
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multimodal imaging of undecalcified tissue sections by MALDI MS and μXRF†
Anastasiya
Svirkova
 a,
Anna
Turyanskaya
b,
Lukas
Perneczky
b,
Christina
Streli
b and
Martina
Marchetti-Deschmann
*a
a,
Anna
Turyanskaya
b,
Lukas
Perneczky
b,
Christina
Streli
b and
Martina
Marchetti-Deschmann
*a
aInstitute of Chemical Technology and Analytics (CTA), TU Wien, Vienna, Austria. E-mail: martina.marchetti-deschmann@tuwien.ac.at; Fax: +43 158801-915162; Tel: +43-158851-15162
bAtominstitut, TU Wien, Vienna, Austria
First published on 16th April 2018
Abstract
Matrix-assisted laser desorption/ionisation mass spectrometric imaging (MALDI MSI) is a technique that provides localized information on intact molecules in a sample. Micro X-ray fluorescence (μXRF) imaging allows the examination of the spatial distribution of elements in a sample without any morphological changes. These methods have already been applied separately to different tissues, organs, plants and bacterial films, but, to the best of our knowledge, they have yet to be coupled in a multimodal analysis. In this proof-of-principle study, we established and tested sample preparation strategies, allowing the multimodal analysis of lipids (sphingomyelin and phosphatidylcholines) and elements relevant to bone structures as calcium, phosphorous and sulphur in the very same sample section of a chicken phalanx without tissue decalcification. The results of the investigation of such parameters as adhesive tapes supporting tissue sections, and the sequence of the imaging experiments are presented. We show specific lipid distributions in skin, cartilage, muscle, nail, and the intact morphology of bone by calcium and phosphorus imaging. A combination of molecular and elemental imaging was achieved, thus, providing now for the first time the possibility of gathering MALDI MSI and μXRF information from the very same sample without any washing steps omitting therefore the analytical artifacts that inevitably occur in approaches using consecutive tissue sections. The proposed combination can benefit in research studies regarding bone diseases, osteoporosis, osteoarthritis, cartilage failure, bone/tendon distinguishing, where elemental and lipid interaction play an essential role.
Introduction
Analytical scientists seek to maximize the amount of information that may be obtained from a single sample. The aggregate spatial visualization of information gathered about the composition of a species by multiple analytical methods would thus be very attractive to an analyst. A large number of methods and instruments with diverse spatial resolution, areas of application and physical principles have been designed to produce an image. Nowadays, different imaging techniques including optical microscopy, radiography, immunostaining, and magnetic resonance exist in order to obtain a deeper understanding of biological samples.1To gain insight into the large variety of processes occurring in biological systems, it is often insufficient to obtain compositional information without further understanding the spatial distribution of molecules and/or elements in the sample. Molecular imaging techniques may provide this information. Although commonly used methods are quantitative, whole-body autoradiography and micro-autoradiography, these techniques are limited by the necessity for radiolabelling.2 Another powerful analytical tool, mass spectrometry imaging (MSI) has been developed to combine such important characteristics as high spatial resolution, down to the subcellular level, and molecular specificity. In spite of the fact that mass spectrometry imaging is well known for biological substrates, the use of this method increased dramatically with the development and application of matrix-assisted laser desorption/ionization mass spectrometry (MALDI) during the last 20 years.3–5 The principle of MALDI MSI could be briefly described as follows: a laser is used to desorb and ionize the analytes of the sample covered by a chemical compound, also called a MALDI matrix, which absorbs energy at the laser wavelength, aiding the process of evaporation and ionisation of the analysed molecules. As the laser is rastered across the specimen, mass spectra are acquired for each x, y position of the sample holder and mass spectra may be combined to produce an artificial image of analyte distributions. A detailed description of the MALDI mechanism is found in the provided ref. 6–8.
Each imaging technique has its own advantages and limitations. MALDI MSI is a label-free method for the analysis of a broad variety of biomolecules, like lipids, proteins, amino acids or metabolites and drug compounds. It is a sensitive and widely used approach to evaluate different types of complex surfaces. Analyte identification usually relies on the accurate mass determination or MS/MS experiments. But MALDI MS is limited by the ionisation potentials of the molecules under study and is influenced by the sample preparation procedure.9 Thus, many aspects must be carefully considered when imaging by the MALDI MS method, especially if multimodal imaging is anticipated.
There are a variety of biological questions which can be effectively addressed using elemental imaging techniques including, but not limited to, the characterization of the metallome (i.e. a set of metal ions in a cell or tissue) under normal and pathological conditions, as well as non-metallic elements within biological objects, pharmacokinetics and the uptake of metallodrugs or nanoparticles and biocompatibility investigations. For the visualisation of elemental distribution, techniques such as laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS), particle-induced X-ray emission (PIXE) or secondary ion mass spectrometry microscopy (SIMS) have been successfully applied.10,11 Micro-beam X-ray fluorescence spectrometry (μXRF) is one of the most popular elemental imaging methods for the analysis of biological samples.12 The technique is based on the excitation of the sample by X-rays, which results in the emission of characteristic radiation by each particular element, provided that the excitation energy is above the absorption edge of the element of interest. The fluorescence radiation emitted is then registered using an energy dispersive detector – thus, the information about multiple elements present in the sample may be obtained simultaneously. The resultant characteristic spectrum can be deconvoluted by factoring in spectral overlaps and artefacts, as well as the background to extract information about a specific element. Similarly, to perform the above-described procedure for MALDI, the sample can be raster-scanned, yielding elemental maps. The spatial resolution of such a scan/map depends on the size of the X-ray beam. In the case of μXRF, the beam is focussed to a few tens of micrometres in size, thus yielding a resolution in the range of micrometers. The ultimate advantage of the μXRF compared with other elemental imaging techniques is its non-destructive nature.13
Biological samples represent complex molecular and elemental arrangements. A correlation of data obtained by different modalities (often also called “sensors”) could combine molecular spatial distribution with morphological features or elemental mapping of the same object. This combination has high potential to allow a deeper understanding of biologically relevant interactions. Questions that might be solved are drug distribution in the bone and associated soft tissues (e.g. cartilage/bone) after medical treatment, finding features correlating with the tissue and bone for differentiation but also osteon elasticity where soft tissue and calcium distributions in this tissue have to be well characterized to ultimately better understand osteoporosis and osteoarthritis. Even cancer research may ultimately benefit from the new proposed combination because the break-down of bone structures during tumour cell invasion can help to better understand tumour progression. These examples are only a few potential fields of application.
Previously published studies of the fusion of MSI with other imaging techniques include combinations with magnetic resonance imaging (MRI),14 computer tomography (CT),15 Raman spectroscopy,16 fluorescence labelling and staining, and a combination of different mass spectrometry methods.17–20 Due to its non-destructive nature and analyte specificity, μXRF is a favourable choice for coupling with other imaging/non-imaging techniques. Routinely, μXRF has been combined with X-ray absorption near edge structure (XANES) for chemical speciation analysis,21 CT,22,23 quantitative backscattered electron imaging (qBEI)24,25 and other elemental imaging techniques, including LA-ICP-MS26 and SIMS.27 Although the combination of MALDI with μXRF has been proposed,28,29 to the best of our knowledge, the experimental coupling of the two methods remains unknown.
To arrange an adequate combination of different modalities, a lot of questions need to be answered prior to empirical research. One critical concern is the selection of a proper spatial resolution. Choosing modalities with the same step size simplifies a composite imaging experiment. From this point of view, merging MALDI MSI with μXRF imaging seems a good possibility. Another important aspect is that the sample preparation procedure should yield a specimen appropriate for all imaging methods concerned. Thus, a fitting section supporting material, sample thickness, sequence of the experiments and analytical conditions must all be carefully considered. Special attention must also be paid if the analysed sample contains tissues of different physical nature (density, rigidity, etc.), e.g., skin, muscle, bone.
In this work, two imaging techniques have been combined to gain comprehensive information about molecular and elemental distributions both in hard and soft tissues of the same section. Micro-XRF imaging was utilized to examine the spatial distribution of the elements without destroying the sample, by mapping calcium (Ca), phosphorus (P) and, when applicable, sulphur (S). Also “structural” information (absorbance mapping) from the samples was obtained by means of region of interest (ROI) imaging or plotting silicon signals, originating from the supporting material. MALDI MSI was used to gather data on sphingomyelins and phosphatidylcholines throughout the specimen. Assessment of the sample backing material and its influence on imaging analysis has been carried out and will be further discussed together with the explanation of the lateral resolution choice and certain aspects of multimodal workflow.
Materials and methods
Materials
All chemicals were of analytical grade if not stated otherwise. The sodium salt of carboxymethyl cellulose (NaCMC) low viscosity, gelatin (Ph. Eur.) and tragacanth, used for hydrogel preparation, and MALDI MS matrices dithranol for imaging experiments and α-cyano-4-hydroxycinnamic acid (CHCA) for supporting material assessment, and a calibrant for mass spectrometry, angiotensin II, and all organic solvents were purchased from Merck (Darmstadt, Germany). Ultra-high-quality water (ddH2O) with a resistivity of <18.2 MΩ cm @ 25 °C was obtained from a Simplicity system (Millipore, Billerica, MA, USA). A peptide mixture for calibration was obtained from LaserBio Laboratories (Sophia-Antipolis, Cedox, France). Glass microscope slides were obtained from Medite GmbH (Burgdorf, Germany) and Peel-A-Way disposable embedding molds (R-40) were acquired from Thermo Scientific (USA).Section preparation
Section preparation was conducted according to a protocol developed in our group. Chicken feet were purchased from a local market and the digits were removed and embedded as described below. As our samples contain tissues of different densities and rigidities, an appropriate embedding medium is necessary to obtain a section without the loss of sample integrity. Thus, chicken digits were embedded separately in a hydrogel containing NaCMC 5% and gelatin 20% (w/v). Embedded samples were slowly frozen on a cryobar (−57 °C) in the chamber of a CryoStar NX50 (Thermo Scientific, Germany) and mounted on a specimen chuck with 10% tragacanth (w/v). Cryosectioning of the phalanx was performed with a non-disposable tungsten carbide knife at −16 °C. Adhesive tape for the accurate handling of the tissue similarly to the Ullberg's method30 was used to transfer tissue sections to microscope slides. Two types of tape, double-sided tape (3M, St Paul, USA) and polyimide one-sided tape made with DuPont™ Kapton® (3M, St Paul, USA), were tested for their suitability. Four consecutive tissue sections were obtained with the thickness of 12 μm and dried under gentle vacuum in a desiccator (Kartell, Italy). Immediately after drying, microscopy images of the sections were taken using a Leica DM2500M (Leica, Germany) optical microscope.μXRF imaging
Micro-XRF imaging was performed using the μXRF setup at the Atominstitut.31,32 The in-house-built μ-XRF spectrometer is specifically designed for the detection of a wide range of elements and its suitability for the analysis of biological samples has been demonstrated.33,34 The instrument is equipped with a Rh-anode low power X-ray tube, and a Si(Li) detector (active area 30 mm2, LN2-cooled) with an ultrathin polymer window to allow the measurement of the low energy fluorescence radiation emitted by light elements. The primary beam was focused by a polycapillary optic (full lens) into a micro-beam with the size 50 × 50 μm2 at Cu-Kα (determined using a standard test sample). For the measurements, the sample was fixed onto an Al-frame, and positioned vertically in the spectrometer chamber using a magnetic stage, so that the standard geometry 45° between the incident radiation beam and the sample, and 90° between the incident beam and the detector – is maintained. For these measurements, the tube voltage and current were set to 50 kV and 0.4 mA, respectively. The measurement conditions were the same for all the scans: steps of 150 μm, with a measurement time of 90 s were performed in a vacuum. Deconvolution of the spectra was performed using the QXAS-AXIL software package,35 ROI imaging was done using the software LP-map.36 The elemental maps were created with X-Ray Lab software,37 as text files, and plotted with ImageJ38 (version 1.50b). Origin (version OriginPro 2015, OriginLab Corporation, Northampton, MA, USA) and PyMCA39 were used for plotting fluorescence sum spectra.MALDI MS and imaging
MALDI MS results were obtained using a Synapt G2 HDMS™ system (Waters, Manchester, UK), operated in positive ion V-mode with a laser energy of 270.0 a.u. The laser operated with a repetition rate of 1 kHz. Profiling MALDI experiments were conducted for evaluating supporting materials. For this purpose angiotensin II (2 pmol μl−1 in acetonitrile/0.1% trifluoroacetic acid [1:1]) was mixed with a CHCA matrix (3 mg ml−1 in acetonitrile) and 2 μl of the mixture was deposited by the dried-droplet method. The scan duration was set at 1 s, at least 50 scans were gathered; the analysed mass range was 100–2000 Da.Directly before MALDI MSI measurements were made, a dithranol matrix used for all the described MALDI imaging experiments was deposited by sublimation at 140 °C with a home-built apparatus40 yielding ∼0.2 mg matrix per cm2 per slide. The choice of the matrix was made in accordance with the literature41 and a protocol developed in-house. The step size (the parameter of the sample carrier movement) was set either to 100 × 100 μm or 150 × 150 μm. The choice of the step size is influenced by the size of the laser ablated area and is explained in detail in the section “Interference from supporting materials studied by MALDI MS” of this manuscript. Data were collected in a mass range of 300–1000 Da. The class of observed lipids was determined by MS/MS analysis (characteristic MALDI MS spectrum of lipids and the corresponding MS/MS spectra are presented in ESI Fig. 1 and 2†) and are in accordance with the literature and previous analyses.42,43 Measuring in positive mode preferentially desorbs and ionizes phosphatidylcholines (PC), while significantly more classes could be distinguished when acquiring data in negative ion mode. Lipids were chosen for visualisation according to the intensity of the m/z values and counter correlated distribution.
All relative spectra were saved in *.txt format and processed using the open source software mMass.44 Images were handled and created with HD Imaging software version 1.0 (Waters, Milford, USA) and normalized by total ion current (TIC). Distribution correlation of lipids was done with BioMap software version 3.8.0.4 (Novartis, Basel, Switzerland).
All methods and represented workflows were tested in at least triplicates.
Results and discussion
Interference from supporting materials studied by MALDI MS
The samples used for this study contain tissues of different densities. As the rigid part of the long bone of a phalanx is surrounded by soft muscle tissue with skin covering the whole digit, the heterogeneity of the sample can cause difficulties during sample preparation. Thus, in order to avoid shifting of the fragments and to maintain integrity of the section, supporting tape is needed for cryocutting. This supporting tape must be chemically inert and stable under the analytical conditions and must not interfere with either MALDI MS or μXRF imaging. Many supporting materials have been used previously for whole body or plant samples, ranging from wet paper to pressure-sensitive adhesive tape.45–47 To guarantee analyte localization for molecules and elements, double-sided tape and single sided Kapton® tape, commonly utilized for XRF analyses, were applied in this study.To assess the potential influences of the supporting tapes on the MALDI measurements, mass spectra in different modes were obtained (ESI Fig. 3†). Angiotensin II was spotted together with the CHCA MALDI matrix on a microscopic glass, on the Kapton® tape and the double-sided tape, both attached to the glass. Both tapes showed no mass spectrometric interferences, yet signal intensities decreased due to the insulating character of the tape. Thus, both were considered applicable for further imaging experiments.
Although lateral resolution lower than 100 μm can be achieved by MALDI MSI, we primarily aimed to combine μXRF and MALDI imaging methods, and therefore, it was considered to choose for MALDI MSI experiments a step size similar to the one used by μXRF imaging (150 × 150 μm), but with the possibility of providing enough structural information. MALDI MSI was performed at 100 × 100 μm and 150 × 150 μm, with lateral resolutions being easily achieved on commercial MALDI instruments with no need for technical improvements and avoiding oversampling. Although the best lateral resolution achieved on the same type of mass spectrometer was 10 μm without oversampling,48 we also considered the critical time factor. Experiments had to be carried out within a reasonable time period for both MALDI MS and μXRF imaging. Measurement times were on average 4 hours for MALDI imaging and 5 days for μXRF imaging for 1 × 1 cm2 areas.
To evaluate the lateral resolution of the MALDI MSI experiments, the dithranol matrix was sublimed on the surface of the supporting tapes and the four square areas (150 × 150 mm2) were measured with a step size of 100 × 100 μm and 150 × 150 μm, starting each line on the left side. The results are depicted in Fig. 1. On the Kapton® tape, we observed that the intensity dramatically decreases after the first pixel of each line when rastering at 100 × 100 μm step size. Evaluation of the sample surfaces after MALDI MSI measurements showed that the area actually ablated by the laser was larger than the step size (about 200 μm, Fig. 2), thus resulting in an oversampling effect that decreases the intensity for every pixel but the first of each line. We found that MSI at a lateral resolution of ≥150 μm is suitable for measurements on the Kapton® tape. For the double-sided tape, this oversampling issue was not observed and may occur only at spatial resolutions below 100 μm.
 | ||
| Fig. 1 MALDI images of square areas (150 × 150 mm2) showing homogeneous desorption/ionization behaviour of dithranol over each area with the exception of the sample on polyimide tape at a step size of 100 × 100 μm (C) showing reduced intensity after the first line. Dithranol was applied by sublimation on double-sided tape (A and B) and polyimide tape (C and D). Step size 100 × 100 μm (A and C) and 150 × 150 μm (B and D). Images are TIC normalized. Intensity scale bar shows relative intensities. | ||
 | ||
| Fig. 2 Microscopic image of the laser ablated areas of the embedding medium attached to the double-sided tape (A) and the polyimide tape (B) after covering with dithranol as a matrix and rastering at 500 × 500 μm step size. | ||
Interference from supporting materials studied by μXRF
The sample preparation and, especially, the choice of the supporting/embedding material can play a pivotal role in the experiment. In principle, μXRF does not require any specific sample treatment, but in the case of non-self-supporting thin sections, the cuts should be flattened and mounted onto a substrate. Ideally, the material should be as thin as possible, as XRF suffers from scattering of the exciting radiation by the sample as well as the sample carrier. Scattering is the main reason for any spectral background.The characteristic lines of the element of interest are superimposed on the background, and the detection limits are strongly influenced by the spectral background, as well as the intensity of the spectral lines. The chemical constituents of the supporting material are also important, as these will be excited along with the sample.
Polyimide Kapton® films (8 μm thick) are relatively “transparent” to X-rays and are quite commonly used.21 Kapton® tape can be used as long as the constituents of the supporting material do not interfere with the elements of interest.27,49 An alternative for biological samples is the even thinner silicon nitride membrane windows (with a thickness of some 100 nm), usually used for small objects (cells, microorganisms), and samples which can easily adhere to the surface (soft tissues). Alternatively, the samples can be mounted directly on a glass microscope slide.21
The measurements of backing materials were conducted prior to the actual sample measurements; the respective fluorescence spectra are shown in Fig. 3. A larger spectral background is observed in the case of the double-sided tape and glass (Fig. 3A). Here, the peaks of sulphur (S), calcium (Ca), iron (Fe) and strontium (Sr) could be confirmed to be originating from the supporting materials. Sulphur, iron and strontium were further excluded from the analysis for this type of backing material. As the content of calcium in the sample tissues, namely, bone, by far exceeds its amount in the backing material, it was subsequently used for mapping.
 | ||
| Fig. 3 XRF single spectra: double-sided tape and glass without (black) and with a MALDI matrix (green) (A), 3M tape made with DuPont™ Kapton® without (black) and with the MALDI matrix (red) (B). Acquisition time was 90 s, in a vacuum. | ||
In contrast, 3M tape made with DuPont™ Kapton® is free from impurities and the spectral background here is lower, which can be seen in Fig. 3B. Of particular importance is the knowledge about MALDI matrix interference with μXRF measurements. As can be seen in Fig. 3, no difference is observed for experiments with and without the MALDI matrix. Therefore, we can conclude that the MALDI matrix does not influence μXRF measurements.
Finally, the μXRF measurements of phalanx tissues samples were performed, and Fig. 4 demonstrates the sum of the fluorescence spectra over all measurement points, specifically for each type of backing material used. The spectra presented were obtained for the samples measured by μXRF first (i.e. samples were not covered with the MALDI matrix) and do not differ considerably from those measured after MALDI-MSI (for further details see next section). In the case of a tissue sample mounted on the double-sided tape and glass (Fig. 4A), several overlapping peaks are found between 1.5 and 3.2 keV (Fig. 4B), which are caused by scattering from the glass-tape support, as well as the sample-itself. This energy range was used as the ROI for all μXRF imaging experiments (i.e. all counts in the ROI were summed up without fitting), as the deconvolution by AXIL was associated with a large error. This uncertainty was caused principally by the overlap of the Rh-L elastic scattering peak with other peaks in this energy region (Si-Kα, P-Kα, S-Kα, and Cl-Kα and the elastic scatter peak of the exciting Rh-L radiation). Calcium (Ca) and phosphorus (P) in the bone tissue could be measured. As for the sample on Kapton® polyimide tape (Fig. 4C), a distinct sulphur (S) signal from soft tissues could be obtained in addition to those from Ca and P. We could also use the silicon peak, which originates from the glue on the Kapton® tape, to gain structural (absorption) information about the sample.
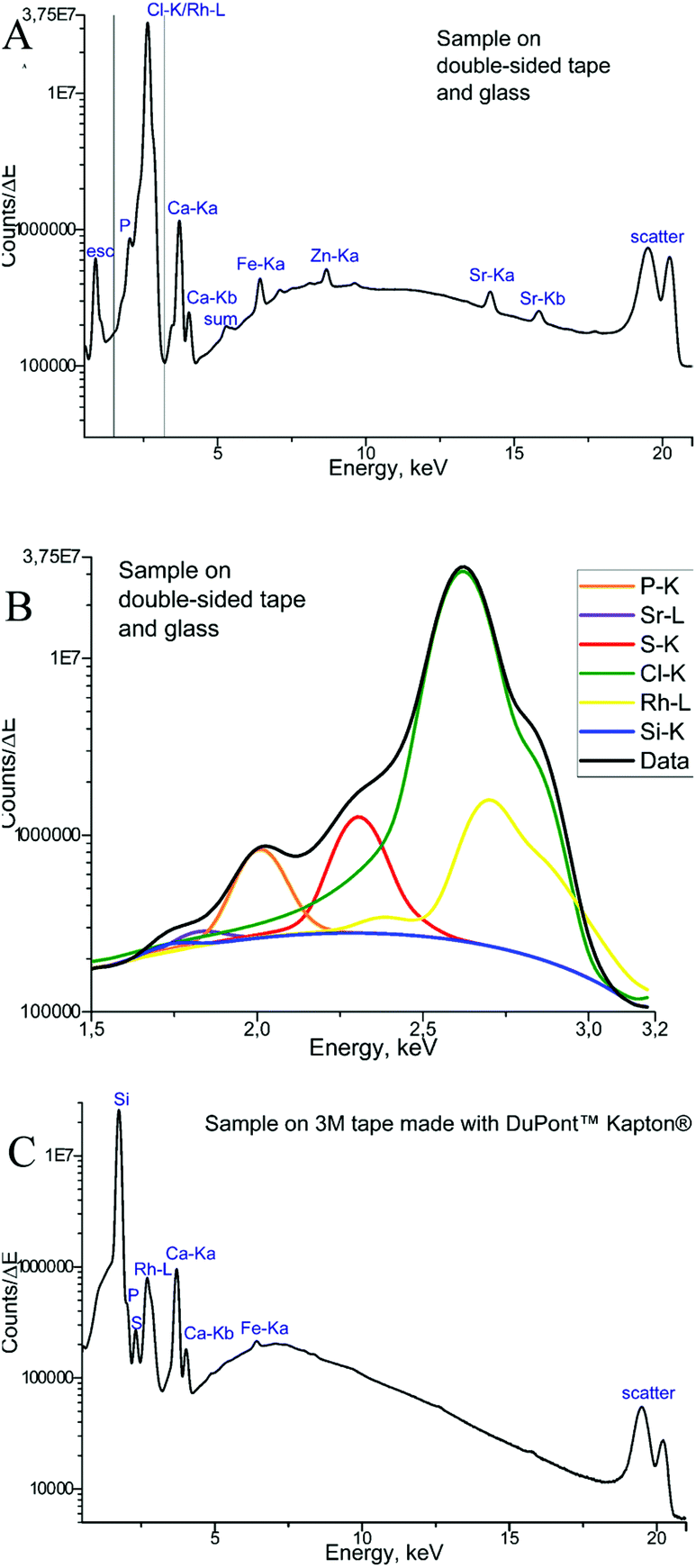 | ||
| Fig. 4 XRF sum spectra: sample prepared on the double-sided tape and glass (A), with the marked energy region of interest (ROI); zoom into the marked ROI showing details for biologically relevant elements (B); sample prepared on 3M tape made with DuPont™ Kapton® (C). | ||
Consecutive sections measured by both MALDI and μXRF on the same samples
While carrying out research with biological material, some issues need to be solved. One of them is the restricted amount of sample material, sometimes just one tissue section is available for analysis, but multiple investigations are needed (e.g. small biopsies). Additionally, it has to be considered that the analyses of the consecutive tissue sections are definitely prone to interpretation errors as two consecutive sections cannot be considered as identical. Considering this, measurements from the very same sample are highly demanded allowing much easier data merging by correct image correlation. In the presented study, this means the co-localization of molecular and elemental distributions. Yet, sample preparation has been predicted to be a major challenge in the development of multimodal imaging methods.29 To provide an in-depth comparison of the experimental conditions for multimodal imaging, four consecutive sample sections were investigated by MALDI MSI and μXRF imaging and special emphasis was laid on the appropriate supporting materials, and the sequence of the imaging experiments. Each section was measured by both techniques, either first by μXRF and subsequently by MALDI MSI (in this case, the MALDI matrix was applied after the μXRF analysis, so μXRF was performed on “intact” samples) or by MALDI MSI first followed by μXRF (in this case, the MALDI matrix was applied from the very beginning but the MALDI process partially ablates material). The sections of the Kapton® tape were fixed either on a microscope slide with Shimadzu conductive tape for MALDI MSI or in the window of the aluminium sample carrier for μXRF imaging. Phalanx sections obtained on the double-sided tape were fixed to a microscope slide by the tape for both types of measurements.Fig. 5 shows that based on the XRF spectral information, images showing the distribution of Ca, P, and ROI data (according to Fig. 4) could be obtained for samples fixed to a glass slide with the double-sided tape. For samples mounted on the Kapton® tape, images showing the distribution of Ca, P, S and Si (latter showing clearly the outline of the sample) could be acquired. Please note that the differences in sulphur maps in Fig. 5(B and D) are due to the extremely high intensity hotspot on 4D, otherwise the sulphur distribution and intensities are comparable (for a rescaled map with the hotspot removed, please refer to ESI Fig. 4†).
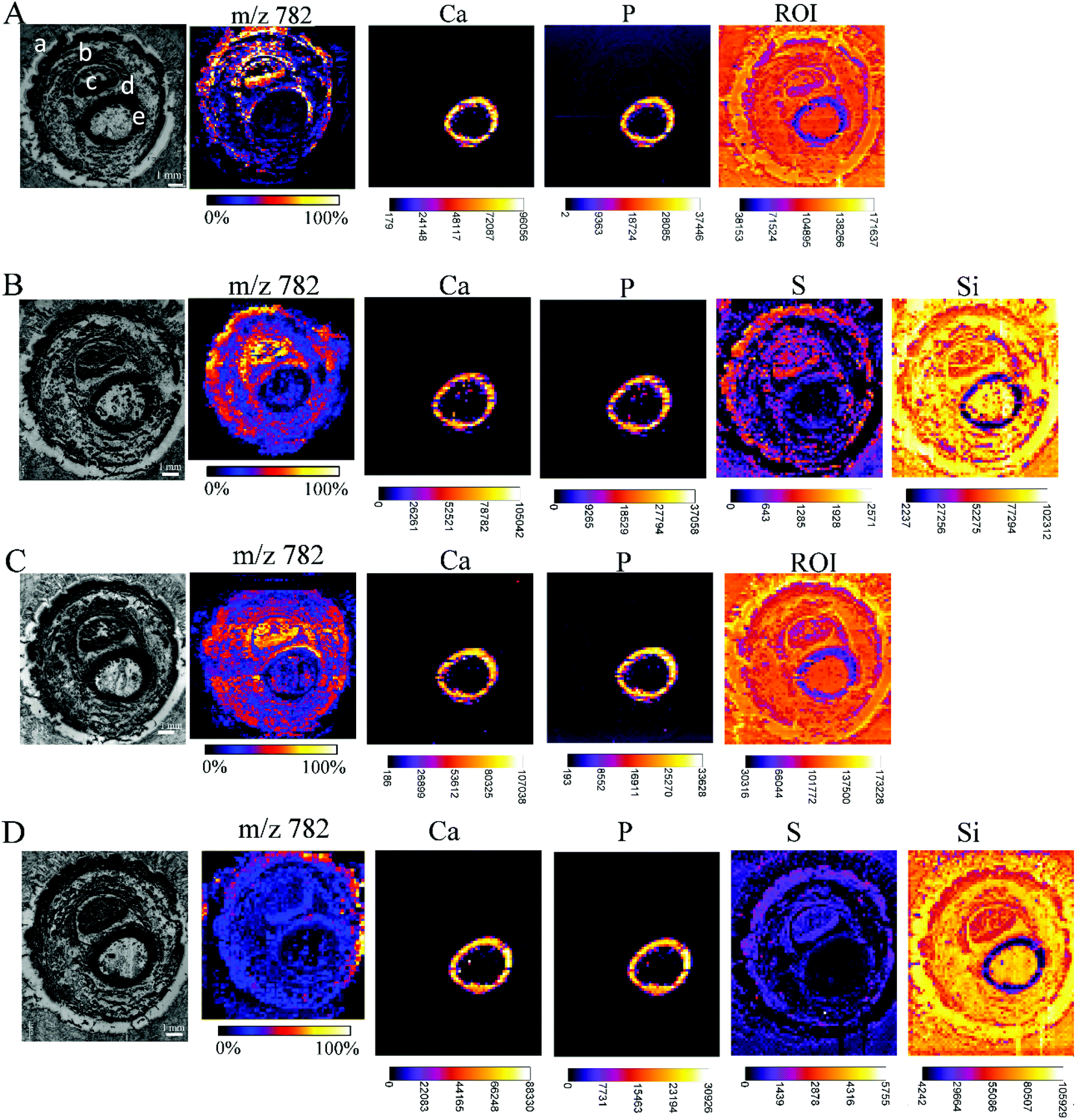 | ||
| Fig. 5 Each array starts with a microscopic image of the tissue sections mounted with different supporting tape, which were measured by MALDI MSI and μXRF, before covering the tissue with dithranol as the MALDI matrix, if μXRF measurements were performed first, or with a dithranol layer, if MALDI MSI were performed first: double-sided tape, measured first by μXRF, then by MALDI (A); Kapton® tape, measured first by μXRF, then by MALDI (B); double-sided tape, measured first by MALDI, then by μXRF (C); Kapton® tape, measured first by MALDI, then by μXRF (D). Lipid distributions detected by MALDI MSI are exemplified by the distribution of m/z 782, elemental distributions measured by μXRF from the same sample preparation are exemplified by calcium (Ca), phosphorus (P) and sulphur (S) distributions, extracted from energy ROI as shown in Fig. 3B, and silicon (Si). All MALDI images were normalized to total ion current (TIC) for each ion across the section and an intensity scale bar is placed under the images. Units for XRF elemental maps are counts. (a: skin, b: muscle, c: nail, d: cartilage, e: undecalcified bone). | ||
The image quality, intensities measured or the number of analytes measurable by MALDI MSI were unaffected by prior μXRF measurements. Conversely, the images obtained by μXRF following MALDI MSI measurements are also unaffected by the application of other analytical techniques. Therefore, we can conclude that the MALDI matrix and the desorption/ionization process do not affect μXRF measurements if the matrix is applied by sublimation, and neither MALDI nor μXRF can cause significant mechanical damage to the sample. Imaging experiments can be carried out in any convenient order without additional handling steps. The only limitation that should be considered is the measuring time. K. Sköld et al.50 demonstrated that proteins and neuropeptides undergo post-mortem degradation. As μXRF imaging is very time-consuming in comparison with MALDI, and one run can take 7 days (or even longer), it is recommended to make this measurement after the MALDI MS imaging to avoid the degradation of the biological substances. Synchrotron radiation-induced μXRF (SR-μXRF) can shorten the μXRF measurement time dramatically, as well as improve the limits of detection for trace elements.
Samples prepared on glass (microscope slide) supported with double-sided tape yielded good results in the MALDI MSI imaging. Contrarily for μXRF, this thick SiO2 glass support caused significant scattering resulting in an elevated background, as well as the contribution of trace elements in support of the resultant spectrum. However, we could demonstrate, that in certain cases, i.e. in the imaging of bone tissue, the μXRF measurements made directly on the glass carrier give satisfactory results. Prospectively, a confocal μXRF setup could be used for the measurement allowing restriction of the element information to a well-defined voxel. Unwanted contributions from the support are thus avoided, as well as the obstruction of the elements of interest due to scattering by backing materials.
The Kapton® polyimide film is a tape commonly used for μXRF measurements due to its high elemental purity, but has not, to date, been used for MALDI-MSI. This tape allows for the imaging of soft tissue (sulphur) by μXRF in addition to bone (calcium and phosphorus). On the other hand, we observed different laser beam behaviour after ablating a specimen covered with a matrix and attached to the Kapton® tape during MALDI MS analyses. This effect leads to a limitation of the spatial resolution for experiments under given conditions of not better than 150 μm step size, which could be critical for small area investigations.
The choice of tape to be used should be taken after considering the purpose of the investigation. If a researcher is interested in combining knowledge about widely distributed Ca and P with a lipid pattern and lateral resolution is of great importance, it is recommended to utilize double-sided tape. If special precision of biological molecules plays a minor role, but it is important to obtain such information as S distribution, together with organic imaging over a large sample, the Kapton® tape must be the sample backing material of choice.
Combination of the imaging information
Images obtained by different modalities on the same section were combined into a single illustration (Fig. 6) for double-sided and Kapton® tape backing materials. | ||
| Fig. 6 Optical microscopy of sample sections (left) and the superimposition of the μXRF and MALDI MS imaging experiments (right) conducted on double-sided tape (A) and Kapton® polyimide tape (B). Calcium is shown in blue and lipids in green (m/z 725) and red (m/z 760). | ||
The structure of the sample resolved by MALDI MSI experiments could be recognised in μXRF images giving the possibility of visual association. Thus, we are able to merge the data of elemental distribution in predominantly undecalcified tissues with the lipid content of the soft tissues for the very same section. During this study, we performed visual fusion without any automated image registration, but software supporting the combination of μXRF and MALDI imaging modalities is of great importance for us and remains a point of interest for future investigation.
Conclusions
To the best of our knowledge, this is the first time when MALDI-MSI and μXRF imaging techniques have been combined giving lipid and elemental information from one tissue section to gain comprehensive structural information in both undecalcified bone and the surrounding soft tissues. Conversely to most previous multimodal (multisensor) investigations, our imaging experiments are carried out on the very same samples without any additional steps, thus, avoiding analytical artefacts. We showed that we are able to cut difficult samples properly and visualize lipid distributions together with biologically relevant elements like Ca, P or S. We performed imaging experiments with similar lateral resolution facilitating the correlation of the images and gaining morphological information that was not possible from either analytical methods independently. We are now ready to apply this method to research questions like tendon wear-off, osteoarthritis, osteoporosis, osteosarcoma or cartilage failure.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Parts of this project were funded by the PhD program “MEIBio (Molecular and Elemental Imaging in Biosciences)” provided by TU Wien. The authors acknowledge the COST Action BM1104 (Mass Spectrometry Imaging: New Tools for Healthcare Research). The authors wish to thank T. Bretschneider for XRF measurements of supporting materials, J. M. Welch for helping with the manuscript preparation and G. Allmaier for supporting the project through access to instrumentation.References
- J. L. Norris and R. M. Caprioli, Chem. Rev., 2013, 113, 2309–2342 CrossRef CAS PubMed.
- E. G. Solon, A. Schweitzer, M. Stoeckli and B. Prideaux, AAPS J., 2010, 12, 11–26 CrossRef CAS PubMed.
- L. A. McDonnell and R. Heeren, Mass Spectrom. Rev., 2007, 26, 606–643 CrossRef CAS PubMed.
- R. M. Caprioli, T. B. Farmer and J. Gile, Anal. Chem., 1997, 69, 4751–4760 CrossRef CAS PubMed.
- M. Stoeckli, P. Chaurand, D. E. Hallahan and R. M. Caprioli, Nat. Med., 2001, 7, 493–496 CrossRef CAS PubMed.
- T. W. Jaskolla and M. Karas, J. Am. Soc. Mass Spectrom., 2011, 22, 976–988 CrossRef CAS PubMed.
- R. Zenobi and R. Knochenmuss, Mass Spectrom. Rev., 1998, 17, 337–366 CrossRef CAS.
- M. Karas and R. Krüger, Chem. Rev., 2003, 103, 427–440 CrossRef CAS PubMed.
- A. Thomas and P. Chaurand, Bioanalysis, 2014, 6, 967–982 CrossRef CAS PubMed.
- C. Petibois, Anal. Bioanal. Chem., 2010, 397, 2051–2065 CrossRef CAS PubMed.
- K. Jurowski, B. Buszewski and W. Piekoszewski, Crit. Rev. Anal. Chem., 2015, 45, 334–347 CrossRef CAS PubMed.
- A. Taylor, N. Barlow, M. P. Day, S. Hill, M. Patriarca and M. White, J. Anal. At. Spectrom., 2017, 32, 432–476 RSC.
- K. H. Janssens, F. Adams and A. Rindby, Microscopic X-ray fluorescence analysis, John Wiley & Sons Inc, 2000 Search PubMed.
- J. Oetjen, M. Aichler, D. Trede, J. Strehlow, J. Berger, S. Heldmann, M. Becker, M. Gottschalk, J. H. Kobarg and S. Wirtz, J. Proteomics, 2013, 90, 52–60 CrossRef CAS PubMed.
- E. Schioppa Jr., S. Ellis, A. Bruinen, J. Visser, R. Heeren, J. Uher and E. Koffeman, J. Instrum., 2014, 9, C04029 CrossRef.
- T. Bocklitz, K. Bräutigam, A. Urbanek, F. Hoffmann, F. von Eggeling, G. Ernst, M. Schmitt, U. Schubert, O. Guntinas-Lichius and J. Popp, Anal. Bioanal. Chem., 2015, 407, 7865–7873 CrossRef CAS PubMed.
- C. Seaman, B. Flinders, G. Eijkel, R. M. Heeren, N. Bricklebank and M. R. Clench, Anal. Chem., 2014, 86, 10071–10077 CrossRef CAS PubMed.
- R. Van de Plas, J. Yang, J. Spraggins and R. M. Caprioli, Nat. Methods, 2015, 12, 366–372 CrossRef CAS PubMed.
- Y. N. Ho, L. J. Shu and Y. L. Yang, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2017, 9, e1387 CrossRef PubMed.
- R. M. Heeren, A. L. Bruinen, N. E. Mascini, G. L. Fisher, T. Porta and S. R. Ellis, Microsc. Microanal., 2015, 21(S3), 2235–2236 CrossRef.
- O. Hachmöller, A. G. Buzanich, M. Aichler, M. Radtke, D. Dietrich, K. Schwamborn, L. Lutz, M. Werner, M. Sperling and A. Walch, Metallomics, 2016, 8, 648–653 RSC.
- B. De Samber, G. Silversmit, K. De Schamphelaere, R. Evens, T. Schoonjans, B. Vekemans, C. Janssen, B. Masschaele, L. Van Hoorebeke and I. Szalóki, J. Anal. At. Spectrom., 2010, 25, 544–553 RSC.
- S. Cagno, D. A. Brede, G. Nuyts, F. Vanmeert, A. Pacureanu, R. Tucoulou, P. Cloetens, G. Falkenberg, K. Janssens, B. Salbu and O. C. Lind, Anal. Chem., 2017, 89, 11435–11442 CrossRef CAS PubMed.
- N. Zoeger, C. Streli, P. Wobrauschek, C. Jokubonis, G. Pepponi, P. Roschger, J. Hofstaetter, A. Berzlanovich, D. Wegrzynek and E. Chinea-Cano, X-Ray Spectrom., 2008, 37, 3–11 CrossRef CAS.
- B. Pemmer, A. Roschger, A. Wastl, J. Hofstaetter, P. Wobrauschek, R. Simon, H. Thaler, P. Roschger, K. Klaushofer and C. Streli, Bone, 2013, 57, 184–193 CrossRef CAS PubMed.
- D. S. Gholap, A. Izmer, B. De Samber, J. T. van Elteren, V. S. Šelih, R. Evens, K. De Schamphelaere, C. Janssen, L. Balcaen and I. Lindemann, Anal. Chim. Acta, 2010, 664, 19–26 CrossRef CAS PubMed.
- K. L. Moore, Y. Chen, A. M. Meene, L. Hughes, W. Liu, T. Geraki, F. Mosselmans, S. P. McGrath, C. Grovenor and F. J. Zhao, New Phytol., 2014, 201, 104–115 CrossRef CAS PubMed.
- B. A. Boughton, D. Thinagaran, D. Sarabia, A. Bacic and U. Roessner, Phytochem. Rev., 2016, 15, 445–488 CrossRef CAS PubMed.
- M. J. Pushie, I. J. Pickering, M. Korbas, M. J. Hackett and G. N. George, Chem. Rev., 2014, 114, 8499 CrossRef CAS PubMed.
- S. Ullberg, Sci. Tools, 1977, 2–29 Search PubMed.
- P. Wobrauschek, B. Frank, N. Zöger, C. Streli, N. Cernohlawek, C. Jokubonis and H. Hoefler, Adv. X-Ray Anal., 2005, 48, 229–235 CAS.
- S. Smolek, C. Streli, N. Zoeger and P. Wobrauschek, Rev. Sci. Instrum., 2010, 81, 053707 CrossRef PubMed.
- L. Borgese, F. Bilo, A. Zacco, E. Bontempi, M. Pasquali, S. Federici, J. Prost, M. Rauwolf, A. Turyanskaya and C. Streli, ECS Trans., 2016, 75, 167–175 CrossRef CAS.
- A. Turyanskaya, M. Rauwolf, T. A. Grünewald, M. Meischel, S. Stanzl-Tschegg, J. F. Löffler, P. Wobrauschek, A. M. Weinberg, H. C. Lichtenegger and C. Streli, Materials, 2016, 9, 811 CrossRef PubMed.
- I. A. E. AGENCY, Quantitative X Ray Analysis System, International Atomic Energy Agency, Vienna, 2009 Search PubMed.
- L. Perneczky, Master thesis (in preparation), TU Wien, 2018.
- S. Smolek, Master thesis, TU Wien, 2014.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- V. Solé, E. Papillon, M. Cotte, P. Walter and J. Susini, Spectrochim. Acta, Part B, 2007, 62, 63–68 CrossRef.
- M. Holzlechner, K. Strasser, E. Zareva, L. Steinhäuser, H. Birnleitner, A. Beer, M. Bergmann, R. Oehler and M. Marchetti-Deschmann, J. Proteome Res., 2017, 16, 65–76 CrossRef CAS PubMed.
- C. H. Le, J. Han and C. H. Borchers, Anal. Chem., 2012, 84(19), 8391–8398 CrossRef CAS PubMed.
- C. Cheng, M. L. Gross and E. Pittenauer, Anal. Chem., 1998, 70, 4417–4426 CrossRef CAS PubMed.
- S. M. Fröhlich, V.-M. Archodoulaki, G. n. Allmaier and M. Marchetti-Deschmann, Anal. Chem., 2014, 86, 9723–9732 CrossRef PubMed.
- M. Strohalm, D. Kavan, P. Novak, M. Volny and V. Havlicek, Anal. Chem., 2010, 82, 4648–4651 CrossRef CAS PubMed.
- T. Kawamoto, Arch. Histol. Cytol., 2003, 66, 123–143 CrossRef PubMed.
- J. Nissanov, L. Bertrand and O. Tretiak, Microsc. Res. Tech., 2001, 53, 239–240 CrossRef CAS PubMed.
- R. J. Goodwin, A. Nilsson, D. Borg, P. R. Langridge-Smith, D. J. Harrison, C. L. Mackay, S. L. Iverson and P. E. Andrén, J. Proteomics, 2012, 75, 4912–4920 CrossRef CAS PubMed.
- H. Kettling, S. Vens-Cappell, J. Soltwisch, A. Pirkl, J. r. Haier, J. Müthing and K. Dreisewerd, Anal. Chem., 2014, 86, 7798–7805 CrossRef CAS PubMed.
- E. Lombi, E. Smith, T. H. Hansen, D. Paterson, M. D. De Jonge, D. L. Howard, D. P. Persson, S. Husted, C. Ryan and J. K. Schjoerring, J. Exp. Bot., 2010, 62, 273–282 CrossRef PubMed.
- K. Sköld, M. Svensson, M. Norrman, B. Sjogren, P. Svenningsson and P. E. Andren, Proteomics, 2007, 7, 4445–4456 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an00313k |
| This journal is © The Royal Society of Chemistry 2018 |