescence of E. coli cells expressing MAO-N was measured for 60 minutes after substrate (AMBA) addition (Fig. 3a). A linear increase in mean fluorescence was observed over the first 30 minutes, followed by a slower rate of increase up to 1 hour after substrate addition. No further increase in fluorescence was observed after 1 hour (data not shown). By contrast, no change in fluorescence of non-induced cells was observed over 60 minutes, further confirming the specificity of the fluorescence in the induced samples.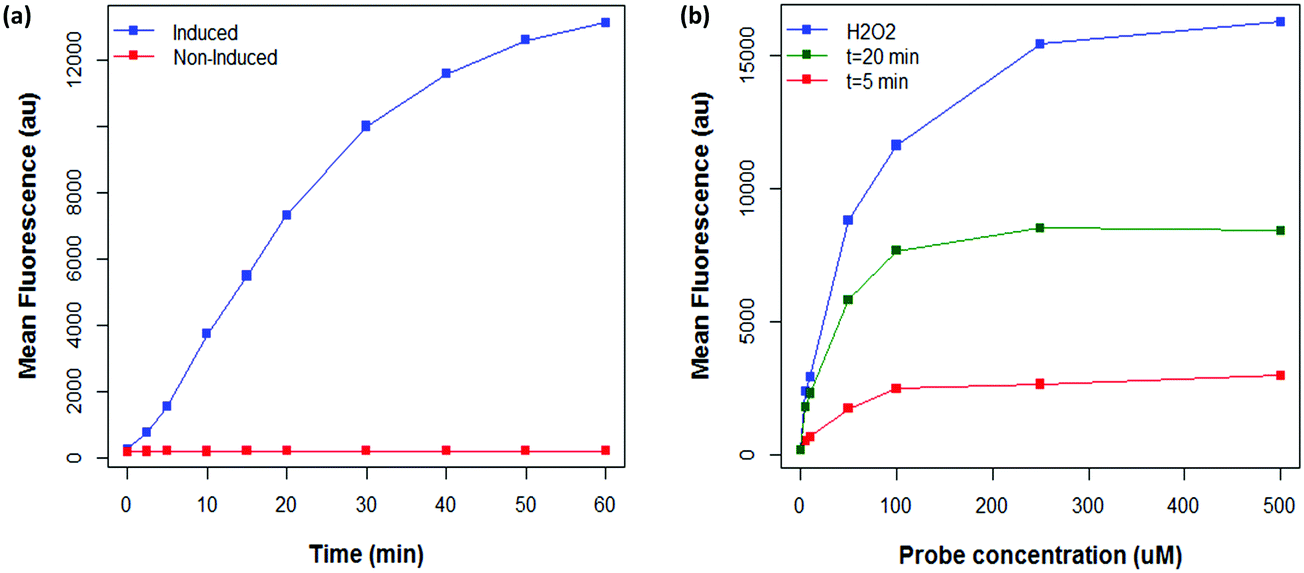 | ||
| Fig. 3 (a) Effect of substrate incubation time on fluorescence of E. coli cells expressing MAO-N D5 stained with 50 μM C-H2DCFDA and incubated with AMBA, compared to a non-induced control. (b) Effect of probe concentration on the maximum fluorescence of E. coli cells stained with C-H2DCFDA determined by the addition of 10 mM H2O2 (blue line) prior to analysis, in comparison to the fluorescence of C-H2DCFDA stained cells after incubation with AMBA for 5 (red line) and 20 (green line) minutes. | ||
It was hypothesised that the linear range of the assay may be increased by using a higher concentration of C-H2DCFDA, enabling faster mutants to be detected before the system became saturated. To this end, the maximum fluorescence of E. coli cells expressing MAO-N stained with a range of concentrations of C-H2DCFDA was measured by spiking samples with exogenous H2O2, which rapidly saturates the available probe such that cells exhibit the maximum possible fluorescence. Additionally, cells stained with each concentration of C-H2DCFDA were analysed after 5 and 20 minutes incubation time with the substrate AMBA (Fig. 3b). These data revealed 250 μM C-H2DCFDA to provide ∼2 fold higher maximum fluorescence than the initial conditions (50 μM C-H2DCFDA). At this higher probe concentration, cells were within the linear range of the assay after 20 minutes incubation with AMBA.
Given the time sensitivity of the assay, we were motivated to screen ROS scavenging agents to stop the assay prior to analysis, thereby improving operational simplicity (i.e. time taken to analyse samples) and increasing accuracy. Based on related publications, both ascorbic acid and sodium pyruvate were investigated.50,51 Out of these, ascorbic acid at a final concentration of 10 mM was found to quench the assay effectively at the desired time-point, such that the cells could be analysed up to an hour after quenching with no change in fluorescence.
Analysis of the same expression construct (MAO-N pBbE2K) in different host strains under the optimised assay conditions identified E. coli K12 cells to have high heterogeneity and exhibit an increase in fluorescence for the non-induced sample. In contrast, B strain E. coli cells showed less heterogeneity and lower background fluorescence and therefore were used in all further experiments.
We found that a freeze–thaw step was crucial for successful reproduction of the assay. When the freeze–thaw step was omitted, no positive population was observed for induced cells under the assay conditions. Furthermore, no shift in fluorescence was observed for induced or non-induced cells after spiking with exogenous H2O2. Based on these data, we consider that the C-H2DCFDA enters the cells via holes in the cell membrane caused by the freeze–thaw cycle (rather than via any transporter proteins in the cell membrane52,53). Cell permeability was improved by addition of butan-1-ol in the staining buffer used to resuspend cells after the freeze–thaw,54 with a final concentration of 1% (v/v) being found to give optimum results (data not shown).
Next, we investigated whether the assay could detect varying levels of activity of MAO-N D5 with a panel of primary amine substrates. To this end, the fluorescence of E. coli expressing MAO-N D5 under the assay conditions and incubated with each of the five amine substrates was analysed (Fig. 4). Consistent with the findings on substrate specificity seen in previous reports,33,36 induced cells incubated with CHA had the lowest fluorescence, followed by AMBA and then AA. As predicted from the in vitro kcat values, induced cells incubated with BTA and BZA had the highest fluorescence. These data validate this method as a simple and sensitive method for screening enzyme activity against a panel of substrates, which will have uses in mapping structure–activity relationships in future studies.
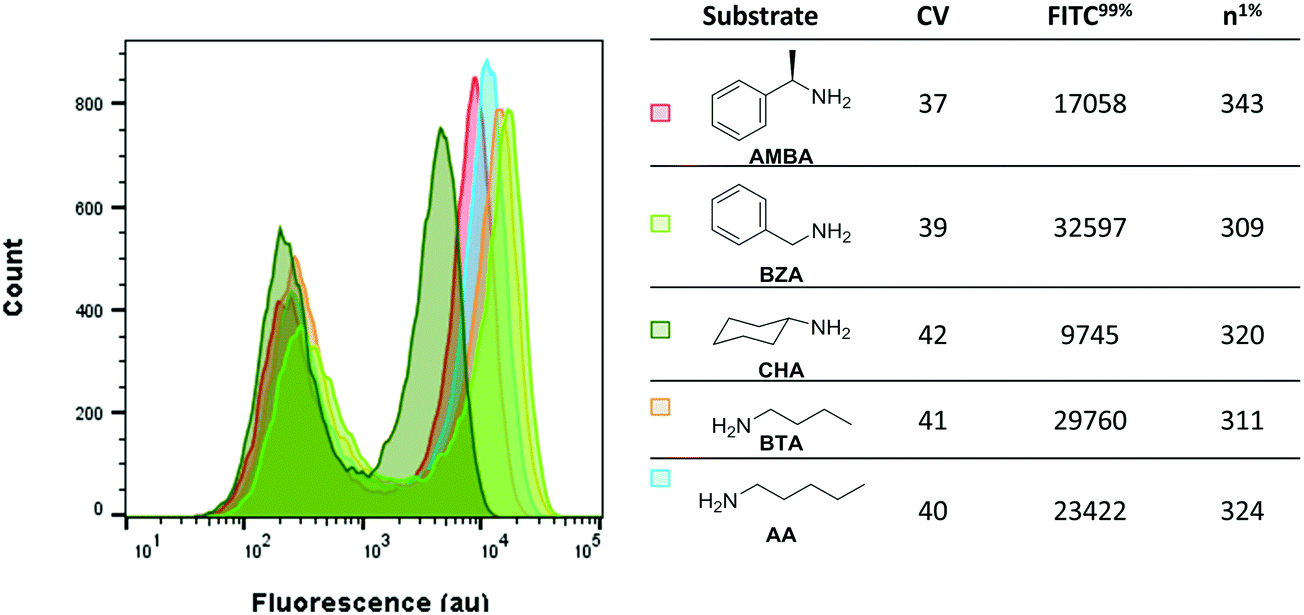 | ||
| Fig. 4 Fluorescence of C-H2DCFDA stained E. coli incubated with a library of amine substrates. Data were collected after 10 minutes incubation time in each case and was gated on FSC-A vs. SSC-A. n1%: Number of cells with fluorescence in the 99% percentile. | ||
Screening and identification of improved enzyme variants
A major advantage of the flow cytometric assay to analyse oxidase activity in comparison to the solid phase assay is the ability to quickly analyse large numbers of variants (our instrument measures up to 10000 cells per second). This is particularly useful in directed evolution projects, where it is necessary to screen large variant libraries in order to navigate vast and rugged sequence spaces efficiently.14 Coupling flow cytometry to FACS enables the isolation of cells with high fluorescence and, as MAO-N variants with a higher turnover number are expected to display higher fluorescence under the assay conditions, FACS can be utilised to isolate improved variants. To test this hypothesis, a combinatorial active site testing (CASTing) library of 40000 variants was prepared as previously described,39,55–58 and screened for activity using the flow cytometric (FCM) assay using the secondary amine N-(4-methoxybenzyl)ethenamine (MBEA) as a substrate, which is not oxidised by MAO-N D5, D9 or D11.50 Using the D5 variant as the negative baseline, cells from the library with elevated fluorescence were sorted and subjected to a second round of screening (Fig. 5a).
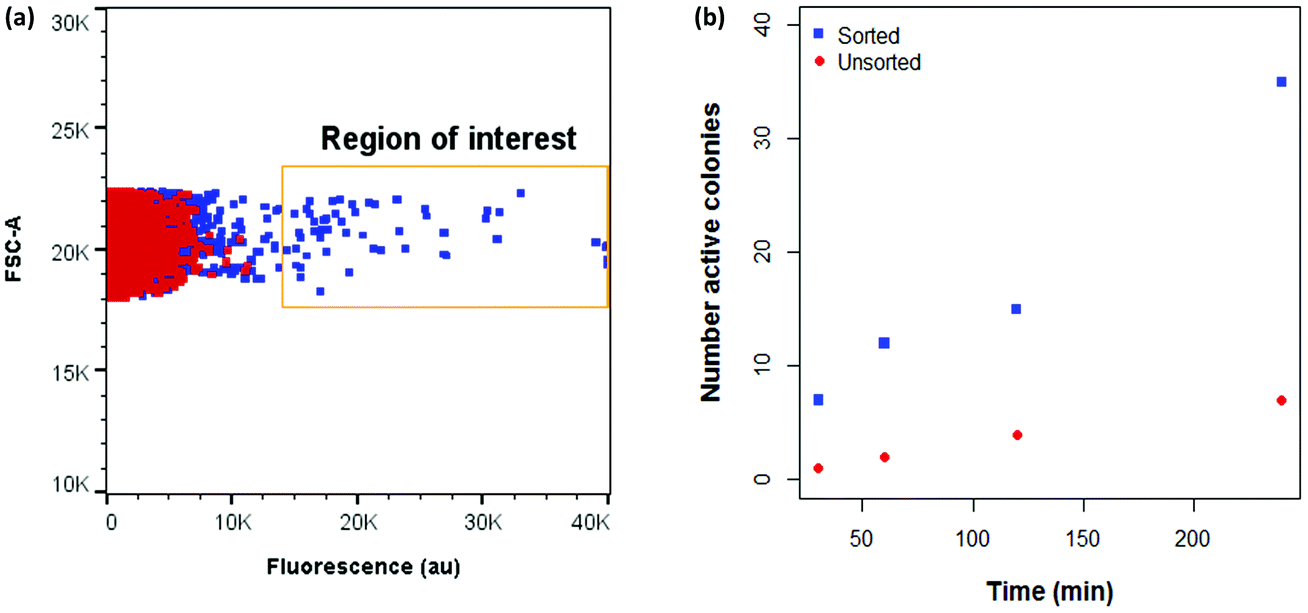 | ||
| Fig. 5 (a) Comparison between fluorescence of CASTing library and MAO-N D5 with MBEA under FCM assay conditions. (b) Functional enrichment of a library of MAO-N variants incubated with MBEA after two rounds of sorting. ROI: Region of interest. | ||
In initial experiments, sorted cells were out-grown in LB medium and the resultant culture used as inoculant for an expression culture to prepare samples of the sorted variants. However, in all cases, a single, negative population was observed on the second round of analysis. This suggests that cells harbouring active MAO-N variants cannot grow at the same rate as negative cells following FACS, thus leading to a dominance of negative variants in the culture. Further experiments revealed that for efficient recovery of improved variants it was necessary to amplify the sorted MAO-N variants’ DNA by polymerase chain reaction (PCR), ligate into the expression vector and transform into fresh E. coli cells for expression and re-analysis using the FCM assay.
After two iterative rounds of sorting, sorted variants were analysed by the previously described solid phase screen37,39 and the number of active colonies compared to that for the unsorted CASTing library was counted. As shown in Fig. 5b, ∼5–7× more colonies from the sorted displayed oxidase activity towards MBEA than from the unsorted library, indicating functional enrichment through the FACS assay. The most active clone from the solid phase screen of the sorted CASTing library was identified as the double mutant MAO-N D5 T93G/F466T, with both mutations being proximal to the cofactor FAD and to the putative substrate binding site (Fig. 6a). Finally, the original CASTing library, MAO-N D5 and MAO-N D5 T93F/466T, were analysed under the assay conditions after incubation with MBEA. Due to low levels of substrate uptake by the cells, 1% (v/v) butan-1-ol was added at the time of substrate addition to increase the permeability of the E. coli cell membranes to the substrate during the assay, resulting in sufficient uptake of MBEA to observe a marked increase in fluorescence of cells expressing MAO-N D5 T93F/466T relative to the original CASTing library and MAO-N D5 (Fig. 6b).
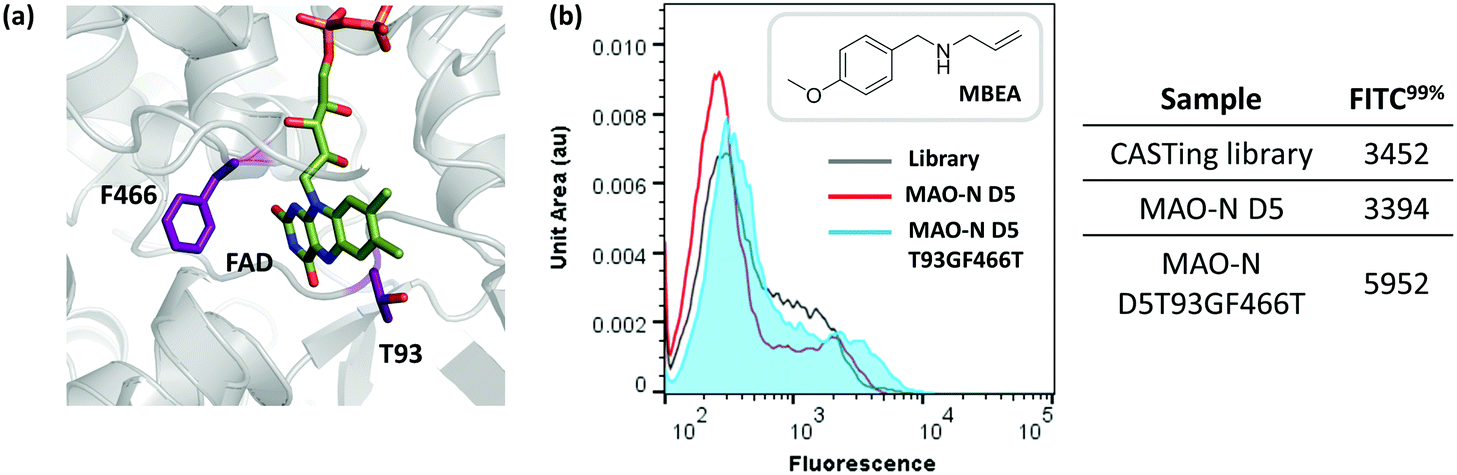 | ||
| Fig. 6 (a) Flow cytometric analysis of MAO-N D5 T93GF466T activity with MBEA in comparison to MAO-N D5 and the variant library under assay conditions. (b) Position of T93 and F466 relative to the cofactor, FAD, in the active site of MAO-N D5 (PBD accession code: 2VVM). | ||
Discussion and conclusion
As a reactive oxygen species (ROS), hydrogen peroxide (H2O2) is generated naturally through numerous cellular processes. In eukaryotes, production of H2O2 can be indicative of oxidative stress and, as such, multiple fluorescent probes have been developed to act as oxidative stress indicators.47,59 In this study, we utilise one such probe (C-H2DCFDA) to assay directly the activity of a recombinantly expressed amine oxidase within an E. coli cell. Under the optimised assay conditions, it is clear that cells expressing MAO-N D5 have a substrate and time-dependent fluorescence. As with most bacterial cultures, there is considerable heterogeneity, and more than can be ascribed merely to variation in the cell cycle.60–65 Nonetheless, through iterative rounds of FACS, it was possible to enrich libraries with amine oxidase variants with improved function. We have demonstrated the utility of this assay by identifying a novel, double mutant of MAO-N D5, which shows oxidase activity toward a secondary amine substrate which was not oxidised by the parent enzyme. By testing relevant ROS probes we could rapidly identify those capable of detecting intracellular MAO-N activity. However, considerable optimisation of the assay conditions and cell treatment was required to detect, sort and recover enzyme variants with improved catalytic activity reliably. Conditions such as E. coli strain, buffer constituents, reaction quenching and PCR recovery of sorted populations proved pivotal in obtaining a robust protocol capable of rapid detection of oxidase activity. By identifying a new MAO-N variant with activity towards a novel substrate we demonstrate its use in a directed evolution study, providing a quick and sensitive means to screen large variant libraries and enrich variants exhibiting a desired catalytic activity. While oxidase activity has been detected using yeast in emulsion droplets,43,44,66 to our knowledge this is the first demonstration that oxidase activity from recombinant protein can be assayed within a single prokaryotic cell and used to enrich enzyme variants exhibiting improved activity.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Biotechnology and Biological Sciences Research Council for support of this work via the BioCatNet, as well as through SYNBIOCHEM (BBSRC BB/M017702/1).References
- F. H. Arnold, Directed Evolution: Bringing New Chemistry to Life, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Engineering the third wave of biocatalysis, Nature, 2012, 485, 185–194 CrossRef PubMed.
- C. A. Denard, H. Ren and H. Zhao, Improving and repurposing biocatalysts via directed evolution, Curr. Opin. Chem. Biol., 2015, 25, 55–64 CrossRef PubMed.
- G. Li, J. Wang and M. T. Reetz, Biocatalysts for the pharmaceutical industry created by structure-guided directed evolution of stereoselective enzymes, Bioorg. Med. Chem., 2018, 26, 1241–1251 CrossRef PubMed.
- M. C. Bassalo, R. Liu and R. T. Gill, Directed evolution and synthetic biology applications to microbial systems, Curr. Opin. Biotechnol., 2016, 39, 126–133 CrossRef PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture, Science, 2010, 329, 305–309 CrossRef PubMed.
- Z. Sun, R. Lonsdale, A. Ilie, G. Li, J. Zhou and M. T. Reetz, Catalytic Asymmetric Reduction of Difficult-to-Reduce Ketones: Triple-Code Saturation Mutagenesis of an Alcohol Dehydrogenase, ACS Catal., 2016, 6, 1598–1605 CrossRef.
- J. Lian, Y. Li, M. HamediRad and H. Zhao, Directed evolution of a cellodextrin transporter for improved biofuel production under anaerobic conditions in Saccharomyces cerevisiae, Biotechnol. Bioeng., 2014, 111, 1521–1531 CrossRef PubMed.
- S. Atsumi and J. C. Liao, Directed evolution of Methanococcus jannaschii citramalate synthase for biosynthesis of 1-propanol and 1-butanol by Escherichia coli, Appl. Environ. Microbiol., 2008, 74, 7802–7808 CrossRef PubMed.
- L. Vojcic, C. Pitzler, G. Körfer, F. Jakob, R. Martinez, K.-H. Maurer and U. Schwaneberg, Advances in protease engineering for laundry detergents, Nat. Biotechnol., 2015, 32, 629–634 Search PubMed.
- A. V. Shivange, A. Serwe, A. Dennig, D. Roccatano, S. Haefner and U. Schwaneberg, Directed evolution of a highly active Yersinia mollaretii phytase, Appl. Microbiol. Biotechnol., 2012, 95, 405–418 CrossRef PubMed.
- A. V. Shivange, D. Roccatano and U. Schwaneberg, Iterative key-residues interrogation of a phytase with thermostability increasing substitutions identified in directed evolution, Appl. Microbiol. Biotechnol., 2016, 100, 227–242 CrossRef PubMed.
- M. Campàs, B. Prieto-Simón and J.-L. Marty, A review of the use of genetically engineered enzymes in electrochemical biosensors, Semin. Cell Dev. Biol., 2009, 20, 3–9 CrossRef PubMed.
- A. Currin, N. Swainston, P. J. Day and D. B. Kell, Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently, Chem. Soc. Rev., 2015, 44, 1172–1239 RSC.
- D. B. Kell, Scientific discovery as a combinatorial optimisation problem: how best to navigate the landscape of possible experiments?, Bioessays, 2012, 34, 236–244 CrossRef PubMed.
- G. Yang and S. G. Withers, Ultrahigh-throughput FACS-based screening for directed enzyme evolution, ChemBioChem, 2009, 10, 2704–2715 CrossRef PubMed.
- T. H. Yoo, M. Pogson, B. L. Iverson and G. Georgiou, Directed Evolution of Highly Selective Proteases by Using a Novel FACS-Based Screen that Capitalizes on the p53 Regulator MDM2, ChemBioChem, 2012, 13, 649–653 CrossRef PubMed.
- S. J. Kwon, R. Petri, A. L. DeBoer and C. Schmidt-Dannert, A high-throughput screen for porphyrin metal chelatases: application to the directed evolution of ferrochelatases for metalloporphyrin biosynthesis, ChemBioChem, 2004, 5, 1069–1074 CrossRef PubMed.
- J. M. Duarte, I. Barbier and Y. Schaerli, Bacterial Microcolonies in Gel Beads for High-Throughput Screening of Libraries in Synthetic Biology, ACS Synth. Biol., 2017, 6, 1988–1995 CrossRef PubMed.
- A. Espargaró, R. Sabate and S. Ventura, Thioflavin-S staining coupled to flow cytometry. A screening tool to detect in vivo protein aggregation, Mol. Biosyst., 2012, 8, 2839 RSC.
- A. Chandrasekaran, K. Deng, C.-Y. Koh, T. Takasuka, L. F. Bergeman, B. G. Fox, P. D. Adams and A. K. Singh, A universal flow cytometry assay for screening carbohydrate-active enzymes using glycan microspheres, Chem. Commun., 2013, 49, 5441–5443 RSC.
- P.-Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvönen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Ultrahigh-throughput discovery of promiscuous enzymes by picodroplet functional metagenomics, Nat. Commun., 2015, 6, 10008 CrossRef PubMed.
- F. Gielen, R. Hours, S. Emond, M. Fischlechner, U. Schell and F. Hollfelder, Ultrahigh-throughput-directed enzyme evolution by absorbance-activated droplet sorting (AADS), Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7383–E7389 CrossRef PubMed.
- B. Chen, S. Lim, A. Kannan, S. C. Alford, F. Sunden, D. Herschlag, I. K. Dimov, T. M. Baer and J. R. Cochran, High-throughput analysis and protein engineering using microcapillary arrays, Nat. Chem. Biol., 2016, 12, 76–81 CrossRef PubMed.
- H. Xiao, Z. Bao and H. Zhao, High Throughput Screening and Selection Methods for Directed Enzyme Evolution, Ind. Eng. Chem. Res., 2015, 54, 4011–4020 CrossRef PubMed.
- K. E. Atkin, R. Reiss, V. Koehler, K. R. Bailey, S. Hart, J. P. Turkenburg, N. J. Turner, A. M. Brzozowski and G. Grogan, The Structure of Monoamine Oxidase from Aspergillus niger Provides a Molecular Context for Improvements in Activity Obtained by Directed Evolution, J. Mol. Biol., 2008, 384, 1218–1231 CrossRef PubMed.
- H. Gaweska and P. F. Fitzpatrick, Structures and Mechanism of the Monoamine Oxidase Family, Biomol. Concepts, 2011, 2, 365–377 Search PubMed.
- K. M. Andrea, C. Michal, R. Anton, G. Nicholas and J. T. M. Rebroš, Cloning and upscale production of monoamine oxidase N (MAO-N D5) by Pichia pastoris, Biotechnol. Lett., 2018, 40, 127–133 CrossRef PubMed.
- K. R. Bailey, A. J. Ellis, R. Reiss, T. J. Snape and N. J. Turner, A template-based mnemonic for monoamine oxidase (MAO-N) catalyzed reactions and its application to the chemo-enzymatic deracemisation of the alkaloid (±)-crispine A, Chem. Commun., 2007, 35, 3640–3642 RSC.
- P. Zajkoska, M. Rosenberg, R. Heath, K. J. Malone, R. Stloukal, N. J. Turner and M. Rebroš, Immobilised whole-cell recombinant monoamine oxidase biocatalysis, Appl. Microbiol. Biotechnol., 2015, 99, 1229–1236 CrossRef PubMed.
- G. Li, P. Yao, R. Gong, J. Li, P. Liu, R. Lonsdale, Q. Wu, J. Lin, D. Zhu and M. T. Reetz, Simultaneous engineering of an enzyme's entrance tunnel and active site: the case of monoamine oxidase MAO-N, Chem. Sci., 2017, 8, 4093–4099 RSC.
- Z. Chen, Y. Ma, M. He, H. Ren, S. Zhou, D. Lai, Z. Wang and L. Jiang, Semi-rational Directed Evolution of Monoamine Oxidase for Kinetic Resolution of rac-Mexiletine, Appl. Biochem. Biotechnol., 2015, 176, 2267–2278 CrossRef PubMed.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, Engineering an enantioselective amine oxidase for the synthesis of pharmaceutical building blocks and alkaloid natural products, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef PubMed.
- D. Ghislieri and N. J. Turner, Biocatalytic Approaches to the Synthesis of Enantiomerically Pure Chiral Amines, Top. Catal., 2014, 57, 284–300 CrossRef.
- I. Rowles, K. J. Malone, L. L. Etchells, S. C. Willies and N. J. Turner, Directed Evolution of the Enzyme Monoamine Oxidase (MAO-N): Highly Efficient Chemo-enzymatic Deracemisation of the Alkaloid (±)-Crispine A, ChemCatChem, 2012, 4, 1259–1261 CrossRef.
- S. Herter, F. Medina, S. Wagschal, C. Benhaïm, F. Leipold and N. J. Turner, Mapping the substrate scope of monoamine oxidase (MAO-N) as a synthetic tool for the enantioselective synthesis of chiral amines, Bioorg. Med. Chem., 2018, 26, 1338–1346 CrossRef PubMed.
- Y. Ahn, S. B. Ko, M. J. Kim, J. Park, M. Alexeeva, A. Enright, M. J. Dawson, M. Mahmoudian, N. J. Turner, M. Carson, D. H. Johnson, H. McDonald, C. Brouillette, L. J. DeLucas, E. T. Farinas, U. Schwaneberg, A. Glieder, F. H. Arnold, C. E. Hoben, L. Kanupp, J.-E. Bäckvall, A. M. G. Meeting, B. Schilling, K. Lerch, T. P. Singer, V. Yankovskaya, S. Bernard, C. Cronin, S. O. Sablin, B. Soediono, K. D. Wittrup, L. Y. Yampolsky, E. Tennessee, J. City, K. E. Atkin, R. Reiss, V. Koehler, K. R. Bailey, S. Hart, J. P. Turkenburg, N. J. Turner, A. M. Brzozowski, G. Grogan, M. I. Youshko, F. Van Rantwijk, R. a. Sheldon, J. S. M. Samec, N. Hermanns, J. Ba, H. E. Alida, O. Pàmies, A. H. Éll, J.-E. Bäckvall, R. Carr, M. Alexeeva, M. J. Dawson, V. Gotor-Fern ndez, N. J. Turner, C. E. Humphrey, A. Enright, T. S. C. Eve, M. J. Dawson, N. J. Turner, M. Stirling, J. Blacker, M. I. Page, C. Chen, Y. Fujimoto, G. Girdaukas, C. J. Sih, C. J. Dunsmore, R. Carr, T. Fleming, N. J. Turner and R. Berg, Deracemization of α-methylbenzylamine using an enzyme obtained by in vitro evolution, Tetrahedron Lett., 2008, 43, 1247–1250 Search PubMed.
- J. H. Schrittwieser, B. Groenendaal, V. Resch, D. Ghislieri, S. Wallner, E.-M. Fischereder, E. Fuchs, B. Grischek, J. H. Sattler, P. Macheroux, N. J. Turner and W. Kroutil, Deracemization By Simultaneous Bio-oxidative Kinetic Resolution and Stereoinversion, Angew. Chem., Int. Ed., 2014, 53, 3731–3734 CrossRef PubMed.
- A. Currin, N. Swainston, P. J. Day and D. B. Kell, SpeedyGenes: An improved gene synthesis method for the efficient production of error-corrected, synthetic protein libraries for directed evolution, Protein Eng., Des. Sel., 2014, 27, 273–280 CrossRef PubMed.
- A. Nobili, Y. Tao, I. V. Pavlidis and T. Van Den Bergh, Simultaneous Use of in Silico Design and a Correlated Mutation Network as a Tool To Efficiently Guide Enzyme Engineering, ChemBioChem, 2015, 16, 805–810 CrossRef PubMed.
- R. Fox, Directed molecular evolution by machine learning and the influence of nonlinear interactions, J. Theor. Biol., 2005, 234, 187–199 CrossRef PubMed.
- S. A. Higgins and D. F. Savage, Protein Science by DNA Sequencing: How Advances in Molecular Biology Are Accelerating Biochemistry, Biochemistry, 2018, 57, 38–46 CrossRef PubMed.
- R. Ostafe, R. Prodanovic, J. Nazor and R. Fischer, Ultra-high-throughput screening method for the directed evolution of glucose oxidase, Chem. Biol., 2014, 21, 414–421 CrossRef PubMed.
- R. Prodanovic, R. Ostafe, M. Blanusa and U. Schwaneberg, Vanadium bromoperoxidase-coupled fluorescent assay for flow cytometry sorting of glucose oxidase gene libraries in double emulsions, Anal. Bioanal. Chem., 2012, 404, 1439–1447 CrossRef PubMed.
- J. C. Sadler, L. Green, N. Swainston, D. B. Kell and A. Currin, Fast and Flexible Synthesis of Combinatorial Libraries for Directed Evolution, Methods Enzymol., 2018 DOI:10.1016/BS.MIE.2018.04.006.
- C. I. Wooddell and R. R. Burgess, Use of asymmetric PCR to generate long primers and single-stranded DNA for incorporating cross-linking analogs into specific sites in a DNA probe, Genome Res., 1996, 6, 886–892 CrossRef PubMed.
- S. G. Rhee, T.-S. Chang, W. Jeong and D. Kang, Methods for detection and measurement of hydrogen peroxide inside and outside of cells, Mol. Cells, 2010, 29, 539–549 CrossRef PubMed.
- J. P. Robinson, V. Patsekin, C. Holdman, K. Ragheb, J. Sturgis, R. Fatig, L. V. Avramova, B. Rajwa, V. J. Davisson, N. Lewis, P. Narayanan, N. Li and C. W. Qualls, High-Throughput Secondary Screening at the Single-Cell Level, J. Lab. Autom., 2013, 18, 85–98 CrossRef PubMed.
- B. Kalyanaraman, M. Hardy, R. Podsiadly, G. Cheng and J. Zielonka, Recent developments in detection of superoxide radical anion and hydrogen peroxide: Opportunities, challenges, and implications in redox signaling, Arch. Biochem. Biophys., 2017, 617, 38–47 CrossRef PubMed.
- S. Hosaka, M. Obuki, J. Nakajima and M. Suzuki, Comparative study of antioxidants as quenchers or scavengers of reactive oxygen species based on quenching of MCLA-dependent chemiluminescence, Luminescence, 2005, 20, 419–427 CrossRef PubMed.
- R. Franco, M. I. Panayiotidis and J. A. Cidlowski, Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation, J. Biol. Chem., 2007, 282, 30452–30465 CrossRef PubMed.
- D. B. Kell and S. G. Oliver, How drugs get into cells: tested and testable predictions to help discriminate between transporter-mediated uptake and lipoidal bilayer diffusion, Front. Pharmacol., 2014, 5, 231–263 Search PubMed.
- D. B. Kell, N. Swainston, P. Pir and S. G. Oliver, Membrane transporter engineering in industrial biotechnology and whole cell biocatalysis, Trends Biotechnol., 2015, 33, 237–246 CrossRef PubMed.
- E. Fletcher, T. Pilizota, P. R. Davies, A. McVey and C. E. French, Characterization of the effects of n-butanol on the cell envelope of E. coli, Appl. Microbiol. Biotechnol., 2016, 100, 9653–9659 CrossRef PubMed.
- A. Currin, N. Swainston, P. J. Day and D. B. Kell, in Methods in molecular biology, Clifton, N.J., 2017, vol. 1472, pp. 63–78 Search PubMed.
- M. T. Reetz, J. D. Carballeira, J. Peyralans, H. Höbenreich, A. Maichele and A. Vogel, Expanding the Substrate Scope of Enzymes: Combining Mutations Obtained by CASTing, Chem. – Eur. J., 2006, 12, 6031–6038 CrossRef PubMed.
- M. T. Reetz, L. W. Wang and M. Bocola, Directed evolution of enantioselective enzymes: Iterative cycles of CASTing for probing protein-sequence space, Angew. Chem., Int. Ed., 2006, 45, 1236–1241 CrossRef PubMed.
- S. H. Ke and E. L. Madison, Rapid and efficient site-directed mutagenesis by single-tube ‘megaprimer’ PCR method, Nucleic Acids Res., 1997, 25, 3371–3372 CrossRef PubMed.
- A. Gomes, E. Fernandes and J. L. F. C. Lima, Fluorescence probes used for detection of reactive oxygen species, J. Biochem. Biophys. Methods, 20005, 65, 45–80 CrossRef PubMed.
- A. S. Kaprelyants and D. B. Kell, Rapid assessment of bacterial viability and vitality by rhodamine 123 and flow cytometry, J. Appl. Bacteriol., 1992, 72, 410–422 CrossRef.
- A. S. Kaprelyants and D. B. Kell, Dormancy in Stationary-Phase Cultures of Micrococcus luteus: Flow Cytometric Analysis of Starvation and Resuscitation, Appl. Environ. Microbiol., 1993, 59, 3187–3196 Search PubMed.
- D. B. Kell, H. M. Ryder, A. S. Kaprelyants and H. V. Westerhoff, Quantifying heterogeneity: flow cytometry of bacterial cultures, Antonie Van Leeuwenhoek, 1991, 60, 145–158 CrossRef PubMed.
- J. R. S. Newman, S. Ghaemmaghami, J. Ihmels, D. K. Breslow, M. Noble, J. L. DeRisi and J. S. Weissman, Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise, Nature, 2006, 441, 840–846 CrossRef PubMed.
- B. M. Martins and J. C. Locke, Microbial individuality: how single-cell heterogeneity enables population level strategies, Curr. Opin. Microbiol., 2015, 24, 104–112 CrossRef PubMed.
- D. Kell, M. Potgieter and E. Pretorius, Individuality, phenotypic differentiation, dormancy and ‘persistence’ in culturable bacterial systems: commonalities shared by environmental, laboratory, and clinical microbiology, F1000Research, 2015, 4, 179 Search PubMed.
- T. W. Johannes, R. D. Woodyer and H. Zhao, in Enzyme Assays, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2006, pp. 77–93 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an00851e |
| This journal is © The Royal Society of Chemistry 2018 |