
An integrated strategy for highly sensitive phosphoproteome analysis from low micrograms of protein samples†
 *ac
*ac
Abstract
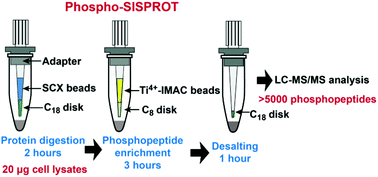
Phosphoproteomics has become a popular proteomic technology for exploring cellular signaling networks. However, current approaches often require milligrams of protein samples which hamper their applications for translational studies with limited starting materials. In this study, we aimed to challenge the lowest starting material limit for phosphoproteome profiling. By carefully optimizing the well-established high-pH reversed-phase (RP) fractionation plus Ti4+-IMAC enrichment strategy, we achieved the identification of 15 260 and 8936 unique phosphopeptides from only 500 μg and 250 μg predigested peptides, respectively. To further improve the sensitivity of phosphoproteome analysis for low micrograms of protein samples, we developed an integrated strategy, termed Phospho-SISPROT. This technology integrates three tips in tandem for protein digestion by the simple and integrated spintip-based proteomics technology (SISPROT), phosphopeptide enrichment by the Ti4+-IMAC tip, and desalting by the StageTip, respectively, which could dramatically reduce the phosphoproteome analysis time from a couple of days to only 6 hours and improve the system sensitivity. The flow through of Phospho-SISPROT could be reused for the global protein identification, which is very helpful for accurate phosphoproteome analysis with limited starting materials. More than 5500 and 600 unique phosphopeptides were respectively identified from 20 μg and 1 μg pervanadate treated HEK 293T cell lysates processed by the Phospho-SISPROT. To the best of our knowledge, this performance is the highest reported to date by using the standard LC-MS/MS setup. We expect that the Phospho-SISPROT and the optimized high-pH RP fractionation plus Ti4+-IMAC enrichment strategy will be well suited for highly sensitive phosphoproteome analysis of rare biological samples.
- This article is part of the themed collection: Next wave advances in single cell analyses
 Please wait while we load your content...
Please wait while we load your content...

