
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel, fast and sensitive supercritical fluid chromatography-tandem mass spectrometry (SFC-MS/MS) method for analysis of arachidonic acid metabolites
S. J.
Kumari A. Ubhayasekera†
 *,
Santosh R.
Acharya†
and
Jonas
Bergquist
*
*,
Santosh R.
Acharya†
and
Jonas
Bergquist
*
Department of Chemistry-BMC, Analytical Chemistry and Neurochemistry, Uppsala University, Sweden. E-mail: kumari.ubhayasekera@kemi.uu.se; Jonas.bergquist@kemi.uu.se
First published on 20th June 2018
Abstract
The development of a rapid, sensitive and reliable method for the quantification of bioactive arachidonic acid metabolites (AA-metabolites) in biological samples is quite challenging due to the minute concentration, short half-life and their structural complexity arising from different isomers. In this study, a simple, fast and environmentally friendly supercritical fluid chromatography-tandem mass spectrometry (SFC-MS/MS) method was developed and validated for simultaneous measurement of five (PGD2, PGE2, PGF2α, 6KetoPGF1α and LTB4) AA-metabolites in biological samples. These analytes were extracted by protein precipitation followed by separation and quantification. The analysis was completed within 3 minutes. The matrix matched linear calibration ranged from 0.5–100 ng mL−1 (r2 ≥ 0.995), whilst, the limit of quantification of PGD2, PGE2, PGF2α, and LTB4 was 0.5 ng mL−1 and was 2.5 ng mL−1 for 6KetoPGF1α. The interday and intraday precisions of the method were less than 15% while the accuracy of most of the analytes varied between 83 and 109%. Finally, as a proof of concept, the method was successfully applied for the determination of eicosanoids in human samples, which expands the possibility to explore physiological states, disease phenotypes, and novel biomarkers.
Introduction
The arachidonic fatty acid (AA, C20:4n-6) is profusely present in biological cell membranes which can be oxidized forming a series of biologically active lipid metabolites mainly through cyclooxygenase (COXs) and lipoxygenase (LOXs) pathways (Fig. 1). AA-metabolites commonly known as eicosanoids include various prostaglandins (PGs), leukotrienes (LTs), thromboxanes (TXs) and prostacyclin (PGI2).1,2 Different eicosanoids: prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), prostaglandin F2α (PGF2α), thromboxane A2 (TXA2), prostacyclin (PGI2), and leukotriene A4 (LTA4) are produced from intermediate PGH2 or hydroperoxy eicosatetraenoic (HPETE) by specific synthases. Most of the eicosanoids are stable whereas TXA2, PGI2, and LTA4 are highly susceptible to nonenzymatic hydrolysis and form TXB2, 6KetoPGF1α and LTB4, respectively under physiological conditions. AA-metabolites can be esterified to phospholipids and then released in free form by phospholipases, which can be detected in various bio-fluids including cerebrospinal fluid (CSF), plasma, broncho-alveolar lavage fluid, amniotic fluid, (etc.).2,3 These bioactive lipids play important roles in regulating cell proliferation, apoptosis, tissue repair, inflammation, blood clotting and immune cell behavior. They are also linked to conditions such as heart and neurodegenerative diseases, migraine, epilepsy, diabetic vascular complication, atherosclerosis, cancers, and allergies.1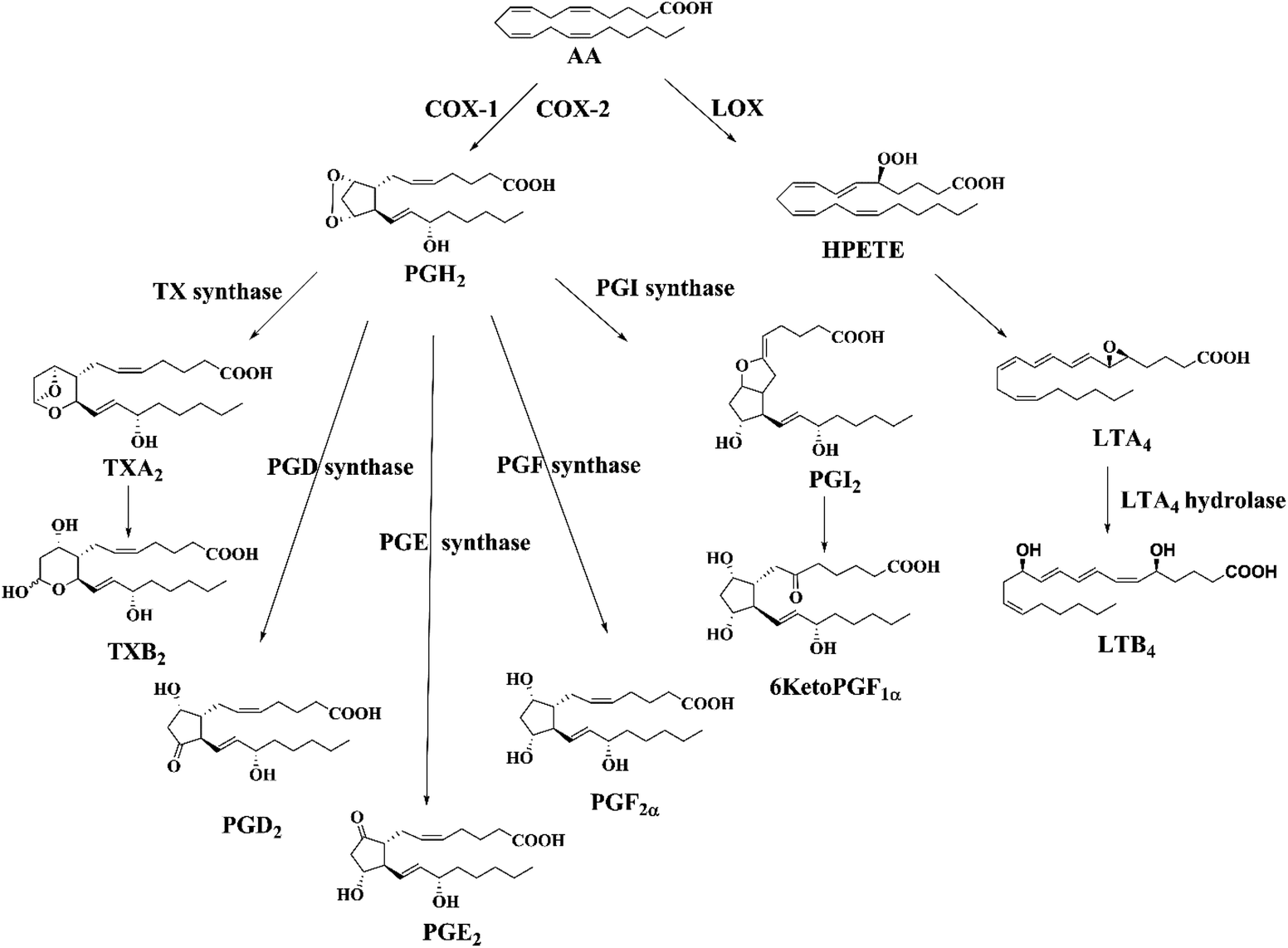 | ||
| Fig. 1 Biosynthetic pathway of AA-metabolites (eicosanoids) through cyclooxygenase (COX) and lipoxygenase (LOX) pathways. | ||
The development of a sensitive, specific, reliable and rapid method for the determination of eicosanoids levels will facilitate the understanding of their complex biological functions, especially lipid peroxidation and oxidative stress.2 The absolute quantification of these metabolites in biological matrix is challenging due to their minute concentration, short half-life and their structural complexity arising from different isomers. Therefore, targeted analytical approaches are advantageous in the investigation of AA-metabolism, especially the formation of different isomeric eicosanoids in vivo.2,4
Currently, various analytical techniques have been developed and validated for the quantification of AA-metabolites including, immunoassays, such as radioimmunoassay (RIA) or enzyme immunoassay (ELISA),5 gas chromatography-mass spectrometry (GC-MS),6 liquid chromatography-mass spectrometry (LC-MS)1,2,4,7 and capillary electrophoresis-ultra violet (CE-UV).8 The antibody based measurements agonizes from cross-reactivity owing to poor selectivity among the analytes and thereby generating misleading results.4 GC-MS and LC-MS techniques often require laborious sample preparation including derivatization, pre-concentration of the sample and extensive cleanup either by solid phase extraction (SPE) or by liquid–liquid extraction (LLE) due to the complex nature of the matrix as well as very low levels of endogenous metabolites.2,4 Thus, developing a reliable quantitative method to assess the eicosanoids levels in the biological fluids is challenging and also a fundamental step owing to pre-analytical and analytical demands for accurate and precise measurements as well as the potential pitfalls.
Recent advances in chromatography and mass spectrometry have extensively facilitated the application of supercritical fluid chromatography mass spectrometry (SFC-MS) for lipidomics research.9,10 SFC utilizes supercritical CO2 as the primary mobile phase which is non-polar in nature. High diffusivity and low viscosity of supercritical CO2 allow higher flow rates with sub-2 μm particles and subsequent shrinking of the time taken for the analysis. Further, the addition of a small amount of modifier (2–40%, v/v) like methanol enhances the limited polarity, solvating power and elution strength of pure CO2, making it suitable for the analysis of both non-polar and polar analytes.9,11 Over the years, many studies have demonstrated successful application of SFC for the separation of diverse species of lipids including phospholipids, lysophospholipids, sphingolipids, ceramides, glycerolipids,10,12 vitamins,13 fatty acids,14 oxylipins,15 and steroids.16 More importantly, the use of SFC has enabled the comprehensive separation of isomers in very short run time.11,13,14 Under such context, the aim of this study was to develop a fully validated supercritical fluid chromatography-tandem mass spectrometry method (SFC-MS/MS) for the rapid identification and quantification of a number of endogenous eicosanoids. Thus, the developed method adopts a simple liquid extraction with protein precipitation overcoming the main limitations of previously published methods e.g. analyte losses, degradation and laboriousness. Finally, the validated method has been employed to determine the levels of targeted analytes in human plasma and CSF samples. According to our knowledge, this is the first simultaneous determination of AA-metabolites (at least one in each class) within three minutes using SFC-MS/MS.
Experimental section
Reagents, standard solution and samples
All ultrapure LC-MS grade solvents were purchased from Thermo Fisher Scientific (Stockholm, Sweden) unless otherwise stated. Water was deionized and distilled with a Milli-Q purification system (Millipore, Bedford, MA, USA). All the standards and internal standards (IS); PGD2, PGD2-d4, PGE2, PGE2-d4, PGF2α, PGF2α-d4, 6KetoPGF1α, 6Keto PGF1α-d4, LTB4, LTB4-d4, TXB2 and TXB2-d4 were purchased from Cayman Chemicals (Michigan 48108, USA). LC-MS grade tert-butyl methyl ether (MTBE), formic acid, ammonium acetate, butylated hydroxyl toluene (BHT) and indomethacin were purchased from Sigma-Aldrich (Stockholm, Sweden).The stock solutions of PGD2, PGE2, PGF2α, 6KetoPGF1α, LTB4, TXB2 and all the IS were prepared by dissolving accurately weighed amounts in methanol with 0.02 mg mL−1 BHT to obtain a final concentration of 5 μg mL−1. All stock solutions were stored at −80 °C until further use.
The human plasma sample from healthy blood donor (healthy control plasma) was obtained from the Academic Hospital (“blodcentralen UAS”), Uppsala, Sweden. The plasma and CSF samples of the chronic pain patients (ten plasma samples and one CSF) were kindly provided by Professor Torsten Gordh, Surgical Science, Anesthesiology and Intensive care, Academic Hospital, Uppsala, Sweden. The study was conducted in accordance with the Declaration of Helsinki. Human plasma and CSF samples were included in this study, which was approved by the Regional Ethical Review Board of Uppsala, Sweden, and undertaken with the written consent of the individual donors. Blood was collected from each participant by venipuncture into EDTA vacutainer tubes and centrifuged at 3500g for 15 min. The CSF, visually free of contamination by blood was collected into polypropylene vacuum collection tubes by lumbar puncture, sedimented at 1000g for 10 min. Aliquoted plasma and CSF samples were frozen immediately at −80 °C until further analysis.
Separation of AA-metabolites by supercritical fluid chromatography (SFC)
The supercritical fluid chromatography system was a Waters ACQUITY® UPC2™ (Milford, MA, USA), equipped with a binary solvent delivery pump, an autosampler, and a column oven. SFC was connected with the mass spectrometer by the commercial interface kit (Waters) composed of two T-pieces enabling the back pressure control and post column infusion with a make-up solvent. The preliminary column screening was performed using fixed initial chromatographic condition where CO2 (99.99%) was the mobile phase ‘A’ and MeOH with 10 mM ammonium acetate was the mobile phase ‘B’. Gradient elution was started at 5% B with a flow rate of 1.5 mL min−1 (except for DIOL and 1-AA column where flow rate was set at 1.2 mL min−1). The content of B mobile phase was increased linearly to 30% over 0.2–7.5 min before reaching the initial condition in 8 min. The column oven, back-pressure, and make-up flow were set at 40 °C, 1500 psi, and 0.2 mL min−1 (B) respectively.We studied 7 different column materials to find the best stationary phase for the separation of target analytes; HSS C18 SB (3 mm × 100 mm, 1.8 μm), BEH 2EP column (3 mm × 100 mm, 1.7 μm), BEH column (3 mm × 150 mm, 1.7 μm), DIOL column (3 mm × 150 mm, 1.7 μm), 2-PIC column (3 mm × 150 mm, 1.7 μm), FP column (2.1 mm × 100 mm, 1.7 μm) and 1-AA column (2.1 mm × 100 mm, 1.7 μm). All columns were purchased from Waters (Milford, MA, USA).
The optimized quantitative analysis was performed using a 2-PIC column at 40 °C. The mobile phase flow rate was maintained at 1.5 mL min−1 with a gradient elution eluent A, CO2; eluent B, MeOH with 0.1% formic acid and 10 mM of ammonium acetate. The gradient program was started with 5% of B, then, a linear gradient of B was programmed from 5% to 20% for 0.2–1 min, followed by a linear gradient up to 25% B for 2–3 min, and finally, the linear gradient step was set down to 5% in 4 min and then it was held for 1.0 min for equilibration before the next injection. Same, eluent B was used as the make-up solvent at a flow rate of 0.2 mL min−1. The back pressure was set at 2000 psi and the injection volume was 3.0 μL.
Identification and quantification of AA-metabolites by tandem mass spectrometry (XEVO® TQ-S)
AA-metabolites were identified by using a Waters Xevo® TQ-S mass spectrometer (Milford, MA, USA). The data acquisition was in the negative electrospray ionization (−ve ESI) mode with unit mass resolution. The desolvation gas was nitrogen, and the collision gas was argon (0.25 mL min−1). The data acquisition range was m/z 50–500. The capillary voltage was 2.3 kV and the source offset was 30 V. The source temperature was 150 °C and the desolvation temperature was 400 °C with the desolvation gas flow rate of 800 L h−1. The collision energy and cone voltage were optimized for each compound to generate the most abundant product ions to develop multiple reaction monitoring (MRM) (Table 1). The cone gas flow rate was 150.0 L h−1. The nebulizer gas flow pressure was maintained at 7.0 bar. Data were acquired and analyzed with Waters MassLynx ™ 4.1 software (Waters, Milford, MA, USA).| Analyte/IS | Precursor ion (m/z) | Product ion (m/z) | D t (s) | |||||
|---|---|---|---|---|---|---|---|---|
| [M − H]− | Quantifier | CV(V) | CE (eV) | Qualifier | CV (V) | CE (eV) | ||
| CV; cone voltage, CE; collision energy, Dt; dwell time. | ||||||||
| PGE2 | 351.22 | 315.22 | 20 | 11 | 189.13 | 35 | 16 | 0.005 |
| PGE2-d4 | 355.22 | 319.13 | 16 | 16 | 193.13 | 20 | 16 | 0.005 |
| PGD2 | 351.16 | 271.24 | 20 | 15 | 189.12 | 25 | 20 | 0.005 |
| PGD2-d4 | 355.10 | 275.37 | 15 | 25 | 193.13 | 20 | 23 | 0.005 |
| PGF2α | 353.30 | 193.20 | 38 | 26 | 308.95 | 20 | 18 | 0.005 |
| PGF2α-d4 | 357.30 | 197.20 | 20 | 18 | 312.95 | 20 | 18 | 0.005 |
| LTB4 | 335.12 | 195.10 | 25 | 16 | 317.20 | 25 | 14 | 0.005 |
| LTB4-d4 | 339.12 | 197.20 | 25 | 17 | 320.90 | 25 | 14 | 0.005 |
| TXB2 | 369.16 | 195.11 | 25 | 13 | 169.15 | 25 | 16 | 0.005 |
| TXB2-d4 | 373.37 | 199.20 | 25 | 12 | 173.12 | 25 | 17 | 0.005 |
| 6KetoPGF1α | 369.40 | 163.16 | 20 | 25 | 245.18 | 20 | 25 | 0.005 |
| 6KetoPGF1α-d4 | 373.40 | 167.16 | 20 | 25 | 249.18 | 20 | 25 | 0.005 |
Sample preparation
In brief, human plasma (100 μL) and CSF (200 μL) samples were mixed with 15 μL of IS mixture (50 ng mL−1 in MeOH). To this solution, 100 μL MeOH containing 10 μM of indomethacin was added to deactivate the COX enzymes. The samples were extracted with 4 mL of cold MeOH containing 0.02 mg mL−1 of BHT (as an antioxidant). It was then gently vortexed for 5 min and was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min at 4 °C. The pooled MeOH extracts were dried under N2 and reconstituted in 50 μL of MeOH prior to the SFC-MS/MS analysis.
000g for 10 min at 4 °C. The pooled MeOH extracts were dried under N2 and reconstituted in 50 μL of MeOH prior to the SFC-MS/MS analysis.
Method validation
Method performance was validated using human plasma to demonstrate the reliability of the analyte measurement in its biological matrix. The method was evaluated for carry over, limit of quantification (LOQ), selectivity, sensitivity, precision, accuracy, linearity, recovery and stability. Slightly modified guidelines of EU Commission Decision/657/EEC were implemented during method validation.17 Matrix-matched calibration was used in this study. A total of nine calibrators were prepared in the human plasma with concentrations of 0.5, 1, 2.5, 5, 10, 20, 40, 80, and 100 ng mL−1. Blank and double blank (blank without IS) were also included while preparing the calibrators. Linearity was evaluated by comparing the measured and known concentration of calibration solutions on different days. Linear regression with 1/x2 weights was used for creating calibration curves. Matrix matched quality control (QC) samples were prepared from different stock solutions at three levels 3 ng mL−1 (low), 20 ng mL−1 (medium) and 60 ng mL−1 (high) in order to assess the method performance. Briefly, carry over was determined by injecting the blank sample immediately after the highest calibration sample injection. The LOQ was determined in the lowest analyte concentration injected that yielded a signal-to-noise (S/N) ratio of ≥10 in six replicates. Within run and between run accuracy and precision were evaluated by running a batch of QC samples on two different days. For accuracy, the mean concentration should be within ±15% of the nominal values for the QC samples. Similarly, the precision expressed as coefficient of variation (CV %) of measurement should be within ±15%. Matrix effect was assessed by comparing the post extraction spiked plasma samples with the neat solvent spiked samples at two different levels, 3 ng mL−1 (low) and 60 ng mL−1 (high). If the observed value is greater or lower than 100% it indicates ionization enhancement or suppression, respectively. Recovery experiments were performed by comparing the analytical results for extracted samples at three concentrations with un-extracted standards that represent 100% recovery. Finally, stability on auto sampler was determined by comparing the response of the sample on auto sampler at 8 °C for 12 and 24 hours with the response of freshly prepared samples.Results and discussion
The novel SFC-MS/MS method was developed and validated to measure five targeted AA-metabolites (PGD2, PGE2, PGF2α, 6KetoPGF1α, and LTB4) within 3.0 minutes. Method validation results are shown in Tables 2–4. This method has been applied to the development of stable isotope dilution assays for patient plasma and CSF samples for the simultaneous analysis of AA-metabolites. To the best of our knowledge, there are no previously published SFC-MS/MS data to compare with our method.| Analyte | R t (min) | Calibration range (ng mL−1) | r 2 | LOQ (ng mL−1) |
|---|---|---|---|---|
| PGE2 | 2.24 | 0.5–100 | 0.9966 | 0.5 |
| PGD2 | 2.34 | 0.5–100 | 0.9999 | 0.5 |
| PGF2α | 2.57 | 0.5–100 | 0.9989 | 0.5 |
| LTB4 | 2.52 | 0.5–100 | 0.9985 | 0.5 |
| 6KetoPGF1α | 2.71 | 2.5–100 | 0.9956 | 2.5 |
| Compound | Level | Accuracy (%) | Precision (CV %) | Recovery (%) | ||
|---|---|---|---|---|---|---|
| Within-run | Between run | Within-run | Between run | |||
| Low; 3 ng mL−1, medium; 20 ng mL−1, and high; 60 ng mL−1. | ||||||
| PGE2 | Low | 98.5 | 94.3 | 9.6 | 8.5 | 104.5 |
| Mid | 101.0 | 103.7 | 6.4 | 7.5 | 114.3 | |
| High | 103.2 | 98.2 | 9.9 | 5.7 | 115.0 | |
| PGD2 | Low | 98.0 | 98.2 | 3.4 | 6.9 | 115.1 |
| Mid | 98.7 | 99.7 | 6.6 | 8.0 | 92.8 | |
| High | 91.6 | 97.3 | 8.4 | 6.0 | 95.0 | |
| PGF2α | Low | 94.7 | 97.3 | 13.5 | 6.0 | 112.5 |
| Mid | 100.7 | 97.8 | 10.7 | 9.7 | 98.3 | |
| High | 102.8 | 90.3 | 10.2 | 6.8 | 92.6 | |
| LTB4 | Low | 108.7 | 106.6 | 1.2 | 3.5 | 110.8 |
| Mid | 102.2 | 101.3 | 3.7 | 4.3 | 104.8 | |
| High | 99.5 | 96.9 | 5.8 | 7.0 | 99.3 | |
| 6KetoPGF1α | Low | 83.3 | 96.5 | 4.5 | 8.5 | 101.0 |
| Mid | 89.9 | 67.6 | 6.7 | 12.6 | 70.2 | |
| High | 91.3 | 63.0 | 12.4 | 6.8 | 75.4 | |
| Compound | Stability % (SD %) | IS normalized matrix factor % (CV %) | |||
|---|---|---|---|---|---|
| Low | Mid | High | Low | High | |
| Low; 3 ng mL−1, medium; 20 ng mL−1, and high; 60 ng mL−1. | |||||
| PGE2 | 105.7 (0.4) | 102.9 (0.2) | 100.7 (2.8) | 119.9 (1.6) | 108.1 (4.0) |
| PGD2 | 86.9 (2.3) | 85.2 (0.5) | 88.2 (1.6) | 106.7 (6.8) | 126.9 (6.1) |
| PGF2α | 100.0 (3.3) | 96.4 (1.3) | 94.0 (6.6) | 127.2 (7.2) | 96.6 (7.3) |
| LTB4 | 95.9 (0.5) | 111.2 (1.6) | 102.0 (4.4) | 112.2 (6.1) | 105.1 (7.9) |
| 6KetoPGF1α | 67.3 (0.8) | 70.2 (5.1) | 75.4 (5.5) | 98.8 (4.2) | 104.3 (3.9) |
All the AA-metabolites contain a carboxylic acid group, where more intense and abundant deprotonated ions [M − H]− can be generated during the negative electrospray ionization (−ve ESI).18 The multiple reactions monitoring (MRM) is the most commonly used mode in the quantification of targeted analytes. In order to enhance the selectivity of the target analyte; two product ions were chosen, out of which one was the quantifier and the other was the qualifier. Selection of the quantifier was based upon the specificity of the ion for the target molecule as well as signal intensity. Although, [M − H − H2O]−, [M − H − 2H2O]−, [M − H − 2H2O − CO2]− ions were readily formed from all the AA-metabolites, some fragments were specific to certain metabolites, such as [M − H − hexanal − H2O]− was specific to PGE2/PGD2. Similarly, ions m/z 309, 291, 273 and 193 were indicative of the F2-ring, which was specific to PGF2α.4,18 The product ion MS spectra of PGE2 and LTB4 are shown in Fig. 2. Such ions were included for individual analytes either as qualifiers or quantifiers based on S/N ratio. The MRM dwell times were adjusted to ensure that at least 12 data points were acquired for each peak during the analytical time to acquire reproducible peaks. The absolute quantification of AA-metabolites was achieved using a stable-isotope dilution strategy where the signature transition of corresponding deuterated IS was used in a calibration curve. Our MRM findings are in good agreement with the previously reported values.7,18,19
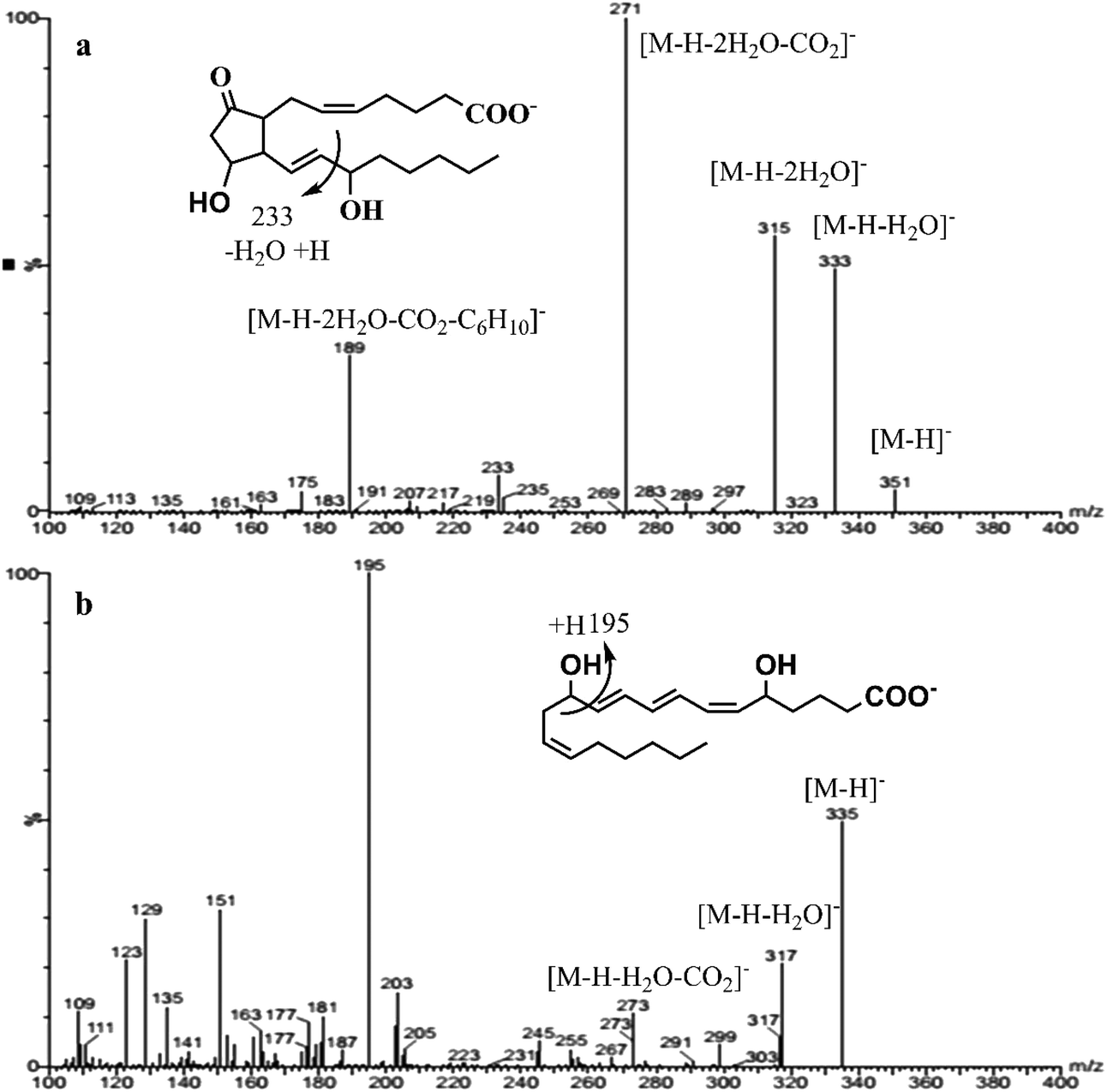 | ||
| Fig. 2 A full scan mass spectrum with detail fragmentation of PGE2 (a) and LTB4 (b) in the negative electrospray ionization (−ve ESI). | ||
The most important parameter influencing the retention of the analytes was the chemistry of the stationary phase.13,16 In spite of the fair availability of different UPC2 columns, with known chemistry of the stationary phases, selecting the best column for the method development still remains challenging.11 Mass spectrometric sensitivity and overall chromatographic resolution of the target analytes including geometrical isomers such as PGD2 and PGE2 provided a basis for the selection of column. In this study, the tested columns have been mentioned in the method section and the chemistry of the selected stationary phases is shown in Fig. 3. After stationary phase screening for AA-metabolites, the best overall separations were obtained with the 2-PIC column, providing highest retention factor for PGE2, PGD2, PGF2α, 6KetoPGF1α and LTB4 following normal-phase chromatography. The TXB2 was excluded owing to its peak broadening. The tested columns produced a similar elution order starting from PGE2, PGD2, PGF2α, LTB4, TXB2, and ending with 6KetoPGF1α. A weak resolution power was observed for HSS C18 SB (Fig. 4a), 1-AA and FP columns where PGE2 and PGD2 could not be resolved. More importantly, HSS C18 SB gave lowest retention for all the analytes, which indicates the need of electrostatic attraction between carboxylic/alcohol groups of analytes with the functional group of the stationary phase for the retention to occur.15 In fact electrostatic attraction does not occurs at all or only with mild effects on HSS C18 stationary phase due to the lack of additional ionizable functional groups beside residual silanols.13 Only DIOL, BEH, BEH 2-EP and 2-PIC columns met the requirement for excellent separation of PGD2/PGE2 and satisfactory peak shape of 6KetoPGF1α. Nevertheless, these columns (except DIOL and FP) could not provide suitable separation of TXB2. Taking all into account; TXB2 was removed from analysis in this study. Notably, DIOL column (Fig. 4b) gave two peaks for the enantiomers of TXB2, which has also been reported with C18 column by Casetta et al.20 while FP gave a single peak. The AA-metabolites containing alcoholic moieties exhibited the greatest retention on the DIOL and 2-PIC columns as these columns are known to have somewhat similar retentions.13 As the analytes possess alcoholic groups, the retention on the polar DIOL column can be attributed to a higher degree of hydrogen bonding.11,13 However, the 2-PIC ligand contains one relatively strong secondary amine group and one weak basic pyridine group (Fig. 3) thus providing attractive effects towards acidic analytes. Further, the presence of aromatic interactions with pyridine group might have resulted a slightly better resolution of analytes on 2-PIC column as compared to DIOl column.11 This resulted in a baseline separation even for the structural enantiomers of PGE2 and PGD2 (Fig. 5). Elution of PGE2 before PGD2 indicates that the position of the carbonyl group at C9 is more favorable for dipole–dipole interaction than the carbonyl group at C11. This shows double bond in the cyclopentane ring has a higher contribution to the separation of isomers. To the best of our knowledge there are no previously published chromatograms to compare our separation pattern on the SFC system.15 However, there are LC-MS/MS methods using BEH C18 column for separation that have shown similar elution pattern to our study.19 In contrast, nano-LC-MS/MS method with C8 column has produced a reverse elution pattern for similar analytes with the elution order of TXB2, LTB4, PGF2α, PGE2 and PGD2.21
 | ||
| Fig. 3 Column chemistries of different stationary phases tested on supercritical fluid chromatography (SFC). | ||
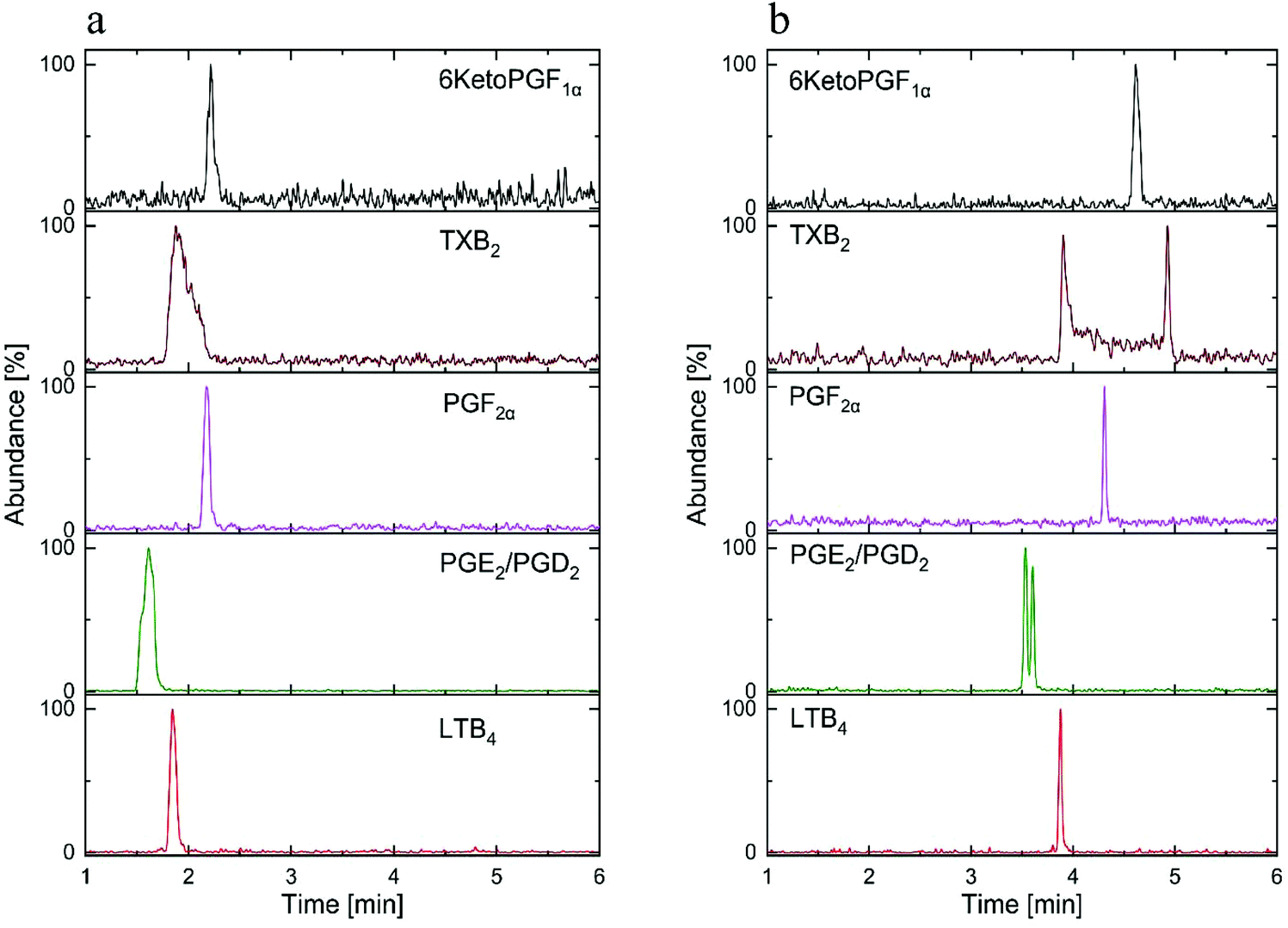 | ||
| Fig. 4 The SFC-MS/MS chromatogram of standard AA-metabolites mixture (30 ng mL−1) on: (a) HSS SB C18 column, (b) DIOL column. | ||
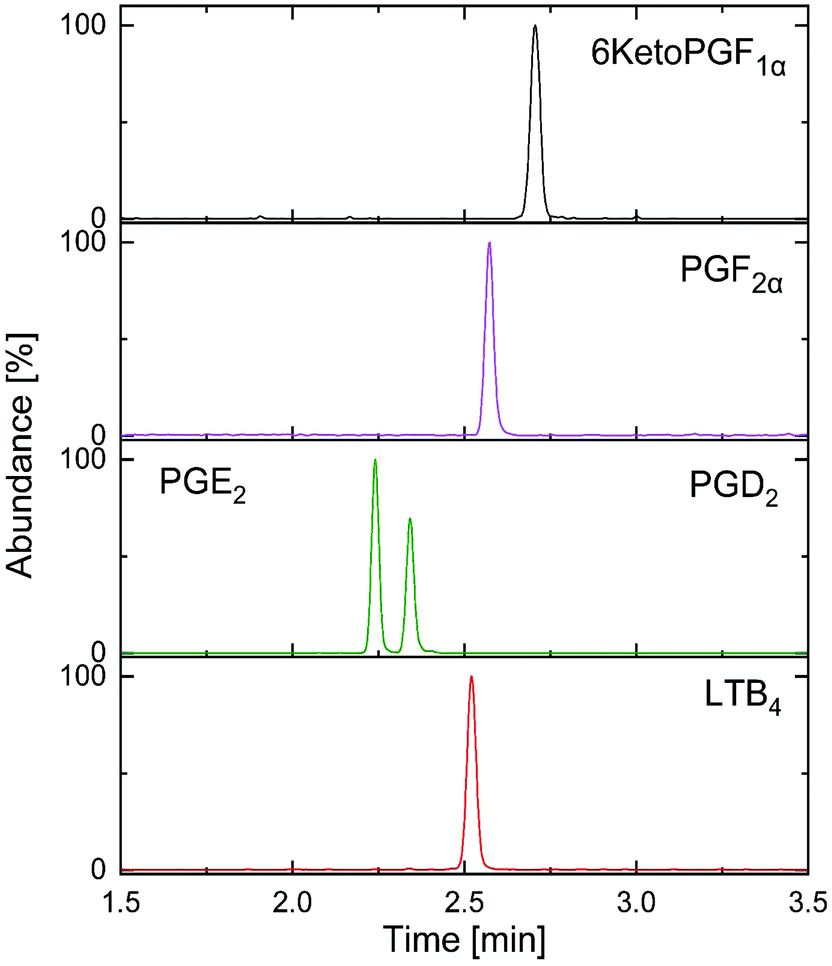 | ||
| Fig. 5 Optimized separation of targeted analysis standard AA-metabolites mixture (30 ng mL−1) on 2-PIC column. | ||
After the generic screening, the method was further developed taking the effect of flow rates, backpressure and column temperature into account to improve the selectivity, sensitivity and the resolution of the AA-metabolites on the 2-PIC column. In general, higher retention time was observed with higher temperature while higher back pressure had opposite influence with little or no effects on resolution of isomers.15 The improved optimized chromatographic and mass spectrometric parameters are described in the method section above. Low concentrations of additives (acids or bases) in the co-solvent increases the solubility of analytes and provides coverage of the active sites, resulting in a beneficial effect on the peak shape as well as mass spectrometric sensitivity.16,22 Therefore, we tested different gradient programs and additives (0.1%, 0.2%, 0.5% formic acid and 10 mM, 20 mM ammonium acetate) to improve the on column separation and peak shape of the analytes. We observed that small amount of formic acid (0.1%) as modifier enhanced the chromatographic separation, particularly for isomers like PGE2 and PGD2 whereas the acetate anion can act as proton acceptor at the −ve ESI thus enhancing the ionization of the AA-metabolites.16 Use of ammonium acetate also improved the peak shape of 6KetoPGF1α in particular. In fact, no advantage was gained by further increase in the concentration of formic acid or ammonium acetate. Taking all these findings into consideration, the final elution was done with 0.1% formic acid and 10 mM ammonium acetate in methanol as the mobile phase ‘B’ and CO2 as the primary mobile phase using the 2-PIC stationary phase. Steepness of gradient has been shown to influence the chromatographic selectivity and sensitivity.15,23 In our case, use of 25% of ‘B’, which was sufficient to elute the most retained polar component (6KetoPGF1α) from the column. Furthermore, we compared very steep gradient from 5% to 20% ‘B’ over 0.2–1 min with slightly less steep gradient from 5% to 20% ‘B’ over 2 min to see the influence in retention and resolution of analytes. As expected, the retention and resolution of analytes decreased with steep gradient.15 For example, resolution of isomers PGE2 and PGD2 decreased from 0.15 min (20% ‘B’ over 2 min) to 0.11 min (20% ‘B’ over 1 min). Nevertheless, we observed peak shape of 6KetoPGF1α improved (more sharper) significantly in later gradient by further increasing ‘B’ up to 25% over 2 min. We then achieved complete baseline separation in less 3.0 min for PGE2, PGD2, PGF2α, 6KetoPFG1α, LTB4 but not for TXB2 which might be explained by the higher affinity to the stationary phase giving a too broad and asymmetric peak.
The extraction of the analytes from the biological samples is often the bottleneck of the analysis. These AA-metabolites are very sensitive to light and temperature. Hence, it became important to develop a simplified method to minimize the total sample preparation time. Several extraction methods have been reported in the literature for the extraction of AA-metabolites from different biological tissues. Our exploration focused on the extensively investigation of different solvents/solvent mixtures including methanol, ethyl acetate, n-hexane:ethyl acetate (1:1 v/v), or hexane:isopropanol (1:1 v/v) and MTBE with spike plasma samples of AA-metabolites (30 ng mL−1).7,21,24 Finally, methanol was selected as an extraction solvent based on the recovery % of each analyte. The mean recovery of analytes ranged from 70–115% on methanol (Table 3). As AA-metabolites are prone to oxidation, antioxidants such as BHT and COX inhibitors (indomethacin) were incorporated into the extraction process to limit any alteration of AA-metabolites during the process.20 In general, our sample preparation technique was fast, reproducible and cost effective and maintained the native nature of the analytes. According to some previous reports, the extraction follows enrichment of analytes by SPE to remove all endogenous interfering substances.1,4 The present method does not need any extra purification steps. The procedure is simple, easy to perform and has the possibility to analyze a large number of samples with low variability per day.
The optimized method was validated for different parameters. The linearity, LOQ, accuracy, precision, recovery, matrix effect and stability of the method were determined and the results were summarized in Tables 2, 3 and 4. The calibration curve comprised of nine points (seven points in case of 6KetoPGF1α) prepared in triplicate, blank and double blank (blank plasma without IS) samples. The linearity was determined by linear regression analysis. The correlation coefficients (r2) of the analytes were >0.995. The back calculated concentration of the calibration samples was within ±15% of the nominal value. Carry-over did not generate any problem, as all analytes were undetectable from blank injections after the highest calibrator. There was no interfering peak during AA-metabolites elution. The linearity range obtained from this study was comparable to those already published LC-MS results. LOQ were 0.5 ng mL−1 (equivalent of 1.5 pg on column) for PGE2, PGD2, PGF2α, LTB4 and 2.5 ng mL−1 (equivalent to 7.5 pg on column) for 6KetoPGF1α which are comparable or better as compared to already existing LC-MS/MS methods for similar analytes (Table 2).4,20,24 Precision and accuracy were assessed by replicate analysis (n = 4) of spiked plasma samples at three different concentrations and data are presented in Table 3. The intra- and inter-day precisions were between 1.2% and 13.5% for all the analytes and the accuracy was within ±10% excluding 6KetoPGF1α. It is worth mentioning that accuracy and precision data of 6KetoPGF1α exceeds the limits probably due to low recovery. The absolute recovery of all other analytes ranged from 93–115% (Table 3). The recovery results of all the analytes in plasma indicate no significant degradation on an auto sampler for 12 hours at 8 °C (data not shown) while both PGD2 and 6KetoPGF1α levels significantly dropped after keeping the samples for 24 hours at 8 °C (Table 4). Degradation of PGD2 and 6KetoPGF1α has been reported previously requiring the sample analysis within 12 hours.24 Evaluation of matrix effects of the analytes for high (60 ng mL−1) and low (3 ng mL−1) concentrations shows that IS-normalized matrix factor for 6KetoPGF1α lie in a very reasonable range of 99–104%, where the signal of the other four analytes appears to be amplified in the plasma (Table 4). Since we employ corresponding deuterated (d4) internal standards and matrix matched calibration in plasma, the signal enhancement issue was easily taken into account.
In order to test the applicability of the validated method in a clinical setting, ten patients plasma (100 μL each) and one CSF (200 μL) samples were extracted and analyzed. Out of the ten plasma samples we could effectively quantify the PGD2 and PGE2 levels in six samples with concentration ranging from 0.49–3.98 ng mL−1. The PGF2α was detected but quantification was not possible as the S/N was below 10 in all the plasma samples. However, LTB4 and 6KetoPGF1α were neither detected nor quantified in any of the plasma samples. There were no quantifiable levels of AA-metabolites in the human CSF sample while PGE2 and PGF2α were present in trace amounts. The variation of AA-metabolites concentrations in plasma was possibly due to the individual metabolic rate and these individuals might have taken nonsteroidal anti-inflammatory drugs which could inhibit the COX enzyme, thus shutting down the AA cascade and preventing the generation of eicosanoids.1,25 Estimated eicosanoid levels in plasma were different than basal levels reported in human metabolome database probably due to the difference in the sample population.1 There is no reported fully validated study on SFC-MS/MS analysis of AA-metabolites to compare with our results. Recently, a method for analysis of oxylipins in plasma, which included eicosanoids, has been reported by Berkecz R. et al.15 However, this study was a preliminary evaluation rather than a fully validated method thus it is not suitable for a clinical study especially when the sample volume is limited. Yet, in a brief survey of published methods on LC-MS/MS during the last 10 years, time for the analysis varies between 6–65 min, due to the different chromatographic and MS conditions as opposed to only 3 minutes (Fig. 5) in our study. A high percentage of the published methods relied on RP-C18 analytical LC-columns while availability of columns of different retention capabilities provides extended range of selectivity in SFC.13 Supercritical CO2 (mobile phase A) is nonpolar in nature (similar to hexane) and the addition of an organic solvent (mobile phase B) makes it suitable for separation of analytes with wide range of polarities.11,16 As only small portion of organic modifier is needed, SFC allows decrease in waste generation, low cost and environmental friendly alternative.11 Further, the SFC based chromatographic system provides an excellent solution for separation of closely related isomeric and isobaric metabolites (structural and positional) which is often challenging with other analytical tools.11,14,15
Conclusions
A novel SFC-MS/MS-based analytical platform was successfully developed and validated for the simultaneous profiling of five AA-metabolites (PGE2, PGD2, PGF2α, LTB4 and 6Keto PGF1α) in human biological samples for the first time. All variables relative to SFC-MS/MS were optimized and a separation based on 2-PIC column was developed to provide sensitive and quantitative analysis of target analytes within 3 minutes. Results from method validation demonstrate that this method is sensitive to perform comprehensive analysis of eicosanoids in large sample sets in clinical studies with limited sample volumes. Further, this study also opens a door to expand the use of SFC for polar isomeric compounds in the complex lipidome. Future work will be focused on using the method on a larger human cohort and also expand the method to other analytes in AA cascade.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Vetenskapsrådet (Swedish Research Council) 2015-4870, the Erasmus Mundus Joint Programme EACH (Excellence in Analytical Chemistry) and Uppsala Berzelii Center for Neurodiagnostics. The authors would like to thank Prof. Torsten Gordh and Dr Anne-Li Lind, Department of Neuroscience, Uppsala University, Sweden and Dr Riin Rebane, Department of Analytical Chemistry, University of Tartu, Estonia for their kind support.References
- D. D. Shinde, K.-B. Kim, K.-S. Oh, N. Abdalla, K.-H. Liu, S. K. Bae, J.-H. Shon, H.-S. Kim, D.-H. Kim and J. G. Shin, J. Chromatogr. B: Biomed. Sci. Appl., 2012, 911, 113–121 CrossRef PubMed.
- I. Unterwurzacher, T. Koal, G. K. Bonn, K. M. Weinberger and S. L. Ramsay, Clin. Chem. Lab. Med., 2008, 46, 1589–1597 Search PubMed.
- B. Samuelsson, M. Goldyne, E. Granstrom, M. Hamberg, S. Hammarstrom and C. Malmsten, Annu. Rev. Biochem., 1978, 47, 997–1029 CrossRef PubMed.
- M. Masoodi and A. Nicolaou, Rapid Commun. Mass Spectrom., 2006, 20, 3023–3029 CrossRef PubMed.
- B. Hegyi, G. Kudlik, É. Monostori and F. Uher, Biochem. Biophys. Res. Commun., 2012, 419, 215–220 CrossRef PubMed.
- D. Tsikas, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 717, 201–245 CrossRef.
- S. A. Brose, A. G. Baker and M. Y. Golovko, Lipids, 2013, 48, 411–419 CrossRef PubMed.
- V. A. VanderNoot and M. VanRollins, Anal. Chem., 2002, 74, 5866–5870 CrossRef PubMed.
- T. Bamba, N. Shimonishi, A. Matsubara, K. Hirata, Y. Nakazawa, A. Kobayashi and E. Fukusaki, J. Biosci. Bioeng., 2008, 105, 460–469 CrossRef PubMed.
- M. Lísa and M. Holčapek, Anal. Chem., 2015, 87, 7187–7195 CrossRef PubMed.
- L. Nováková, A. G.-G. Perrenoud, I. Francois, C. West, E. Lesellier and D. Guillarme, Anal. Chim. Acta, 2014, 824, 18–35 CrossRef PubMed.
- J. W. Lee, T. Yamamoto, T. Uchikata, A. Matsubara, E. Fukusaki and T. Bamba, J. Sep. Sci., 2011, 34, 3553–3560 CrossRef PubMed.
- V. Desfontaine, J.-L. Veuthey and D. Guillarme, J. Chromatogr., A, 2016, 1438, 244–253 CrossRef PubMed.
- M. Ashraf-Khorassani, G. Isaac, P. Rainville, K. Fountain and L. Taylor, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 997, 45–55 CrossRef PubMed.
- R. Berkecz, M. Lísa and M. Holčapek, J. Chromatogr., A, 2017, 1511, 107–121 CrossRef PubMed.
- M. Doué, G. Dervilly-Pinel, K. Pouponneau, F. Monteau and B. Le Bizec, Anal. Bioanal. Chem., 2015, 407, 4473–4484 CrossRef PubMed.
- I. C. Directive, Off. J. Eur. Communities, 2002, 221, 8–36 Search PubMed.
- S. A. Brose, B. T. Thuen and M. Y. Golovko, J. Lipid Res., 2011, 52, 850–859 CrossRef PubMed.
- M. Y. Golovko and E. J. Murphy, J. Lipid Res., 2008, 49, 893–902 CrossRef PubMed.
- B. Casetta, G. Vecchione, M. Tomaiuolo, M. Margaglione and E. Grandone, J. Mass Spectrom., 2009, 44, 346–352 CrossRef PubMed.
- D. Thomas, J. Suo, T. Ulshöfer, H. Jordan, N. de Bruin, K. Scholich, G. Geisslinger and N. Ferreirós, Anal. Bioanal. Chem., 2014, 406, 7103–7116 CrossRef PubMed.
- C. West, J. Melin, H. Ansouri and M. M. Metogo, J. Chromatogr., A, 2017, 1492, 136–143 CrossRef PubMed.
- A. G.-G. Perrenoud, J.-L. Veuthey and D. Guillarme, J. Chromatogr., A, 2012, 1266, 158–167 CrossRef PubMed.
- H. Cao, L. Xiao, G. Park, X. Wang, A. C. Azim, J. W. Christman and R. B. van Breemen, Anal. Biochem., 2008, 372, 41–51 CrossRef PubMed.
- B. von Jeinsen, J. Watrous, M. Henglin, J. Rong, T. J. Niiranen, R. S. Vasan, M. G. Larson, M. Jain and S. Cheng, Circulation, 2017, 136, A19156 Search PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2018 |