
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReporter bacteriophage T7NLC utilizes a novel NanoLuc::CBM fusion for the ultrasensitive detection of Escherichia coli in water
T. C.
Hinkley
 a,
S.
Garing
b,
S.
Singh
c,
A-L. M.
Le Ny
b,
K. P.
Nichols
c,
J. E.
Peters
d,
J. N.
Talbert
c and
S. R.
Nugen
*a
a,
S.
Garing
b,
S.
Singh
c,
A-L. M.
Le Ny
b,
K. P.
Nichols
c,
J. E.
Peters
d,
J. N.
Talbert
c and
S. R.
Nugen
*a
aDepartment of Food Science, Cornell University, Ithaca, NY 14853, USA. E-mail: snugen@cornell.edu
bIntellectual Ventures Laboratory/Global Good, Bellevue, WA 98007, USA
cDepartment of Food Science and Human Nutrition, Iowa State University, Ames, IA 50011, USA
dDepartment of Microbiology, Cornell University, Ithaca, NY 14853, USA
First published on 2nd August 2018
Abstract
Rapid detection of bacteria responsible for foodborne diseases is a growing necessity for public health. Reporter bacteriophages (phages) are robust biorecognition elements uniquely suited for the rapid and sensitive detection of bacterial species. The advantages of phages include their host specificity, ability to distinguish viable and non-viable cells, low cost, and ease of genetic engineering. Upon infection with reporter phages, target bacteria express reporter enzymes encoded within the phage genome. In this study, the T7 coliphage was genetically engineered to express the newly developed luceriferase, NanoLuc (NLuc), as an indicator of bacterial contamination. While several genetic approaches were employed to optimize reporter enzyme expression, the novel achievement of this work was the successful fusion of the NanoLuc reporter to a carbohydrate binding module (CBM) with specificity to crystalline cellulose. This novel chimeric reporter (nluc::cbm) bestows the specific and irreversible immobilization of NanoLuc onto a low-cost, widely available crystalline cellulosic substrate. We have shown the possibility of detecting the immobilized fusion protein in a filter plate which resulted from a single CFU of E. coli. We then demonstrated that microcrystalline cellulose can be used to concentrate the fusion reporter from 100 mL water samples allowing a limit of detection of <10 CFU mL−1E. coli in 3 hours. Therefore, we conclude that our phage-based detection assay displays significant aptitude as a proof-of-concept drinking water diagnostic assay for the low-cost, rapid and sensitive detection of E. coli. Additional improvements in the capture efficiency of the phage-based fusion reporter should allow a limit of detection of <10 CFU per 100 mL.
Introduction
Drinking water contaminated with pathogenic bacteria is a major public health concern, both in the United States1–4 and worldwide.5Escherichia coli (E. coli) is a major cause of global morbidity and mortality. While the WHO estimates approximately 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 annual deaths are due to foodborne E. coli infections, there is an added consequence of 5 million years of life lost (YLLS) and 5 million disability adjusted life years (DALYS).5 In addition to gastrointestinal infections, E. coli is the most common causative agent for urinary tract infections, 8.9% of sepsis cases, and 29% of early onset neonatal sepsis cases.6,7 “Generic” E. coli is often used as an indicator for contamination or improper sanitation of water or food.8,9 Found in high concentrations in the feces of most mammals, the presence of E. coli is considered the best biological indicator for fecal contamination in drinking water.10
000 annual deaths are due to foodborne E. coli infections, there is an added consequence of 5 million years of life lost (YLLS) and 5 million disability adjusted life years (DALYS).5 In addition to gastrointestinal infections, E. coli is the most common causative agent for urinary tract infections, 8.9% of sepsis cases, and 29% of early onset neonatal sepsis cases.6,7 “Generic” E. coli is often used as an indicator for contamination or improper sanitation of water or food.8,9 Found in high concentrations in the feces of most mammals, the presence of E. coli is considered the best biological indicator for fecal contamination in drinking water.10
While the identification of indicators or potential pathogens often involves the culturing of serological, food, or environmental samples, new technologies have been introduced with the promise of bringing assay times from days, to hours, and even minutes. While some of these advanced technologies (e.g. optical nanostructures, surface enhanced Raman spectroscopy) have shown promise as sensitive detection methods, these methods typically require a relatively clean sample in a small volume. It is clear that the true bottleneck to rapid detection remains with the separation, concentration, and cleanup steps of the initial sample preparation. An ideal separation method should (1) remove the analyte from the matrix, (2) remove any possible inhibitors to a downstream detection system, and (3) reduce the sample size while maintaining a high capture efficiency.11 Unfortunately, there does not presently exist a pragmatic system that integrates sample preparation and pathogen detection from large volume samples. While the vast majority of research focuses on improving the sensitivity of pathogen detection rather than separation/concentration, an ideal detection platform would incorporate all these components as well as the ability to determine cell viability.
Furthermore, bacterial species causing foodborne disease have been shown to rapidly gain virulence factors as well as antibiotic resistance markers, further necessitating the need for rapid detection methods to successfully mitigate the population's exposure to potentially harmful pathogens. The increasing prevalence of E. coli strains that are resistant to all known antibiotics is considered one of the most serious risks to public health in the near future.12 This relatively rapid rise of antibiotic resistance in pathogens has been confirmed and is actively monitored by the Centers for Disease Control (CDC) and the National Antimicrobial Resistance Monitoring System (NARMS).13,14
Bacteriophage (phage) based detection methods have demonstrated a high potential to detect, mitigate and control the causative bacterial agents of foodborne illness.15–21 Phages are a natural viral predator of bacteria whose total population outnumbers any other biological entity on the planet.22 Infection begins with binding to host cell surface receptors and then injecting their genome into the cell through specific and localized degradation of the bacterial cell wall.23 This highly specific and irreversible interaction is the first major determinant of a phage's range of potential hosts. While other genetic factors contribute to a phage's ability to infect a host (restriction enzymes, CRISPR, etc.), directed genetic modifications to phage tail components involved in the initial binding event have successfully expanded the host range.24
Upon infection, the injected phage DNA has been shown to dramatically alter bacterial gene expression and metabolism. For example, some phages encode factors that alter the promoter specificity of the host RNA polymerase to selectively force overexpression of phage DNA.25 Furthermore, other phages have been shown to cause the complete cessation of bacterial macromolecular synthesis, allowing for the specialized allocation of cellular resources for phage expression and reproduction.26
Reporter phages are created when an exogenous gene is added to a phage genome, causing expression of a readily detectable enzyme concurrent with phage infection.19 The concentration of enzyme in solution can be correlated with the susceptible bacterial population within the sample. Commonly used phage reporters include alkaline phosphatase (phoA),27–29 beta-galactosidase (lacZ),28,30,31 green fluorescent protein (gfp),32,33 and bacterial luciferases (luxAB or luxCDABE),34–36 among others.23,37,38
Recently engineered and commercialized by Promega, a highly active luciferase (NanoLuc or NLuc) and substrate (furimazine) system have generated luminescent signals orders of magnitude greater than that of other commonly used luciferases.39 Furthermore, the small size of NanoLuc (19 kDa), coupled with its high activity, makes it an ideal candidate for a phage reporter where any genetic insertion must be small enough to fit the expanded genome into the phage capsid.40,41
In order to concentrate and purify engineered proteins, numerous epitope tags such as His-tag,42 AviTag,43 and FLAG,44 among others45 are commonly used. These tags bind to cobalt,46 biotin,43 and/or antibody substrates, respectively, all of which are expensive, complex or both. The type of affinity tag used in this work, a Carbohydrate Binding Module (CBM), has been widely employed as protein fusions47 to provide immobilization onto low-cost, widely available substrates. Carbohydrate binding modules are commonly found within carbohydrate active enzymes that fold independently of the larger protein structure and display specific binding to carbohydrate substrates.47 The specific ligand used in this work (CBM2a from Cellulomonas fimi) displayed irreversible binding to crystalline cellulose when expressed as a fusion protein.48 A CBM is a crucial component of many carbohydrate active enzymes, especially for insoluble substrates where enzyme diffusion would limit substrate availability.
In this paper, we present a novel recombinant phage that has been constructed and implemented into a detection system capable of detecting low concentrations of viable E. coli cells. The novel fusion reporter phage T7NLC employs the highly active NLuc luciferase fused to the irreversibly binding carbohydrate binding module CBM2a to sensitively detect E. coli cells in drinking water samples. While a cocktail of phages may be required for the detection of an entire bacterial species, we targeted E. coli BL21 as a proof-of-principle model to demonstrate the potential sensitivity of our system.
This work outlines the successful (i) modification of the T7 phage genome to contain nluc or nluc::cbm reporter genes, (ii) expression of NanoLuc or NanoLuc::CBM in E. coli from infection of T7NL or T7NLC, respectively, (iii) estimation of E. coli concentration required to generate a detectable signal, and (iv) demonstration of a low-cost and rapid method to detect E. coli in 100 mL of drinking water using a phage-based detection strategy.
Experimental
Materials
Unless otherwise noted, all chemicals were reagent grade and purchased from Fisher Scientific (Pittsburgh, PA, USA) or Sigma-Aldrich (St Louis, MO, USA). DNA synthesis was provided by Integrated DNA Technologies (Coralville, Iowa, USA). Restriction enzymes and DNA polymerases were purchased from New England Biolabs (Ipswitch, MA, USA). Escherichia coli BL21 (ATCC® BAA-1025TM) and E. coli Castellani and Chalmers (ATCC 13706), routinely used in this study as hosts for phage T7, were grown aerobically in Luria Bertani (LB) at 37 °C with continuous shaking agitation. Transformation of recombinant phage DNA was performed in electrocompetent E. coli DH10B cells (MegaX, Invitrogen, USA). Genomic phage DNA to be used as a cloning vector was purified from T7Select 415–1 (Novagen, USA) propagated in BL21. Extraction of phage DNA was performed with the Qiagen Genomic Tip 100/G (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Routine DNA purifications were performed using glycogen for nucleation and precipitated using sodium acetate (3 M) and ethanol (100%).49Phage stock preparation
An overnight E. coli BL21 culture (2 mL) was added to LB media (200 mL) and incubated (37 °C, 250 rpm, 2–3 hours) until an OD600 of 0.4–0.6, suggesting steady state growth.50 Recombinant T7 phage was added at a multiplicity of infection (MOI) of 0.1 and incubated (37 °C, 250 rpm, 1.5–2 hours) until a significant decrease in OD600 was observed. Chloroform (200 μL) was added directly to the lysate before centrifugation (12000g, 5 min) to clear bacterial debris. The supernatant was sterile filtered (0.22 μm) and phage titers were determined by standard double overlay plaque assays. Phage samples with titer's exceeding 109 PFU mL−1 were further concentrated with the addition of PEG6000 (0.4%) & NaCl (0.3 M) and incubated (4 °C, 12–16 hours) to precipitate phage particles. The precipitated phage samples were centrifuged (35000g, 2 hours, 4 °C), the supernatant was discarded, the concentrated phage pellet was re-suspended in Tris-HCl pH 7.4 (∼5 mL), sterile filtered (0.22 μm) and stored at 4 °C until needed.
Bacteriophage genome preparation
Aliquots (5 mL) of concentrated phage samples (>1011 PFU mL−1) were mixed with 4% SDS (5 mL) and incubated at 70 °C for 20 minutes. After cooling on ice, sodium acetate (2.55 M, 5 mL) was added before centrifugation (10000g, 10 min). The supernatant was passed onto the Qiagen 100/G Genomic DNA column (Qiagen, Hilden, Germany) and phage genomic DNA was purified according to the manufacturer's instructions. DNA concentration was determined on a NanoDrop One (Thermo Scientific, Wilmington, DE, USA).
Genetic engineering of bacteriophage T7
A reporter enzyme expression construct was inserted into the phage T7 genome to create the reporter phage T7NLC (Fig. 1). In an effort to increase expression levels of NLuc, the strongest wild type T7 promoter sequence51 was used to maximize transcription and a custom ribosome binding site (RBS) was designed to optimize translation.52 An N-terminal leader sequence (pelB) was added immediately upstream of nluc as it has been shown to significantly increase soluble heterologous enzyme expression in E. coli53,54 by directing newly made proteins to the periplasmic space. Cleavage by a signal peptidase in the periplasm eliminates the 22 amino acid pelB sequence from the mature NLuc enzyme and confers no loss in enzymatic activity.55 The nluc and nluc::cbm genes were codon optimized with respect to the E. coli species within the T7 host range.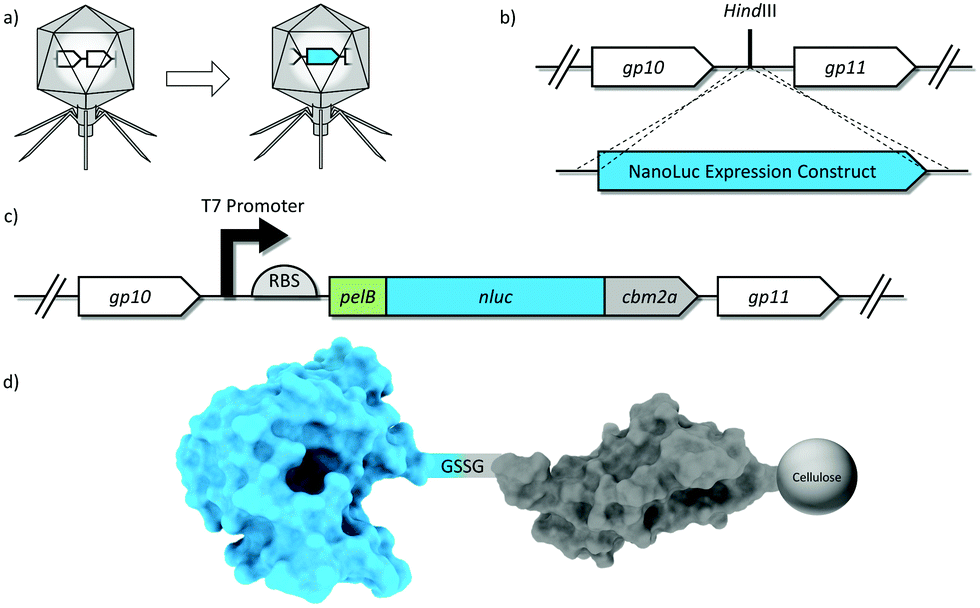 | ||
| Fig. 1 Schematic representation of phage genetic engineering. (A) The ∼1 kb NanoLuc expression construct was successfully integrated into the T7 phage genome (B) at a highly expressed region immediately downstream of the capsid gene. (C) Expression construct details: A T7 promoter (black arrow), a ribosome binding site (RBS; half circle), and a pelB leader sequence. The carbohydrate binding module (cbm2a) was fused to the C-terminus of nluc with a short flexible linker. (D) Schematic of the novel reporter fusion displaying binding affinity to cellulose. Genes/proteins/phages not drawn to scale for purposes of illustration. | ||
The nluc & nluc::cbm expression constructs, used to make T7NL (accession number MH651797) and T7NLC (accession number MH651798) respectively, were expected to add 642 bp and 984 bp to the phage genome, respectively. Based on previous work with T7 reporter phages bearing much larger reporters,31,56 the 1.7% (T7NL) and 2.6% (T7NLC) increases in genome size were expected to bear no significant reduction in fitness.
A standard restriction digest was performed on the purified phage genome using HindIII-HF to create double strand DNA breaks at the insertion site. The NanoLuc reporter enzyme expression cassette was synthesized as a linear double stranded DNA molecule with 50 bp of phage homology at each terminus. The insert was added to the digested phage genome at a 2:1 molar ratio and assembled with NEBuilder® Hifi DNA Assembly Master Mix (NEB, Ipswitch, MA), in accordance with the manufacturer's specifications. Insert confirmation was performed by PCR and DNA was purified using standard methods and resuspended in nuclease free water prior to transformation. The recombinant phage DNA was electroporated into E. coli DH10B cells (MegaX, New England Biolabs, Ipswitch, USA). Sterile SOB media was added, and the transformed cells were incubated (37 °C, 2 hours) until visible lysis occurred. Chloroform (1–2 drops) was added to lyse any remaining cells and the mixture was centrifuged at 10000g for 1 minute to clear bacterial debris. Following sterile filtration (0.22 μm), the transformation lysate was plated on a lawn of E. coli BL21 where individual plaques were isolated, resuspended in broth and insert confirmation done by PCR. Positive plaques were isolated, propagated and submitted for full genome sequencing.
Bacteriophage T7NL & T7NLC stock preparation
Due to their high sensitivity, it was necessary to remove excess reporter enzyme from the phage stock solutions so as to minimize background signal that could significantly impair the assay sensitivity. The concentrated phage sample (∼1011 PFU mL−1) was first diluted in sterile LB to a concentration of 109 PFU mL−1. For phage T7NLC, powdered microcrystalline cellulose (0.5 g) was added and the sample was placed on a rotational shaker for 30 minutes before sterile filtration (0.22 μm). The resulting phage stock was tested for enzymatic activity and the cellulose sequestration procedure was repeated if detectable signal was generated.Reporter enzyme binding to cellulose
The lysates resulting from infections between the engineered phages and E. coli were prepared as described above. In order to test the ability of the CBM to allow binding of the reporter enzyme to cellulose, the lysates were spotted onto regenerated cellulose filters, washed to remove unbound reporters, and then imaged following the addition of the NanoGlo substrate.Detection of log phase E. coli in broth
Log phase E. coli 13706 cultures were obtained by adding overnight culture (200 μL) to fresh broth (5 mL). The culture was incubated (37 °C, 250 rpm, ∼1 h) until an OD600 value of 0.8 was reached. Phage were added to serial dilutions of bacteria and incubated (37 °C, 250 rpm, 90 min) for infection to occur.Following phage infection, an aliquot of each sample was added to a 384 well cellulose filter plate (AcroPrep 384 BioTrace NT, Pall, Port Washington, USA). Vacuum was applied according to the manufacturer's specifications until no liquid remained. NanoGlo substrate was applied to each well (20 μL) and luminescence was measured on a plate reader (BioTek Synergy H1) with a 0.1 s integration time. While the vacuum filter plate provides a highly efficient capture mechanism for small sample volumes (∼200 μL), higher sample throughput becomes prohibitory when the large sample (100 mL) volumes required for regulatory testing are considered.
Detection of stationary phase E. coli in lake water
Lake water was collected from Lake Sammamish (WA) and used as a representative environmental sample matrix. Samples were filtered sterilized (0.22 μm), inoculated with E. coli 13707 (ATCC) and left at room temperature for 2 days. After 48 hours, samples were diluted in LB and incubated (37 °C, 250 rpm) for various time intervals. Following enrichment, T7NLC phage (107 PFU mL−1) was added and luminescence was measured using 384 well filter plates as described previously.Drinking water assay
A phage-based drinking water assay was developed as a proof of principle to detect E. coli in 100 mL of drinking water using the recombinant T7NLC reporter phage (Fig. 2). Drinking water samples (100 mL) were autoclaved and inoculated with various concentrations of stationary phase E. coli BL21. Sterile concentrated 5× LB media (25 mL) was added to the sample and incubated (37 °C, 225 rpm, 60 min) to allow resuscitation of injured or stressed E. coli cells. Following pre-enrichment, phage stock (1 mL of 109 PFU mL−1) and microcrystalline cellulose (0.05 g) were added and incubated (37 °C, 225 rpm, 90 min) to allow for phage infection and reporter enzyme production. NanoLuc-CBM complexed with the microcrystalline cellulose was pelleted by low speed centrifugation (3000g, 5 min). The bulk lysate was decanted, and the cellulose pellet was resuspended directly in NanoGlo substrate and immediately evaluated for luminescence. Bulk lysate luminescence was evaluated for both T7NL and T7NLC phages by adding equal volumes of the corresponding lysate (100 μL) to undiluted NanoGlo substrate (100 μL), in accordance with the manufacturer's specifications. Controls included uninfected bacterial cultures with no phage and phage alone. Measurements were performed in opaque white 96 well plates on a luminescent plate reader (Biotek, Winooski, VT, USA). Luminescent intensity was reported as relative luminescence units (RLU) using an integration time of 0.1 s. Samples were evaluated in triplicate and reported as the mean ± standard deviation. The limit of detection was determined using the sum of the negative control and three standard deviations (0 + 3SD) as the lowest detectable signal.57–60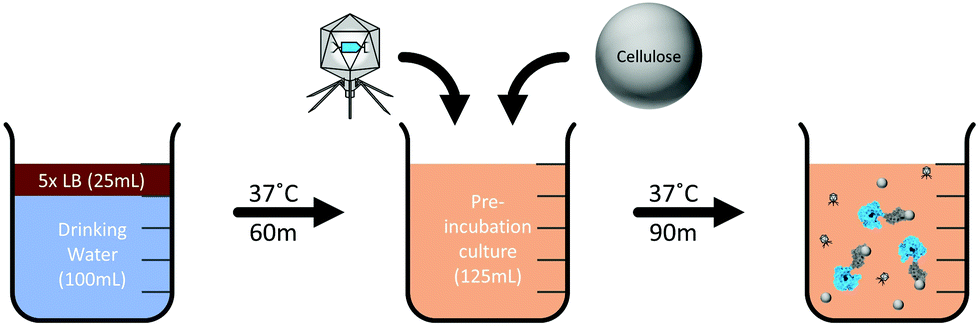 | ||
| Fig. 2 Schematic representation of detection assay. The water sample is first supplemented with concentrated growth media (5× LB) to allow resuscitation of E. coli. After an incubation period, the phage and cellulose are added to the sample and the infection incubation period begins, during which expression of NanoLuc-CBM occurs. The reporter enzyme then binds to the cellulose which is collected for analysis. | ||
Results and discussion
Reporter phage construction & isolation
The nluc & nluc::cbm reporter genes were synthesized within expression constructs and successfully inserted into phage T7 to generate reporter phages T7NL & T7NLC, respectively. Plaques were screened initially by the direct addition of substrate followed by PCR. Plaque morphologies for both recombinant phages similar identical to wild type phages. The burst size and lysis times were also comparable to the wild type, suggesting no measureable loss of fitness (data not shown). Correct insertion of the nluc & nluc::cbm expression constructs were confirmed via PCR using external screening primers. Sanger sequencing results revealed the correct 642 bp & 984 bp insertions, for T7NL and T7NLC respectively, without other significant mutations, insertions or deletions within the insertion site.Characterization of phage infection
Infection of E.coli BL21 with recombinant reporter phage transduced the nluc::cbm gene into the target bacterial cell where the reporter enzyme was successfully expressed. As seen in Fig. 3, a characteristic phage infection was performed to evaluate the phage's ability to lyse target cell populations and express reporter enzyme by measuring luminescence intensity and optical density at regular intervals. Post phage addition, a plateau in the OD600 occurred after approximately 30 minutes. The bioluminescent signal generation was relatively fast following phage addition. While some of the rapid signal generation is due to the carryover of reporter enzyme from the phage lysate, the plateau of the signal occurs 40–50 minutes after phage addition. The final phage stock concentration (107 PFU mL−1) was used to maintain an MOI > 1 and therefore minimize the time required for the assay. Lysis times are shortest when phages outnumber bacteria because phage adsorption times are near instantaneous and co-infections could possibly deliver multiple copies of reporter enzyme DNA.61,62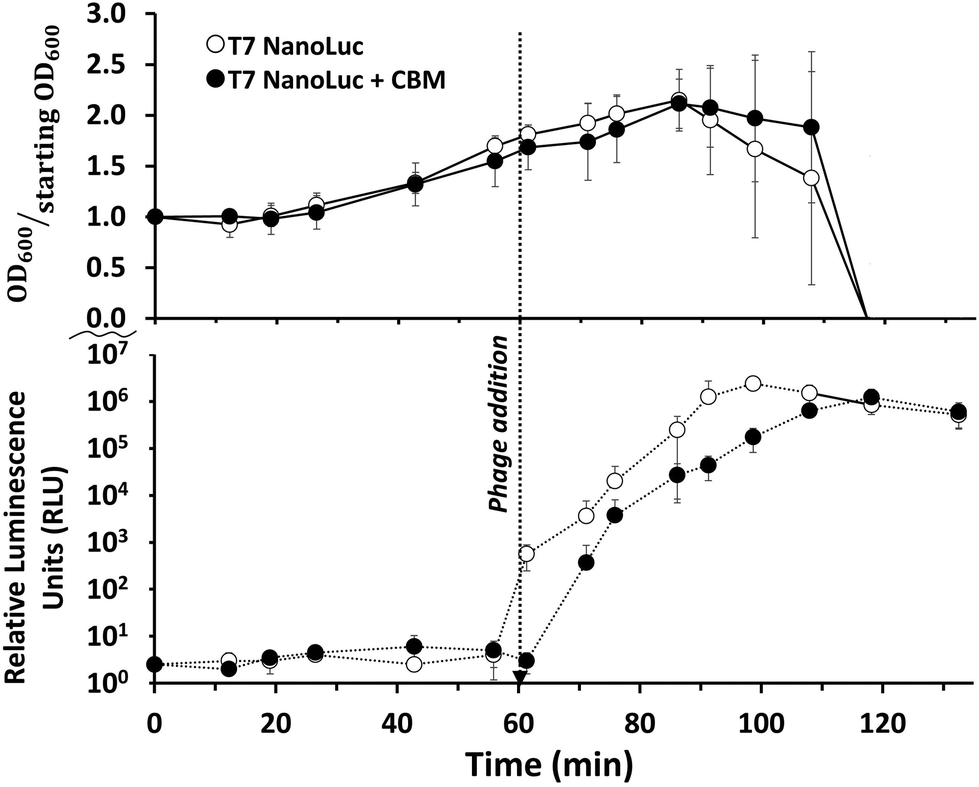 | ||
| Fig. 3 Optical density and luminescence of E. coli samples before and after the addition of the phages. The optical density briefly increased following the addition of reporter phage (indicating steady state growth) then rapidly dropped as cells lysed. Data points represent the average of three replicates and error bars represent the standard deviations. | ||
Reporter enzyme binding to cellulose
In order to confirm the ability of the CBM to immobilize the enzyme onto cellulose, lysate resulting from the respective infections of T7NL and T7NLC with E. coli BL21 were spotted onto regenerated cellulose filter papers. NanoGlo was then directly added to some of the filters and imaged, while another set of filters was washed prior to the addition of substrate. The results demonstrated a spreading of the bioluminescence in relation to the original deposition spot for the T7NL on unwashed filters, while the T7NLC luinesence remained limited to the original lysate spot. Additionally, the washed filters had a significant drop in bioluminescence for the T7NL, while the T7NLC was similar to the unwashed filters (data not shown). This suggested that the CBM facilitated to the immobilization of the reporter enzyme onto cellulose.Performance of T7NLC for E. coli detection
The ability of T7NLC to overexpress reporter enzymes in target bacterial hosts in different stages of growth was evaluated. Exponentially growing cells in broth were infected to evaluate the minimum number of cells required to produce a detectable signal and thus establish a limit of detection. The resulting lysate was vacuum filtered through a cellulose membrane on a filter plate to immobilize the reporter enzyme. Fig. 4 indicates that when coupled with a highly efficient capture mechanism, a detectable signal can be generated from an infection between T7NLC and <10 mid-log phase bacteria. Furthermore, an apparent linear relationship between the E. coli population and the resulting bioluminescent signal was observed, indicating that the T7NLC phage can be used to approximate bacterial concentrations from a single CFU to over 103 CFU mL−1.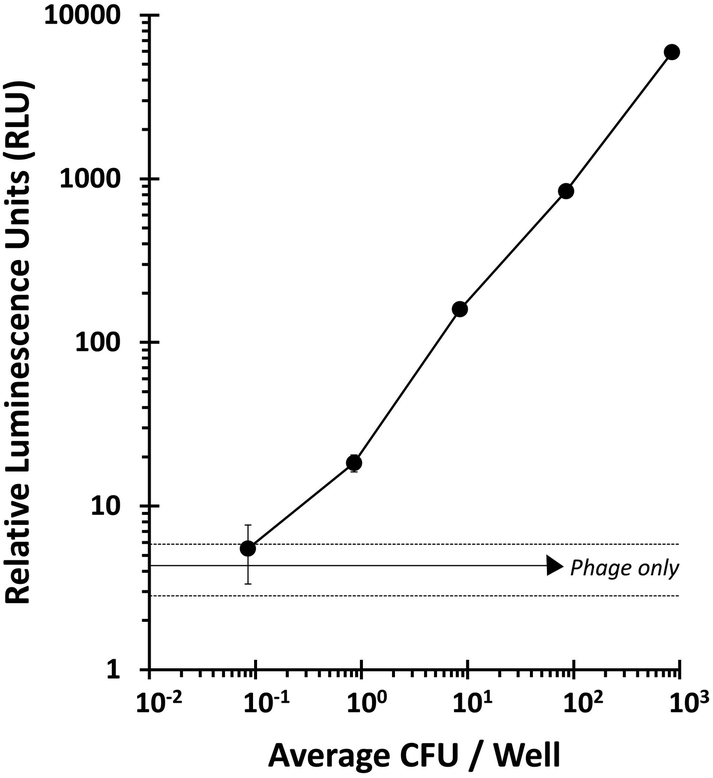 | ||
| Fig. 4 The number of E. coli CFUs needed to produce a detectable signal was determined by phage infecting serial dilutions of E. coli and passing them through a filter plate. The “phage only” negative control (solid horizontal line) is represented ±standard deviations (dashed lines). Data points represent the average of six replicates and error bars represent standard deviation. | ||
To approximate realistic analytical conditions, E. coli were left in sterile filtered lake water for 48 hours to ensure they reached stationary growth phase. Cells were then enriched and infected with T7NLC to evaluate the phage's effectiveness in representative drinking water samples. As seen in Fig. 5, E. coli rapidly reached steady state growth allowing a starting concentration of <10 CFU E. coli to become detectable after only 3 hours of enrichment.
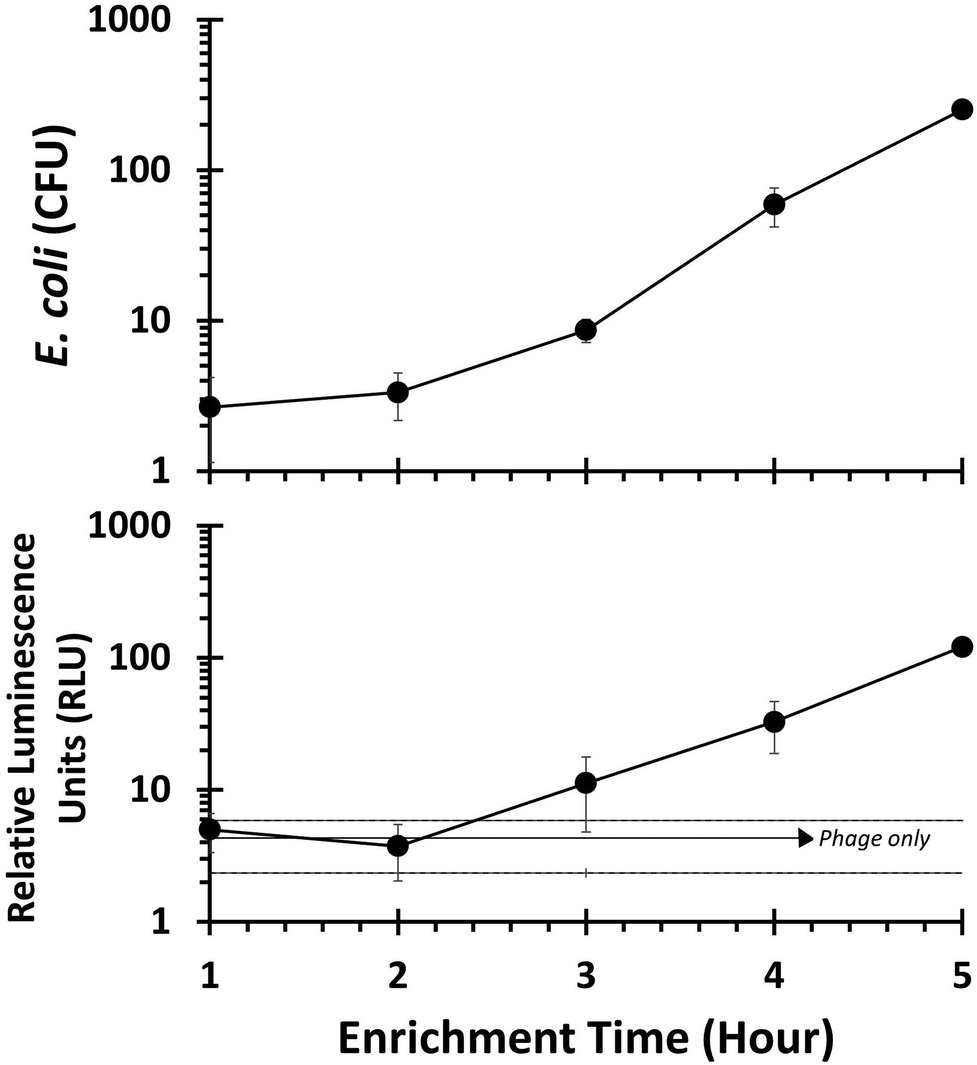 | ||
| Fig. 5 Stationary phase E. coli inoculated into lake water was enriched from 1 to 5 hours prior to being infected with T7NLC. The results indicate that after 3 hours of enrichment, <10 CFU of E. coli are detectable from lake water. The “phage only” negative control (solid horizontal line) is represented as ±standard deviations (dashed lines). Data points represent the average of three replicates and error bars represent the standard deviation. | ||
Phage-based assay conditions for the detection of E. coli in 100 mL water samples
The T7NLC phage was used to detect E. coli from 100 mL samples of water. Water samples were inoculated with varying concentrations of E. coli in order to determine the limit of detection for the assay. Prior to phage/cellulose addition, a 60 minute pre-enrichment step served to resuscitate any bacterial cells that were injured or had reached stationary growth (Fig. 2). While the time can be varied according to host growth rates, the pre-enrichment step is critical for a phage-based assay because phage require an actively growing host for successful infection.63 Although two recombinant phages were generated in this work, only T7NLC, which employed the cellulose affinity tag, was selected for the final detection assay. As seen in Fig. 6, the detectable NanoLuc signal was closely correlated with the bacterial cell population. The detection limit for T7NLC is <10 CFU mL−1 in a total assay time of 3 hours. Furthermore, the results seen in Fig. 4 suggest a lower limit of detection is achievable and therefore it is likely that the demonstrated 100 mL water assay is: (i) not efficiently infecting the E. coli in the larger samples, or (ii) the free cellulose is not efficiently capturing the fusion reporter released from the resulting infections. Additionally, the effect of nonspecific binding must be taken into account as well. It is possible that some signal is lost due to the binding of the reporter enzyme to carbohydrates within the bacterial cell as well as nonspecific interactions with the polypropylene tubes used for the assay.48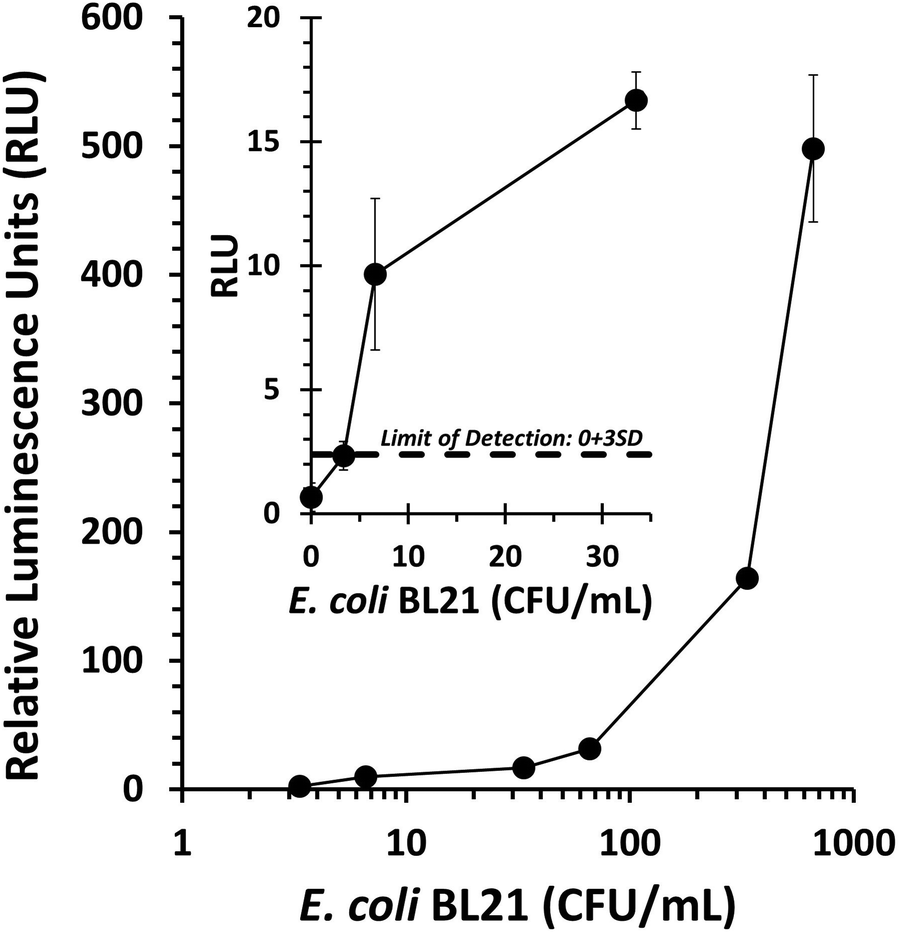 | ||
| Fig. 6 Drinking water (100 mL) containing E. coli BL21 was used to determine the dose response of the phage-based assay. Within the range tested, increasing E. coli concentrations correlated with stronger bioluminescent signal. The limit of detection was defined as the negative control (0 CFU mL−1) plus three standard deviations. Data points represent the average of three replicates and error bars represent the standard deviation. | ||
Conclusions
Current methods for the detection of microorganisms in food samples are largely culture based, requiring trained laboratory personal multiple days before a definite result. Furthermore, many pathogenic strains of E. coli have such low infectious doses (<100 CFU) that the development of rapid and sensitive detection schemes is required to adequately mitigate potential risk to public health. The successful integration of a newly developed, highly active luciferase gene (nluc) into a phage genome, created a reporter phage capable of rapidly detecting low concentrations of E. coli. The nluc gene was synthesized within an expression construct specifically engineered for T7 expression. Overexpression of the nluc gene was achieved by optimization of regulatory sequences as well as by insertion of the reporter into a highly expressed region of the phage genome. The expression construct employed the strongest native T7 promoter sequence, a custom ribosome binding site, a pelB leader sequence, and a codon optimized nluc gene. Finally, the most significant development of this work was the genetic fusion of a carbohydrate binding module (CBM2a) to NanoLuc within the previously described expression construct. As a result, the functionalized reporter was successfully concentrated through immobilization onto inexpensive, inert, insoluble microcrystalline cellulose. Microcrystalline cellulose dispersed into the bacteria-phage complex successfully captured the novel chimeric NanoLuc::CBM produced as a result of target cell infection. Following a full phage infection, the cellulose can be either actively pelleted by centrifugation or passively separated by gravity depending on access to resources. The insoluble cellulose pellet can be resuspended in enzyme substrate for immediate measurement or in a biological buffer for downstream applications.The use of cellulose to collect the reporter probe from the bulk sample solution served to reduce the overall sample volume by over 2 orders of magnitude. This was achieved by separating the 0.05 g cellulose added to the 125 mL sample and resuspending the cellulose-enzyme complex in less than 1 mL of substrate following phage infection. Future studies using cellulose filters to capture the reporter probes may demonstrate a significant improvement in the capture of the NanoLuc::CBM resulting in a lower limit of detection.
The light emitted from NanoLuc is bright blue as evidenced by a sharp emission peak at 450 nm. When images of NanoLuc are separated in RGB channels, over 98% of signal emitted resides within the blue channel.64 As a result, NanoLuc has significant potential to be incorporated into a multiplex recombinant phage biosensor (i.e. NanoLuc + Red and/or Green luciferases inserted into phages with different host ranges) that would be capable of rapid and simultaneous differentiation of specific analytical epitopes.
Areas for further optimization include broadening phage host range, increasing enzyme expression during phage infection, and minimizing the enzymatic loss of activity when genetically fusing affinity binding ligands. Genetic modifications to the tail fibers of T7 have been shown to successfully expand the range of permissive hosts.24 Similar genetic engineering would be straightforward with T7NLC as the NanoLuc expression cassette bears no effect on the tail fibers. Increased enzyme expression will be achieved by further optimization of regulatory and signal sequences as well as the insert location within the phage genome. The optimal leader sequence must be proven empirically, as leader sequences other than pelB have been shown to significantly increase expression as well.55 Furthermore, optimization of the linker sequence between NanoLuc and the CBM could function to fully retain enzymatic activity of our novel chimeric reporter as compared to the standard NanoLuc enzyme. Expression of a reporter enzyme with a CBM causes binding to cellulosic entities within the cell during expression as well as plastic species within standard laboratory tubing and glassware. Blocking of plastics with 1% BSA should lead to additional efficiencies and a lower limit of detection.48
A current limitation to phage-based assays remains the need for cocktails of phages in order to cover the desired host range. This is due to the high specificity of the phages which can often be limited to single strains. Ongoing work by the authors and other labs aims to customize the tail fibers of phages to allow tailored host ranges. Such work will allow a better utilization of phages as both detection and therapy tools.
Our results suggest that our novel recombinant phages are ideal for an ultrasensitive bacterial detection assay. Our proof-of-principle detection assay utilizes recombinant phages that express novel chimeric reporter enzymes to rapidly and sensitively detect less than 10 CFU mL−1E. coli. Lower limits of detection can be reached by increasing the pre-enrichment times thereby allowing the bacteria to reach a higher concentration.
As we demonstrated the successful genetic engineering of a phage to sensitively detect E. coli, similar efforts could provide low cost detection assays to other fields burdened with bacterial threats including food and water safety, medical diagnostics, animal health, and bio threat detection.
Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
The authors gratefully acknowledge the Bill and Melinda Gates Foundation Trust for their sponsorship through Intellectual Ventures’ Global Good Fund.This work is also supported by the AFRI NIFA Fellowships Grant Program: Predoctoral Fellowships, Accession Number 1010727 from the USDA National Institute of Food and Agriculture. The authors would like to acknowledge additional support from the U.S. Department of Agriculture awards (2013-02037) and (2016-67017-26462).
Notes and references
- M. B. Batz, S. Hoffmann and J. G. Morris Jr., J. Food Prot., 2012, 75, 1278–1291 CrossRef PubMed.
- E. Scallan, R. M. Hoekstra, F. J. Angulo, R. V. Tauxe, M.-A. Widdowson, S. L. Roy, J. L. Jones and P. M. Griffin, Emerging Infect. Dis., 2011, 17, 7–15 CrossRef PubMed.
- K. D. Beer, J. W. Gargano, V. A. Roberts, V. R. Hill, L. E. Garrison, P. K. Kutty, E. D. Hilborn, T. J. Wade, K. E. Fullerton and J. S. Yoder, MMWR Morb. Mortal. Wkly. Rep., 2015, 64, 842–848 CrossRef PubMed.
- M. C. Hlavsa, V. A. Roberts, A. M. Kahler, E. D. Hilborn, T. R. Mecher, M. J. Beach, T. J. Wade and J. S. Yoder, MMWR Morb. Mortal. Wkly. Rep., 2015, 64, 668–672 Search PubMed.
- A. H. Havelaar, M. D. Kirk, P. R. Torgerson, H. J. Gibb, T. Hald, R. J. Lake, N. Praet, D. C. Bellinger, N. R. de Silva, N. Gargouri, N. Speybroeck, A. Cawthorne, C. Mathers, C. Stein, F. J. Angulo, B. Devleesschauwer and G. World Health Organization Foodborne Disease Burden Epidemiology Reference, PLoS Med., 2015, 12, e1001923 CrossRef PubMed.
- E. Barantsevich, N. Barantsevich, N. Rybkova, I. Churkina, N. Pestova and M. Karpenko, Crit. Care, 2011, 15, P45–P45 CrossRef.
- B. J. Stoll, N. I. Hansen, P. J. Sanchez, R. G. Faix, B. B. Poindexter, K. P. Van Meurs, M. J. Bizzarro, R. N. Goldberg, I. D. Frantz 3rd, E. C. Hale, S. Shankaran, K. Kennedy, W. A. Carlo, K. L. Watterberg, E. F. Bell, M. C. Walsh, K. Schibler, A. R. Laptook, A. L. Shane, S. J. Schrag, A. Das and R. D. Higgins, Pediatrics, 2011, 127, 817–826 CrossRef PubMed.
- J. Wright, S. Gundry and R. Conroy, Trop. Med. Int. Health, 2004, 9, 106–117 CrossRef PubMed.
- E. I. L. L. C. Office of The Federal Register, Title 40 Protection of Environment Parts 136 to 149 (Revised as of July 1, 2013): 40-CFR-Vol-24, U.S. Government Printing Office, 2014 Search PubMed.
- S. C. Edberg, E. W. Rice, R. J. Karlin and M. J. Allen, Symp. Ser. - Soc. Appl. Microbiol., 2000, 106s–116s CrossRef.
- K. A. Stevens and L. A. Jaykus, Crit. Rev. Microbiol., 2004, 30, 7–24 CrossRef PubMed.
- A. Okoh, A. Adegoke, O. Adesemoye, O. Babalola, I. Igbinosa and F. Aghdasi, J. Pure Appl. Microbiol., 2012, 6, 1069–1083 Search PubMed.
- C. f. D. Control and Prevention, National Antimicrobial Resistance Monitoring System for Enteric Bacteria (NARMS): Human isolates surveillance report for 2014 (final report), CDC, Atlanta, GA, 2016 Search PubMed.
- T. Frieden, Antibiotic Resistance Threats in the United States 2013, DIANE Publishing Company, 2013 Search PubMed.
- C. E. D. Rees, B. M. C. Swift and G. Botsaris, Bacteriophage-Based Techniques for Detection of Foodborne Pathogens, 2014, pp. 194–202, DOI:10.1016/b978-0-12-384730-0.00032-x.
- M. Schmelcher and M. J. Loessner, Bacteriophage, 2014, 4, e28137 CrossRef PubMed.
- A. Singh, S. Poshtiban and S. Evoy, Sensors, 2013, 13, 1763–1786 CrossRef PubMed.
- M. Sharma, Bacteriophage, 2013, 3, e25518 CrossRef PubMed.
- A. E. Smartt and S. Ripp, Anal. Bioanal. Chem., 2011, 400, 991–1007 CrossRef PubMed.
- P. Garcia, B. Martinez, J. M. Obeso and A. Rodriguez, Lett. Appl. Microbiol., 2008, 47, 479–485 CrossRef PubMed.
- S. Hagens and M. J. Loessner, Appl. Microbiol. Biotechnol., 2007, 76, 513–519 CrossRef PubMed.
- F. Rohwer and R. Edwards, J. Bacteriol., 2002, 184, 4529–4535 CrossRef PubMed.
- J. Klumpp, D. E. Fouts and S. Sozhamannan, Briefings Funct. Genomics, 2013, 12, 354–365 CrossRef PubMed.
- H. Ando, S. Lemire, D. P. Pires and T. K. Lu, Cell Syst., 2015, 1, 187–196 CrossRef PubMed.
- D. M. Hinton, S. Pande, N. Wais, X. B. Johnson, M. Vuthoori, A. Makela and I. Hook-Barnard, Microbiology, 2005, 151, 1729–1740 CrossRef PubMed.
- J. F. Koerner and D. P. Snustad, Microbiol. Rev., 1979, 43, 199–223 Search PubMed.
- S. D. Alcaine, L. Tilton, M. A. Serrano, M. Wang, R. W. Vachet and S. R. Nugen, Appl. Microbiol. Biotechnol., 2015, 99, 8177–8185 CrossRef PubMed.
- J. Chen, S. D. Alcaine, A. A. Jackson, V. M. Rotello and S. R. Nugen, ACS Sens., 2017, 2, 484–489 CrossRef PubMed.
- A. A. Jackson, T. C. Hinkley, J. N. Talbert, S. R. Nugen and D. A. Sela, Analyst, 2016, 141(19), 5543–5548 RSC.
- R. Derda, M. R. Lockett, S. K. Tang, R. C. Fuller, E. J. Maxwell, B. Breiten, C. A. Cuddemi, A. Ozdogan and G. M. Whitesides, Anal. Chem., 2013, 85, 7213–7220 CrossRef PubMed.
- J. Chen, S. D. Alcaine, Z. Jiang, V. M. Rotello and S. R. Nugen, Anal. Chem., 2015, 87, 8977–8984 CrossRef PubMed.
- T. Funatsu, T. Taniyama, T. Tajima, H. Tadakuma and H. Namiki, Microbiol. Immunol., 2002, 46, 365–369 CrossRef PubMed.
- Y. Tanji, C. Furukawa, S. H. Na, T. Hijikata, K. Miyanaga and H. Unno, J. Biotechnol., 2004, 114, 11–20 CrossRef PubMed.
- M. J. Loessner, C. E. Rees, G. S. Stewart and S. Scherer, Appl. Environ. Microbiol., 1996, 62, 1133–1140 Search PubMed.
- S. Kim, M. Kim and S. Ryu, Anal. Chem., 2014, 86, 5858–5864 CrossRef PubMed.
- Y. Born, L. Fieseler, V. Thony, N. Leimer, B. Duffy and M. J. Loessner, Appl. Environ. Microbiol., 2017, 83, 0341 CrossRef PubMed.
- S. Hagens, T. de Wouters, P. Vollenweider and M. J. Loessner, Bacteriophage, 2011, 1, 143–151 CrossRef PubMed.
- J. Klumpp and M. J. Loessner, Bacteriophage, 2013, 3, e26861 CrossRef PubMed.
- M. P. Hall, J. Unch, B. F. Binkowski, M. P. Valley, B. L. Butler, M. G. Wood, P. Otto, K. Zimmerman, G. Vidugiris, T. Machleidt, M. B. Robers, H. A. Benink, C. T. Eggers, M. R. Slater, P. L. Meisenheimer, D. H. Klaubert, F. Fan, L. P. Encell and K. V. Wood, ACS Chem. Biol., 2012, 7, 1848–1857 CrossRef PubMed.
- L. Oliveira, P. Tavares and J. C. Alonso, Virus Res., 2013, 173, 247–259 CrossRef PubMed.
- D. Zhang, C. P. Coronel-Aguilera, P. L. Romero, L. Perry, U. Minocha, C. Rosenfield, A. G. Gehring, G. C. Paoli, A. K. Bhunia and B. Applegate, Sci. Rep., 2016, 6, 33235 CrossRef PubMed.
- W. B. Asher and K. L. Bren, Protein Sci., 2010, 19, 1830–1839 CrossRef PubMed.
- P. J. Schatz, Bio/Technology, 1993, 11, 1138–1143 Search PubMed.
- A. Einhauer and A. Jungbauer, J. Biochem. Biophys. Methods, 2001, 49, 455–465 CrossRef PubMed.
- R. J. Giannone and A. B. Dykstra, Protein Affinity Tags: Methods and Protocols, Springer, New York, 2016 Search PubMed.
- J. Arnau, C. Lauritzen, G. E. Petersen and J. Pedersen, Protein Expression Purif., 2006, 48, 1–13 CrossRef PubMed.
- C. Oliveira, V. Carvalho, L. Domingues and F. M. Gama, Biotechnol. Adv., 2015, 33, 358–369 CrossRef PubMed.
- B. W. McLean, M. R. Bray, A. B. Boraston, N. R. Gilkes, C. A. Haynes and D. G. Kilburn, Protein Eng., 2000, 13, 801–809 CrossRef PubMed.
- R. Fregel, A. Gonzalez and V. M. Cabrera, Electrophoresis, 2010, 31, 1350–1352 CrossRef PubMed.
- G. Sezonov, D. Joseleau-Petit and R. D'Ari, J. Bacteriol., 2007, 189, 8746–8749 CrossRef PubMed.
- R. A. Ikeda, J. Biol. Chem., 1992, 267, 11322–11328 Search PubMed.
- T. Tian and H. M. Salis, Nucleic Acids Res., 2015, 43, 7137–7151 CrossRef PubMed.
- H. Sletta, A. Tondervik, S. Hakvag, T. E. Aune, A. Nedal, R. Aune, G. Evensen, S. Valla, T. E. Ellingsen and T. Brautaset, Appl. Environ. Microbiol., 2007, 73, 906–912 CrossRef PubMed.
- H. Sletta, A. Nedal, T. E. Aune, H. Hellebust, S. Hakvag, R. Aune, T. E. Ellingsen, S. Valla and T. Brautaset, Appl. Environ. Microbiol., 2004, 70, 7033–7039 CrossRef PubMed.
- A. Economou, Protein Secretion: Methods and Protocols, Humana Press, 2011 Search PubMed.
- S. D. Alcaine, K. Law, S. Ho, A. J. Kinchla, D. A. Sela and S. R. Nugen, Biosens. Bioelectron., 2016, 82, 14–19 CrossRef PubMed.
- U. Missler, M. Wiesmann, C. Friedrich and M. Kaps, Stroke, 1997, 28, 1956–1960 CrossRef PubMed.
- M. A. Kessler, Anal. Chim. Acta, 1998, 364, 125–129 CrossRef.
- L. L. Kjems, M. E. Roder, B. Dinesen, S. G. Hartling, P. N. Jorgensen and C. Binder, Clin. Chem., 1993, 39, 2146–2150 Search PubMed.
- Y. Wang, C. Fill and S. R. Nugen, Biosensors, 2012, 2, 32–42 CrossRef PubMed.
- M. Villion and S. Moineau, Nature, 2013, 494, 433–434 CrossRef PubMed.
- J. E. Samson, A. H. Magadan, M. Sabri and S. Moineau, Nat. Rev. Microbiol., 2013, 11, 675–687 CrossRef PubMed.
- M. R. J. Clokie and A. M. Kropinski, Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions, Humana Press, 2009 Search PubMed.
- L. Cevenini, M. M. Calabretta, A. Lopreside, G. Tarantino, A. Tassoni, M. Ferri, A. Roda and E. Michelini, Anal. Bioanal. Chem., 2016, 408, 8859–8868 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |