
Histological coherent Raman imaging: a prognostic review
Marcus T.
Cicerone†
 * and
Charles H.
Camp
Jr.
* and
Charles H.
Camp
Jr.
100 Bureau Drive, Mail Stop 8543, Gaithersburg, MD 20899, USA. E-mail: cicerone@nist.gov; Fax: +1 301 975 4977; Tel: +1 301 975 8104
First published on 10th October 2017
Abstract
Histopathology plays a central role in diagnosis of many diseases including solid cancers. Efforts are underway to transform this subjective art to an objective and quantitative science. Coherent Raman imaging (CRI), a label-free imaging modality with sub-cellular spatial resolution and molecule-specific contrast possesses characteristics which could support the qualitative-to-quantitative transition of histopathology. In this work we briefly survey major themes related to modernization of histopathology, review applications of CRI to histopathology and, finally, discuss potential roles for CRI in the transformation of histopathology that is already underway.
1 Introduction
Histopathology has its roots in sixteenth century biology,1 and currently plays a key role in diagnosis of many cancers and other diseases. Nowadays, disease assignments from histology are generally based on the spatial arrangement of cell and tissue features that are highlighted using stains and sample preparation methods that were developed near the beginning of the 20th century.2 The current standard is subjective, consensus-based tissue analysis. This practice has held for many decades, however, it is known to suffer from several important sources of variability,3–7 including differences in staining protocols and tissue analysis approaches among individuals and institutions. Further, while there are general agreements regarding sets of features that indicate a particular diagnosis,8 knowledge of and proficiency in identifying these vary significantly with level and emphasis of training among pathologists. Together, these factors lead to some degree of diagnostic imprecision. Given the central role of histology in influencing diagnosis and subsequent treatment regimens, it is not surprising that efforts are underway to increase objectivity, repeatability, and precision of histopathological analysis.Over the past several decades, a number of approaches have been pursued to increase the amount of quantitative information extracted from histology samples, and thus support improved analytical performance of histopathology approaches. Immunohistochemistry9 (IHC), introduced in the early 1940s, enhances image information content through antibody-based contrast agents for diagnostically valuable targets, such as cell surface markers and structural proteins. This approach can yield information on the spatial distribution of these diagnostic targets and has proven very useful in some cases,10 but also has important limitations.11–13 Other efforts have focused on quantifying image information and presenting it to physicians in the form of computer aided diagnostic (CAD) tools. A requirement for widespread use of CAD methods is that clinical tissue images be digitized. Although histopathology analysis is generally performed directly through the microscope, with no digital image ever acquired, digital recording of whole slide images (WSI) is becoming increasingly common, particularly for training purposes.14
Several label-free contrast modalities are being explored with regards to their utility in histopathology. Among these, vibrational spectroscopies such as Raman scattering and infrared absorption (IR) offer significant potential to increase the information content of histology images. Like IHC, these modalities provide chemically specific and diagnostically relevant information, but without the need of labeling. IR spectroscopy was applied as early as 1952 for qualitative characterization of normal and neoplastic tissue.15 Several IR and Raman histopathological studies in the 1990s through the 2000s utilized multivariate analysis methods to provide probabilities of disease within regions of interest.16,17 Some studies also provided relative abundances of major tissue components having diagnostic value.18 While studies such as these showed impressive sensitivity and specificity, they often did not provide image information that could be easily integrated into a diagnostic decision. IR imaging was not of sufficient spatial resolution, and Raman spectral acquisition is sufficiently slow as to preclude imaging at the desired spatial resolution. These have been important limitations for a field where image interpretation is central.
The recent advent of coherent Raman microscopies19 have facilitated rapid label-free imaging, and bright infrared (IR) sources now available allow for IR imaging with higher spatial resolution.20,21 When spatially resolved, vibrational spectroscopy can provide familiar and diagnostically important information such as cell arrangement, phenotype, cellular and subcellular morphologies, structural proteins, and intracellular lipids. Because they require little or no sample preparation, these modalities can provide this data in situ or with minimized latency between excision and availability of actionable information. Further, the intrinsically digital image format and potential for rich chemical contrast makes vibrational histopathology a strong candidate for strengthening CAD.
Here we review current applications of coherent Raman methods to histopathology, and consider its potential for meeting the needs of this field in the future. To better evaluate this potential, we first provide brief overviews of histopathology workflow, diagnostic criteria & uncertainty, and technology-related efforts underway to improve histopathology, including application of infrared and spontaneous Raman spectroscopy.
2 Histopathology
2.1 Histopathology workflow – a brief outline
Histopathology, the microscopic analysis of tissues for the purpose of identifying disease, may be performed on tissue samples pre-operatively, intra-operatively, or post-operatively. Pre-operative analysis is performed to help establish a detailed diagnosis and treatment regimen. Intra-operative and post-operative histopathology is performed to ensure complete resection of diseased tissue by confirming absence of disease in margins of extracted or in situ tissue. In cases where the benefit outweighs the risk, it is common practice to remove grossly healthy tissue surrounding the tumor to decrease the likelihood of positive margins. When excising a margin of nominally healthy tissue is contraindicated, such as for brain, the surgeon takes care not to remove more tissue than is justified. In such cases, intra-operative histopathology studies are sometimes performed on flash-frozen resected tissue to ensure that the entire tumor has been removed. The tissues are analyzed immediately, but an intra-operative histology analysis typically takes 20 to 30 minutes, and so prolongs the surgery.Pre- and post-operative histopathology studies are typically done in a batch mode, and usually take a day or two to wend their way through the process, requiring approximately 13 hours of actual processing time. In this process, excised tissue is first grossly examined (by eye) for diagnostic information, and prepared for microscopy. Tissue samples are typically fixed and embedded within paraffin wax, and some of the tissue is sliced in 10 to 50 νm thick sections. These formalin-fixed paraffin-embedded (FFPE) sections are then stained (typically with hematoxylin and eosin [H&E]) and mounted for inspection. In a large hospital, a pathologist may inspect 500 histology slides in a day. Diseased regions are frequently small, so the clinician may spend a significant amount of time combing through normal tissue. In order to search more efficiently, pathologists will often identify landmark species such as blood cells and vessels, lymphocytes and ducts, which are frequently found in vicinity of cancerous lesions. Preliminary microscopic analysis and diagnosis are often carried out by a single pathologist. In difficult cases, a panel of experts may review the slides, and additional staining may be requested to identify diagnostically important species that H&E staining does not highlight. Ultimately, analysis and diagnosis are generally reviewed and signed-out by an attending physician.
2.2 Diagnostic criteria
The primary diagnostic criteria for most cancer types are related to intra- and intercellular organization.8 There are a number of additional criteria that are specific for various cancer types. These include cellular features such as number density of mitotic events, infiltration of immune cells, degree of cell differentiation within and surrounding the tumor, the degree of necrosis, abnormal nuclear shape or intracellular lipid levels, the appearance of over-developed mitochondria, and chromatin clumping.8,22–24 Tissue structural features are also important for some cancer types, such as prostatic adenocarcinoma.25 These features may include acinar organization, mineralized tissue, mucin or polysaccharide deposits, the presence or absence of structural proteins, such as laminin, elastin, or collagen. In the course of qualitative tissue classification, the pathologist must evaluate whether observed features fall in or out of the normal range. These judgments are subjective and thus susceptible to variability which is in a sense uncontrolled, because it cannot be quantified on a case-by-case basis.2.3 Diagnostic uncertainty
While it is not currently feasible to assign diagnostic probabilities on a case-by-case basis, most studies address the issue of diagnostic uncertainty through analyzing outcomes in groups of patients. These studies can be classified into several types, and, while the results of these studies vary significantly, a few general trends emerge.One type of study involves mandatory or voluntary review of a wide range of tissues by “peer” physicians (i.e., not necessarily subspecialty experts). In this class, Kronz et al.26 and Raab et al.27 report on inter-institution or institution-wide studies where large numbers of cases (6171 and 6162 respectively) covering many tumor types were reviewed. Kronz et al.26 found 1.5% discordant diagnosis that resulted in a major modification of therapy or prognosis was observed. The majority of these cases involved a change between benign and malignant or a major change in tumor classification, and changes involving only a modification of tumor grade or stage were not included. That study found significant variability in diagnostic discordance with type of tissue. Serosal surfaces and the female reproductive tract tissues had 9.5% and 5.1% diagnostic discordance respectively, the highest found in the study. Raab et al.27 estimated an error rate 6.7% from self-reported discrepancies upon second review at 72 institutions. In that study, only 1.1% of discordant diagnosis resulted in a major modification in therapy or prognosis.
Addressing these types of studies, Ho et al.28 argue that statistics based on second review likely reflect an underrepresentation of errors since the reviewer often has knowledge of the original diagnosis and the sign-out pathologist. They argue that such a priori knowledge has led to biased review in similar situations.29
In another class of study, previously diagnosed cases for a single tissue type are analyzed by a panel of sub-specialists. Higher discordant diagnosis rates are typically found in these types of studies. Lurkin et al.30 report on a review of all sarcoma cases (366) in the Rhone-Alpes region of France over a 12 month period. They found that 19% of cases resulted in change of type or invalidation of diagnosis, and 27% of cases resulted in change of grade or subtype of diagnosed cancer. Bruner et al.31 found similar results for 500 neuropathology cases submitted to a specialist review committee. They found that 9% of reviews resulted in immediate significance for therapy or intervention, and 19% resulted in a change in type or grade of glioma. Similarly, specialist review of 602 prostate adenocarcinoma cases32 led to a change in the Gleason score by at least 1 point in 44% of cases, and patients’ risk category was increased in 11% of cases. Likewise, of 340 patients presenting for second opinions regarding breast cancer, 80% resulted in some diagnostic change, with 8% of reviews leading to altered surgical therapy.33 In another study, 131 bladder carcinoma cases underwent secondary review, with 18% exhibiting significant discrepancies.34 In a gynecological oncology35 study of 295 referred patients, 5%, resulted in diagnostic changes that had major therapeutic or prognostic implications.
Arriving at definitive numbers for diagnostic uncertainty is outside the scope of this review. However, it does seem clear from the studies cited above that the uncertainty is significant for some cancer types. This widely recognized fact was reflected in a recent survey of pathologists,36 which showed that physicians on average expect 10% diagnostic uncertainty from histopathology for cancer, and that lack of sub-specialty expertise is seen as the most important factor contributing to misdiagnoses. Based on these facts, one may be inclined to favor the sub-specialist review literature, and the slightly higher discordant diagnosis numbers found therein. The overall picture is that difficulty in diagnosis varies significantly with tissue and tumor type, and many human factors figure in precision and accuracy of diagnostics.36,37
2.4 Impact of diagnostic uncertainty
Table 1 shows published figures for cancer rates, inter-observer diagnostic variability among pathologists, and cost associated with initial treatment. It appears that approximately 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people each year in the U.S. are either incorrectly informed that they have cancer, and subsequently undergo invasive treatments, or are incorrectly told that they do not have cancer, and miss the opportunity for potentially life-saving treatments. One might assume that half of misdiagnosed cases are false positive and half are false negative. Under this naive assumption, one estimates the cost of unnecessary procedures is $2.5 B annually in the U.S. Of course, the cost of missed treatment is harder to estimate, but misdiagnoses are uniformly the largest reason for medical lawsuits, which cost $55.6 B annually, or 2.4% of all health spending.38
000 people each year in the U.S. are either incorrectly informed that they have cancer, and subsequently undergo invasive treatments, or are incorrectly told that they do not have cancer, and miss the opportunity for potentially life-saving treatments. One might assume that half of misdiagnosed cases are false positive and half are false negative. Under this naive assumption, one estimates the cost of unnecessary procedures is $2.5 B annually in the U.S. Of course, the cost of missed treatment is harder to estimate, but misdiagnoses are uniformly the largest reason for medical lawsuits, which cost $55.6 B annually, or 2.4% of all health spending.38
| Type | New cases39 | Diagnostic uncertainty | Initial cost40 | Unnecessary treatment costa | Raman uncertainty |
|---|---|---|---|---|---|
| a These values are estimates assuming diagnostic uncertainty is evenly distributed between precision and accuracy. | |||||
| Breast | 250k | 20%7 | $23k | $575 M | 5%18 |
| Lung | 225k | 10%3 | $61k | $680 M | 10%41 |
| Prostate | 180k | 30%5 | $20k | $540 M | 5%42 |
| Colon & Rectal | 135k | 10%6 | $52k | $350 M | 5%42 |
| Bladder | 77k | 40%4 | $21k | $320 M | 7%43 |
| Melanoma | 76k | 25%44 | $5.5k | $52 M | 5%44 |
The magnitude and gravity of this problem is not lost on physicians and technologists, and the field of histopathology has been slowly evolving to provide better informed and precise diagnoses. Several innovations have been in process of adoption over the past decades.
2.5 Immunohistochemistry
Immunohistochemistry (IHC) typically uses fluorescently tagged antibodies to provide image contrast specific to proteins of diagnostic value. Most IHC tags are targeted to functional proteins, such as cell surface markers;45 however, soluble46 and structural8,47–49 proteins are also targeted. The IHC approach facilitates mechanism-linked disease detection, and has enjoyed some important successes. For example, a protein (KIT), found to be mechanistically related to gastrointestinal stromal tumors (GISTs), is easily visualized through IHC labeling, and has found widespread use in diagnosing GISTs.10 While it now seems that KIT labeling is also prone to false positives in some cases,12 the use of IHC for KIT detection has nevertheless simplified detection of GISTs, which was notoriously controversial with regard to classification, line(s) of differentiation, and prognostication.10 In spite of successes, IHC in general has some drawbacks including variability of labeling,11 and incompatibility with some stains. Compatibility issues include background fluorescence from a stain that interferes with IHC detection, and stains chemically modifying the affinity antibodies used in IHC. H&E, for example, has both of these issues. Additionally, while IHC images can provide specific chemo-spatial information, they are also interpreted subjectively which can lead to variability in diagnostic outcomes.13Improved diagnostic precision has been achieved by multiplexing classical and IHC stains, where the various contrast agents are imaged together.50 Alternatively, cyclic IHC labeling24,51–53 of samples facilitates registered imaging of many proteins. Such approaches can lead to improved specificity for cancer characterization, but can add considerable complexity and, in the case of serial IHC, require up to a day for each labeling cycle.53
2.6 Whole slide imaging
Pathology slides are typically viewed and analyzed directly through a light microscope for diagnostic purposes. Image digitization is not a common practice for histopathology, but that is now beginning to change. Whole-slide imaging54 (WSI), which entails digitization and storage of entire tissue slides, was introduced in the mid 1990s. Image acquisition speeds were initially too slow to be of practical use, but current WSI instruments have pixel acquisition rates on the order of 3 MHz, allowing them to generate bright-field images of a 1 cm2 tissue sample at 500 nm resolution in about 2 minutes; roughly the time a pathologist might spend on a tissue sample of similar size. WSI is now used widely in teaching environments,14 and has been fully adopted in some hospital systems.55 WSI is also useful for quality assurance, consultation, and telemedicine applications.28,55Fig. 1 shows an example of a prostate adenocarcinoma display from a pedagogical WSI application, allowing simultaneous view of an image thumbnail, and a detailed sub-image section.14 This sort of data presentation has obvious pedagogical and practical benefits, but large, high-resolution images such as this may occupy ≈1 Gb of disk space. Thus, the work of a single pathologist may occupy 0.5 Tb day−1, and storage space requirements are often cited as a potential barrier to widespread acceptance of WSI.56 Another barrier is slightly reduced image quality compared to direct microscopic observation.55 There are further concerns about variation in sample preparation and staining practices, and that image capture and coloration can vary over an important range among WSI instruments. Consideration of these issues is currently underway at the U.S. Food and Drug Administration.57
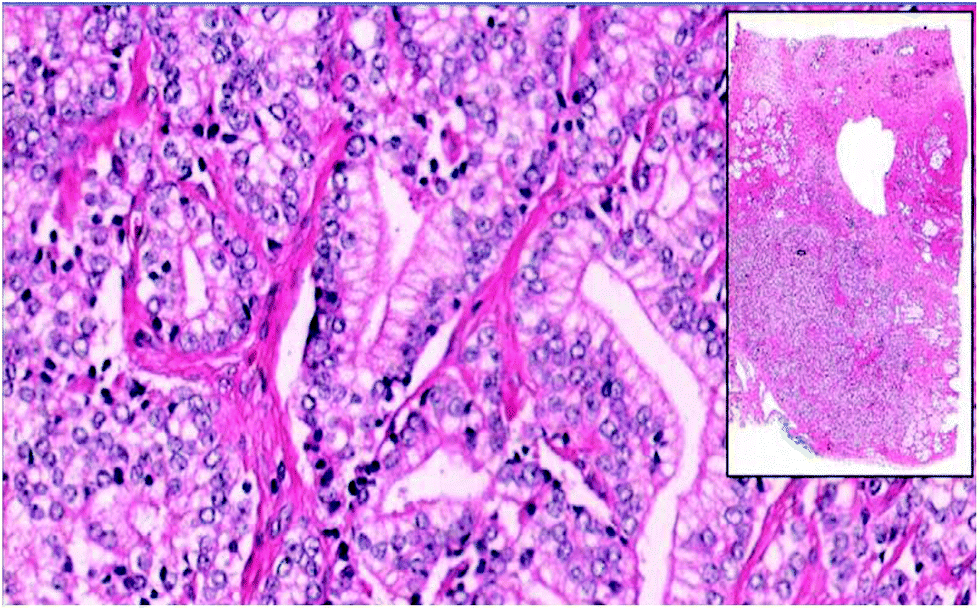 | ||
| Fig. 1 WSI image of prostate adenocarcinoma tissue from set of Genitourinary Block training in the preclinical curriculum at the University of Arizona College of Medicine, in Tucson, AZ.14 | ||
2.7 Computer aided diagnosis (CAD)
CAD approaches support improved diagnostic precision by providing image-derived, diagnostically relevant information to pathologist. Efforts towards CAD began in the late 1990s with detection of malignant masses in radiological images of breast tissue,58 and CAD is now routinely used in radiology for identification of lesions. Further, it seems that the prevalence and sophistication of CAD is likely to grow. In 2015, IBM acquired Merge Healthcare, for its 30 billion radiological and histological images, with the intent of using the image data to train Watson, their physician assistant software.59While radiological CAD is now well-integrated into the diagnostic process, histological CAD is not used routinely. This dichotomy may be due in part to the fact that the former simply provides evidence for the presence of tumors, while the latter must address the more complex questions of tumor type and grade, and deals with images of higher complexity. Numerous algorithms have been proposed for diagnosis of specific cancers,60–62 but there has not been significant clinical adoption. Histological CAD, however, seems to be emerging from the research-only phase, as a number of companies are becoming involved in developing these tools.63
Histological CAD systems may exploit a large range of image features to derive clinically significant information,60 including object size and shape, intensity, color, texture, and inter-object organization. Approaches for extracting these inputs from images have been reviewed recently.64,65 The image processing associate with CAD will generally include some or all of the following steps: (1) preprocessing, such as color and intensity normalization and de-noising. (2) Feature identification using thresholding or segmentation approaches. (3) Quantification of relevant feature characteristics, such as shape, size, spatial arrangement, texture, or color. (4) Reduction of data dimensionality using methods such as principle component analysis. (5) Classification based on the reduced-dimension dataset. Steps 2 through 5 (and sometimes step 1) are collectively referred to as “machine learning”. We briefly discuss these steps below.
Use of blank images66 or reference materials can reduce inter-image variability, but this is often insufficient. In order to minimize these variations, preprocessing steps such as color and intensity normalization are often employed. Color convolution67 quantifies staining densities of a small number of contrast agents based on the fact that light absorptivity of individual stains is linear in log space. This approach continues to be developed, and can be used to map image color and intensities from many samples onto common range,68 assuming each image contains similar ranges of features. However, color and intensity normalization approaches can lead to significant artifacts when based on assumptions that the proportion of pixels or range of stain intensities from each image in a set should be identical.69 In cases where a given stain vector may not be appropriate for all samples, individual vectors can also be normalized to a single image.69
Another approach uses active contours, which are deformable splines that encompass image segments. They are constructed to minimize some energy function that may have contributions dictating the stiffness of the spline, the placement of the spline on the image (e.g., lowest energy at a minimum image gradient location), and a term that encapsulates a priori knowledge of image segment shape or location.65 These splines can be parameterized (SNAKES)76 or unparameterized (level set methods).77 Each approach has strengths and weaknesses, with SNAKES being more robust to noise, and level set methods having greater flexibility to accommodate variable topology. In cases where salient aspects of the image segment shape is known, Bayesian analysis of template comparison78 has been successful, and found robust to image variation. Each of these segmenting approaches may have trouble with overlapping features of interest, and active contours are prone to inclusion of unwanted background objects.64 Also, Bayesian methods are, in general, computationally expensive.
Of course, pixel-level and active contour approaches are not mutually exclusive. It is possible to leverage the strengths of each in a single algorithmic approach.79
Linear discriminant analysis (LDA) is a commonly used supervised feature selection approach that assumes independent variables are normally distributed. It is similar to PCA, except that the feature types are specified by the user. A comparison of PCA and LDA can be found in Martínez et al.,83 who prefer PCA when the training dataset is small, other factors being equal.
Similar to methods mentioned above, manifold learning (ML) approaches project an M dimensional dataset to a space with fewer (N) dimensions, where preferably, N ≪ M. Both supervised and unsupervised ML approaches exist, and these differ from those above in that there is no assumption of linearity in the relationship between the features of interest and the original data.64
Deep learning is another route to classification. These approaches are an evolution of neural networks in which the data vectors are iteratively transformed using nonlinear functions in such a way as to emphasize image aspects having predictive power. Using these approaches, segmentation, feature selection, and classification are all done at once, and all without user intervention. However, the entire process is opaque to the user in that there is no way to know what features the model is using for classification. Thus, there is thus no way to know when the model is likely to fail. In fact, only to the extent that the dataset on which the deep learning model was trained is comprehensive, can the characterization be reliable. By way of apparent counterexample, however, Cirean et al. were able to use a relatively small sampling of mitotic events to create a highly effective deep learning model for detecting mitoses in H&E images.87 Nonetheless, building a comprehensive dataset for deep-learning image recognition seems to be the intent of IBM in acquiring a company with access to 30 billion radiological and histology images.59
Even the most basic level of CAD assistance – that of providing image feature identification, such as of nuclei or regions of potential metastases86 may be quite valuable. In many cases, a pathologist may search for regions of interest (ROIs) that cover only a small fraction of the sample. Identifying such regions up front, or even identifying landmark species that typically signify such regions could provide a significant time savings.53
Feature quantification might be considered the next level of sophistication where CAD approaches could be of high value. We find evidence for this by considering the work of Fuchs and Buhmann,88 who found 10% disagreement on location and size of nuclei in a renal cell carcinoma sample between two sub-specialists, and inter- and intrareader disagreement of 42% and 21% respectively for counts of normal and atypical nuclei among five pathologists who were not sub-specialists. This level of variability is comparable to the overall diagnostic uncertainty discussed in Section 2.3, and suggests that correct identification of relevant features may play a significant role in diagnostic uncertainty for this and other cancers. Image analysis approaches discussed in the previous sections can provide quantitative characterization of key features, such as cell density61 or mitotic event counts87 and nuclear shape metrics78,84 at precision levels similar to that of sub-specialist analysis in the Fuchs and Buhmann work.88
While CAD work is largely done using H&E,87 improved approaches to image contrast would likely lead to increased reliability of CAD outputs.63 Naturally, an expanded contrast pallet serves to put image-derived metrics on a more solid footing.53 Even the simplest pixel-level thresholding methods can be quite reliable when pixel intensity values are related to specific chemical or functional information. For example, pixel-level analysis of vibrational spectra resulted in accuracies of 94%–100% for classification of ten disparate histologic classes,89 98% for positive prostate nuclei from IHC images with contrast for androgen receptor protein,90 and 95% for micro-metastasis from cytokeratin-stained lymph node sections.91
2.8 Emerging contrast modes
While use of classical stains and IHC approaches is well-established in histopathology practice, interactions of light with unlabeled tissue can also be used as contrast to provide important diagnostic information. Elastic interactions between light and tissue are generally characterized through static light scattering or quantitative phase imaging (QPI). Inelastic interactions are characterized through light absorption, autofluorescence, and vibrational spectroscopy. If one could rely exclusively on these label-free contrast modalities, there could be a significant reduction in sample-to-sample contrast variability in addition to reduced costs and handling time of pathology slides. It is unlikely that label-free contrasts alone could, in every case, provide sufficient information for diagnosis. However, these label-free methods do provide a surprising amount of diagnostic information – perhaps sufficient for many or most cases.Refractive index maps and light scattering patterns from tissues both provide information useful for diagnosing aspects of some cancers. In light scattering experiments, light is refracted, or scattered, as passes through materials with discontinuities inrefractive index. Optical coherence tomography is now well developed as a clinical imaging tool, and can be used to determine spatial location of scattering sites.92 Additionally, polarization, wavelength, and scattering angle may be measured directly in an imaging mode to deduce relevant tissue properties. Tumor growth modes produce tissues that scatter light strongly93 and anisotropically,94 and it appears that light scattering may be sufficiently sensitive for very early tumor detection of some epithelial cancers.94 Elastic interactions of light and matter may also be characterized by directly mapping out the refractive index of a sample through quantitative phase imaging.95 QPI reports on refractive index variation from the nano-scale to the micro-scale, and as with qualitative phase imaging, it makes boundaries between tissue regions and cellular compartments readily visible. QPI can also discriminate some tissue and cell types,93,96 locate micro-calcifications,93 and even indicate degree of malignant transformation97 based on values of refractive index. Light scattering pattern maps and phase imaging maps are essentially interchangeable as the former can be calculated from the latter and vice versa, provided the light scatting is spectrally resolved.98,99
Light absorption100 and intrinsic fluorescence101 from ultraviolet102 or visible and infrared light103 can be used to characterize tissues. When specific excitation and emission wavelengths are used, signals deriving primarily from species such as nicotinamide adenine dinucleotide (NAD) and flavin adenine dinucleotide (FAD),100 elastin and collagen,104 tryptophan,105 and possibly porphryns103 can be isolated. Accumulation or deficit of these species is not specific to tumors, but does seem to be a sensitive indicator for the presence of neoplastic masses. In fact, autofluorescence has been used as method to rapidly locate candidate tissue regions for a slower, but more specific mode of characterization, vibrational spectroscopy.106
3 Spectroscopic histopathology
Vibrational spectroscopy characterizes molecular vibrations intrinsic to species of interest in a specimen. This rich source of chemical information has been applied to histopathology, albeit primarily in a research mode. From spectroscopic and spectral imaging studies it appears that much of the information sought through staining, and perhaps more, is available without labeling from techniques such as Raman or IR spectroscopy.It is common to describe vibrations of molecules with N constituent atoms in terms of oscillatory motions along vectors with up to 3N-6 dimensions called “normal modes” (Q). These modes constitute collective intramolecular motions that preserve the overall symmetry of the molecule. The modes have discrete allowed states with energies approximately equal to (ν + 1/2)ħωm, where ν are integer values, and ωm is the ground vibration frequency. The ground state vibrational frequencies and energies vary depending on the strengths of bonds and masses of atoms involved in the normal mode motion. Transitions with Δν = ±1 are most often measured, and those with energies in the range (500 to 3100) cm−1 are typically of interest in biological systems. Signatures in the higher energy range (2800 to 3100) cm−1 arise from transitions between states of modes involving symmetric or asymmetric stretching of C–H bonds. The range (1800 to 2800) cm−1 is referred to as the quiescent region because transitions of natural biological molecules typically do not appear in this range. The greatest variety of vibrational transitions in biological molecules occur in the fingerprint range (500 to 1800) cm−1.
Transitions between these states are usually detected through absorption, emission, or inelastic scattering of light. The probability for photon absorption or emission is significant when ∂μ/∂Q ≠ 0 at Q = 0, where μ is a mode's permanent dipole moment. By contrast, inelastic scattering is important when ∂α/∂Q ≠ 0 at Q = 0, where α is the molecular polarizability. Infrared and Raman spectroscopies are respectively based on absorption and inelastic scattering.
Fig. 2, panels a and b show IR and Raman spectra for major classes of tissue constituents. Since these have distinct signatures in both spectroscopies, they can be identified by either method. On the other hand, IR absorption linesare slightly broader. This is of consequence in the fingerprint region, where spectral lines are quite congested. Panels c and d of Fig. 2 show fingerprint regions of IR and Raman spectra from cells, illustrating that the width of the individual IR peaks makes it difficult to separate them into more than just a few distinct contributions without peak fitting. On the other hand, Raman peaks are more narrow, so that spectra from similar systems contain a larger number of easily resolvable peaks, providing a much higher level of chemical specificity. The differences can be quantified in terms of information content, or entropy, Si = ln(Ωi), where Ωi is the maximum number of distinct spectral states one can discriminate. Here, Ωi = SNRM where M is the number of spectral peaks and SNR is the signal to noise ratio in each peak. We can estimate this value by noting approximately 15 peaks in the IR spectrum, 5 peaks in the Raman CH region, and 45 peaks in the Raman fingerprint region. Assuming SNR = 15 for IR peaks and for CH stretch Raman peaks, and SNR = 3 for Raman fingerprint, we obtain Ωi = 1017, 106, and 1021 for IR, Raman CH, and Raman fingerprint spectra respectively (1027 for the entire Raman spectrum). Partly for this reason, Raman is found to have better specificity for classifying cancer.110
 | ||
| Fig. 2 a & b: Respectively, IR spectra and Raman spectra of major cell components.107 From top to bottom on both panels, spectra are from albumin, globulins, collagen, glycogen, DNA, RNA, and lipid. c: IR fingerprint spectrum of whole cells,108 with ≈12 resolvable peaks. d: Raman fingerprint spectrum of whole cells,109 with ≈45 resolvable peaks. | ||
Given their capacity for encoding chemical information, it is not surprising that these spectroscopies have been applied to many general aspects of disease diagnoses.111,112 They have also been used for guidance in tissue sampling,113 augmented bases for stratified diagnosis,114 and intraoperative guidance during tissue-conserving resection.115,116 Of particular present interest is the considerable body of literature showing that vibrational spectroscopy provides diagnostically significant information for cancer diagnosis in conjunction with histopathology. There have been many excellent reviews on this topic in recent years,107,111,117–121 so here we will only summarize relevant aspects.
Histology-related studies using vibrational spectroscopy began to appear in the 1960s122 for IR, and in the 1980s for Raman,123 and to date, there have been thousands of publications. However, it appears that only in the past decade or so that IR and Raman technology have been sufficiently developed to produce meaningful studies.107 Recent work provides rather convincing evidence that vibrational spectroscopy can detect diagnostically important changes in tissues, routinely yielding objective sensitivity and specificity figures generally in the 85% to 95% range for detection and classification of cancers, even when spectra on which discrimination is based are usually taken at just a few spots from normal and diseased portions of tissue.
In one example, Pence et al.120 discuss 26 large studies of epithelial cancers (>50 patients), among which, the average sensitivity and specificity obtained using Raman spectroscopy were 89 ± 7% and 88 ± 8% respectively. Such precision generally exceeds that possible by subjective morphological analysis. Similar examples are listed in the final column of Table 1, indicating a reduction in diagnostic uncertainty by more than a factor of two in most cases. Table 1 also provides prevalence, histology-based diagnostic uncertainty and initial treatment costs for the six most common cancers in the US. Assuming naively that diagnostic uncertainty is evenly distributed between precision and accuracy, we estimate that using Raman spectroscopy to supplement histology could save >$2B per year in unneeded treatments, and reduce the burden on patients and families associated with missed opportunities for earlier treatment.
3.1 Vibrationally detected cancer markers
The impressive diagnostic value of vibrational spectroscopy accrues from its inherent chemical specificity. It can reveal information that is otherwise available only by methods such as IHC and polymerase chain reaction (PCR) or gene arrays,107 leading to more precise tissue segmentation,89 and providing new prognostic information.133Table 2 gives a partial list of cancer biomarkers that have been used in Raman studies to classify neoplastic and cancerous tissues, and lists spectral peaks used to identify those markers. Most markers have several peaks, facilitating increased robustness in detection, to the extent that noise in separate peaks is uncorrelated, but even correlated noise can be suppressed if peak ratios can be used. The fact that most of the peaks used in these Raman studies are found in the fingerprint region is due to the high spectral density and low peak widths found there, and underscores the importance of this spectral range for chemical specificity.
| Marker | Relevant cancers | Spectral lines [cm−1] |
|---|---|---|
| ν-stretch, δ-scissoring, ρ-rocking, ω-wag, τ-twist. | ||
| Metabolic markers | ||
| DNA (relative abundance) | Breast18 larynx124 | 668, 678, 728, 750, 785νO–P–O 825νO–P–O , 1336, 1488, 1580 , 1336, 1488, 1580 |
| Lipid (structure and relative abundance) | Breast18 colon42,125 bladder43 skin44,126 | 1309τCH2 1445δCH2 1654νC–C 2850νCH2,sym 2880νCH2,asym 2920νCH3,sym 2960νCH3,asym |
| Cholesterol ester | Breast18 brain127 | 430, 702, 1302, 1442, 1740νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
| Glycogen | Bladder43 prostate42 brain127 | 472, 481, 846, 932 |
| Creatinine | Breast128 | 600νN–CH3 678νCO 685τC–S 692δCO2 840δN–CH2 903νC–C–N |
| Structural molecules, etc. | ||
| Protein structure | Skin44 melanoma44 | 1660νC–O (amide I) 2850νCH2,sym 2880νCH2,asym 2920νCH3,sym 2960νCH3,asym |
| Keratin | Epithelial cancers129 | 623νCS, 643νCS, 850δCCHaromatic, 885ρCH2 933, 1002νC–C (ring breathing), 1031 (phenylalanine), 1200 1350 (amide III) 1450τCH 1650νCO (amide I), 2940νCH3 |
| Collagen | Breast18 prostate42 colon42 lung41,130 skin131 | 855, 938, 1004νC–C1260νCN, δNH1314ωCH3CH21445δCH21660νC–O 2940 |
| Elastin | Lung41 | 1442δCH2 1660νC–O |
| Carotenoids | Bladder43 lung41 brain127 | 1159, 1523 |
| Polysaccharides | Brain132 | 856 |
| Others | ||
| Phospholipids | Lung41 brain127 | 719 1442δCH2 |
| CaC2O4, Ca5(PO4)3 | Breast18 brain132 | 912νC–C 960νPO 1477νC–O |
| Phosphatidylcholine | Adenocarcenoma107 | 719, 1666νC–O |
| Trypotphan | Lung41 prostate42 | 650τC–C 1260νCN, δNH |
| Proline | Prostate42 | 939νC–C |
| Tyrosine | Prostate42 | 1176νC–H 1217νC–C6H5 |
Many of the markers listed in Table 2 are commonly visualized in pathology through IHC or classical stains, but, of course those methods require labeling. Not only is vibrational contrast label-free, it can also be semi-quantitative when peak amplitude ratios are used. Accordingly, peak pairs that provide information on protein-to-DNA or protein-to-lipid ratios, or on degree of unsaturation in fatty acids frequently yield highly reliable metrics of disease type and grade.41,124,132,134–137
Macromolecular structure and material phase information are also available from vibrational spectroscopy, and can be useful in cancer detection and diagnosis. For example, degree of calcification is a known marker for many cancers.8 Using classical stains, it is difficult to distinguish between types of calcification, but such discrimination turns out to be important. Haka et al.18 showed that Raman spectroscopy can be used to easily distinguish CaC2O4 from Ca5(PO4)3, and that their relative abundances correlate with malignancies in breast cancer. Raman spectroscopy also contains structural and hydrogen bonding information for lipid, protein, and nucleic acids, which has diagnostic value for many cancers.18,44,126,128
Spectral changes between healthy and diseased tissue can be striking,41,138 or subtle,107 but generally appear in the context of highly complex spectra from the tissue. Thus, while it is sometimes possible to determine identity of diagnostically relevant species though differences between spectra from normal and diseased tissue,127 this is sometimes not possible even when the primary marker species is known.129 For this reason researchers often derive diagnostic information from tissue spectra using spectral pattern recognition approaches.
These unsupervised feature extraction methods frequently serve as the input to unsupervised and supervised classification methods. Unsupervised methods, such as K-means clustering139,146 (KMC), divisive correlation cluster analysis (DCCA), and agglomerative hierarchical cluster analysis (AHCA), link together related spectra based on spectral proximity metrics.139 Supervised methods, such as linear discriminant analysis111 (LDA), partial least squares147,148 (PLS), support vector machines149,150 (SVM), and random forest classifiers,146,151,152 rely on training data to generate a classification model. These models may be conservative or relaxed according to clinical need. For applications such as tissue-conserving resection,115 one may choose a conservative model to minimize false positives. Alternatively, if the aim is to reduce the burden on pathologists, one could design a model that may include false positives, but automatically classifies the most clear-cut cases, leaving the rest for inspection.153 Training these models may be arduous, requiring many datasets and extensive validation; but post-training classification may be rapid.146,148 There are, additionally, ‘semi-supervised’, methods that aim to incorporate known and unknown information into classification problems, such as hybrid linear analysis154 (HLA) semi-supervised PCA-linear discriminant analysis155 (PCA-LDA).
Deep learning156 (DL) and artificial neural networks157,158 (ANN) constitute an important family of methods that have received significant attention recently. These methods rely on layers of interconnected decision units or ‘neurons’; typically 1–2 layers for ANN approaches, and potentially many layers for DL approaches. Whether employed as supervised or unsupervised learning, the weights and the interconnection strengths of the neurons are iteratively modified to optimize a merit function. One of the primary strengths of ANNs and DL is that nonlinear and extremely complex interconnections can be created within the network that may not be possible with traditional linear or nonlinear methods such as those mentioned above. A significant challenge to ANNs and DL, as with all supervised techniques, is the requirement that the training set be comprehensive in that it is representative of all future data. Another challenge is that these approaches are ‘black box’ in that it is not currently clear how to understand what data features are found to be important, and how they are used in the evolved ANN/DL neural connection architecture. Interpretation of ANNs and DL is an active area of research.159
Thanks to recent commercial availability of quantum cascade lasers, approaches20 for IR imaging at spatial resolution as high as 1 μm are now feasible, making it possible to generate gross tissue maps based on IR absorption. Since the chemical specificity of IR spectroscopy is sufficient to easily discriminate among tissue regions such as epithelium and stroma,160 and between benign and malignant tissue,89 IR maps can be merged with H&E images to indicate tissue regions of interest,133 as shown schematically in Fig. 3a. Morphological features extracted directly from such maps have been used to classify tissue samples as cancer or non-cancer, with high accuracy.161 This automated ability could provide considerable value, as mentioned in Section 2.7.6.
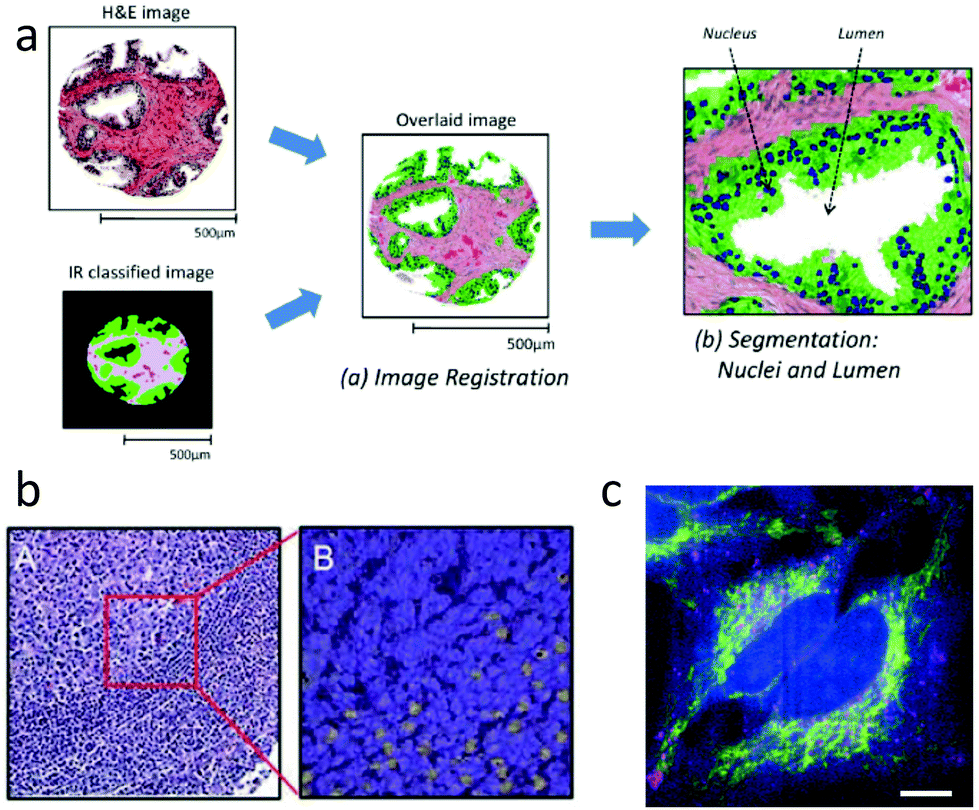 | ||
| Fig. 3 IR and Raman histopathology: (a) Infrared absorption spectra contain sufficient information for classification of tissue, and provide value through indicating regions of interest.161 (b) A: H&E stained section of lymph node germinal and mantle zone, and B: a sub-image with contrast from Raman spectroscopy showing activated lymphocytes in yellow.107 (c) Spatially multiplexed Raman spectroscopy with 30 ms effective spectral acquisition time using 500 mW of 532 nm light. The cytochrome-c distribution is indicated in yellow.164 | ||
From a spatial resolution perspective, Raman imaging has an important advantage over IR in that it uses visible or near infrared wavelengths, so can reach spatial resolution 2× to 3× higher than with IR, making cellular and sub-cellular imaging possible. The combination of high resolution and rich fingerprint spectra allows for significant information in addition to providing classification of tissue regions. For example, Raman imaging can provide label-free identification of cell type,162 distribution139,163 and content of organelles.164 Important functional information is also available, such as DNA to protein ratio of nuclei,165 the cell cycle state,163,166,167 and whether immune cells such as leukocytes are activated.107,168 Examples of cellular function and organelle distribution maps generated with Raman spectroscopy are shown in Fig. 3b and c.
Although IR and spontaneous Raman imaging can provide useful diagnostic information, both have practical drawbacks. For example, conventional glass slides can be problematic for both methods as they emit fluorescent light when excited with visible and near IR light, and absorb IR light at wavelengths of 3.5 μm and longer, which constitute the fingerprint region for IR. Consequently, both approaches often require special substrates – quartz or CaF substrates for Raman studies, and CaF or BaF2 substrates are used in IR. On the other hand, glass slides are transmissive to IR wavelengths in the range λ = (2.8 to 3.5) μm, allowing only the CH stretch spectral region to be used.160 Another consideration is that the intrinsically low Raman scattering levels can be easily masked by fluorescence; thus, in addition to use of specialty substrates, it is often necessary to photobleach samples before acquiring Raman spectra.
The benefits of spectral image acquisition should be considered in context of required imaging time. These considerations differ significantly for IR and Raman spectral imaging. IR absorption cross sections for fingerprint and CH stretch modes are typically on the order of 10−18 cm2, so most of the light will be absorbed for a high density of absorber with (5 to 10) μm path length. Having such a large effect on transmitted light, signal can be acquired very quickly. Yeh et al.21 describe work wherein 128 × 128 pixel wide field IR absorption images were acquired serially on a fixed region of the tissue while the laser wavelength was scanned. In total, 282 frames were acquired, yielding IR absorption images over a 1128 cm−1 range in the fingerprint region, with an effective pixel rate of 1.6 MHz, and spectral acquisition rate of 3 kHz. In cases where fewer spectral points are required, some imaging speed increase can be obtained by using only several discrete frequencies. For example, Tiwari et al.169 found that they could reliably identify diseased cardiovascular tissue regions with on-the-order of 10 discrete spectral points. Allowing for imaging of ∼10 discrete contrast frequencies and a 5 μm pixel size, tissue could be mapped at 0.5 cm2 min−1, similar to WSI instruments currently in use. Further, if the discrete frequencies are exclusively in the CH stretch region, glass slides could be used, and there would be minimal impact on the histology work flow, as IR imaging can be performed on H&E stained slides.160
In contrast to IR absorption, the Raman signal is quite small, and imaging with this contrast mechanism is proportionally slow. The differential scattering cross-section of Raman-scattered light collected of over solid angle (Ω) is given by dσ/dΩ ∝ |∂α/∂Q|2, and is on the order of 10−30 cm2 sr−1 for most modes. Accordingly, spectral acquisition rates typically range from 5 to 0.01 Hz.129,170 Using spontaneous Raman scattering, one cannot excite only selected vibrational frequencies, so there is no benefit to discrete spectral imaging as in IR. On the other hand, some increase in acquisition speed can be achieved through spatial multiplexing. Fig. 3c displays a spontaneous Raman image acquired at a 90 Hz effective spectral acquisition rate164 through line-focused (rather than spot-focused) excitation light. Because practical detector arrays are presently limited to 2 dimensions, and one of those is devoted to spectral variations, spontaneous Raman signal is limited to 1 dimensional multiplexing such as line excitation. Of course, spatially multiplexed signal generation requires light sources with proportionally scaled power.
Given the much slower acquisition speeds for spontaneous Raman scattering, it is difficult to find an obvious application in the histopathology work flow. For example, pixel sizes of 27 μm and a 90 Hz spectral acquisition rate achieved through spatial multiplexing would provide appropriate sample throughput, but with such course resolution, one would give up significant benefit of sub-cellular chemical detail otherwise available from Raman scattering. Spontaneous Raman imaging could be useful in a guided subsampling mode, where only regions of special interest were imaged. On the other hand, even with spatial multiplexing and 90 Hz spectral acquisition, only 0.003% of the total tissue area could be imaged at high resolution in the 2 minutes time frame required to obtain a 1 cm2 whole slide brightfield image.
At this point it is appropriate to consider that the 2 minutes per slide imaging time is the standard for single-contrast image formation, whereas Raman spectral images could contain most or all of the potentially desired contrasts in a single, label-free image. Nonetheless, it seems clear that some trade-off between coverage and resolution would be necessary if spontaneous Raman scattering is used. The severity of such a trade-off could be reduced by use of coherent Raman techniques, which provide equivalent spectroscopic information with imaging throughput that is closer to that of WSI.
4 Coherent Raman imaging
4.1 Introduction and mechanism
Coherent Raman scattering was first predicted in 1962171 and first measured in 1964.172 In 1982, Duncan et al.173 first demonstrated the coherent Raman scattering effect in a microscope, but the cross-beam (phase-matching) optical arrangement they used was challenging. In 1999 Zumbusch et al.19 demonstrated that the CARS effect could be achieved with a simple, collinear beam geometry. This discovery gave rise to the now burgeoning field of coherent Raman imaging (CRI). Currently, the most prominent bioimaging CRI methods are stimulated Raman scattering (SRS) and coherent anti-Stokes Raman scattering (CARS). It is not our purpose to review the CRI field in its entirety; excellent reviews have been given recently.174–178 Instead, we focus on applications of CRI to histopathology, and associated technical aspects that facilitate or hinder such applications.We can gain insight into coherent Raman scattering by considering that an oscillating electric field of light induces a polarization in a substance that can be described as:
| P(t) = Nα(t)Esin(ωt) | (1) |
 | (2) |
In coherent scattering, vibrational modes are driven by external light fields. When the difference in energy between a pump (p) and Stokes (S) field is equal to the energy difference (Ωv) between two adjacent quantum levels of the vibrational mode, i.e., when Ωm = Δω = ωp − ωS, energy can be transferred to that vibrational mode. While the primary fields, E(t) = Eje−iωjt, (j = p,S), ωp > ωS ≫ Ωm, are at much too high a frequency to directly induce significant molecular response, electrons are able to follow the fields adiabatically. Through its nonlinearity, the electron response will contain some amplitude at the difference frequency, Δω. This component of the response will in turn exert an oscillatory force F(t) on the molecule:179
 | (3) |
 | (4) |
 | (5) |
Substituting eqn (5) and a first-order truncation of eqn (2) into eqn (1), we obtain an expression for the polarizability of a given mode in presence of a pump and Stokes field:
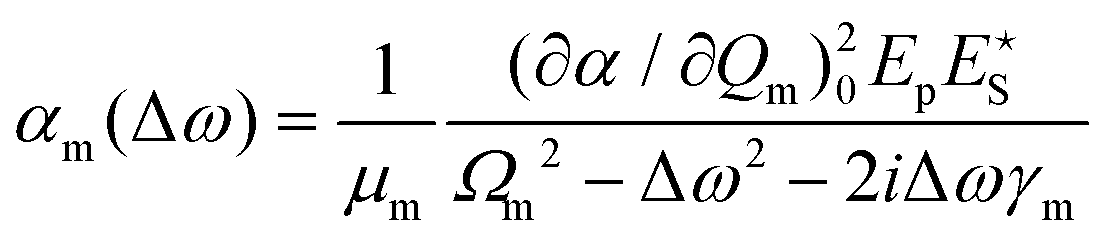 | (6) |
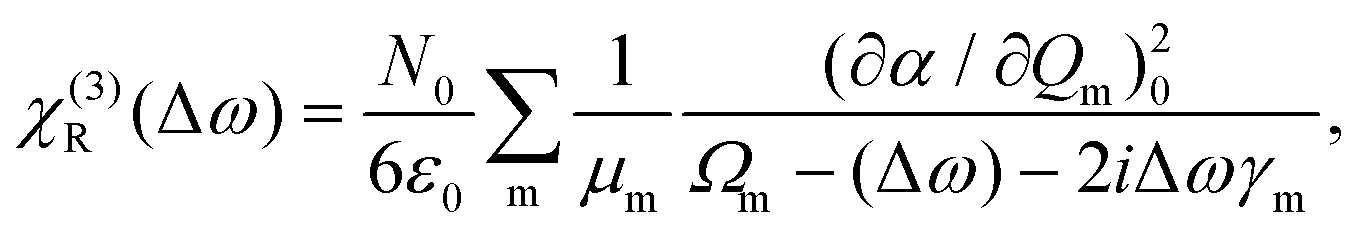 | (7) |
| χ(3)(Δω) = χ(3)NR(Δω) + χ(3)R(Δω) | (8) |
In third order coherent Raman processes, a pump and Stokes field interact with a medium to create a vibrational coherence, given as:
| C(Δω) = χ(3)(Δω)[ES ★ Ep](Δω) | (9) |
4.2 Coherent anti-stokes Raman scattering (CARS)
Coherent anti-Stokes Raman scattering is generated by interaction of a probe field with the vibrational coherence generated through the process described by eqn (9), as represented by the Jablonski diagram in Fig. 4a, and by the following expression: | (10) |
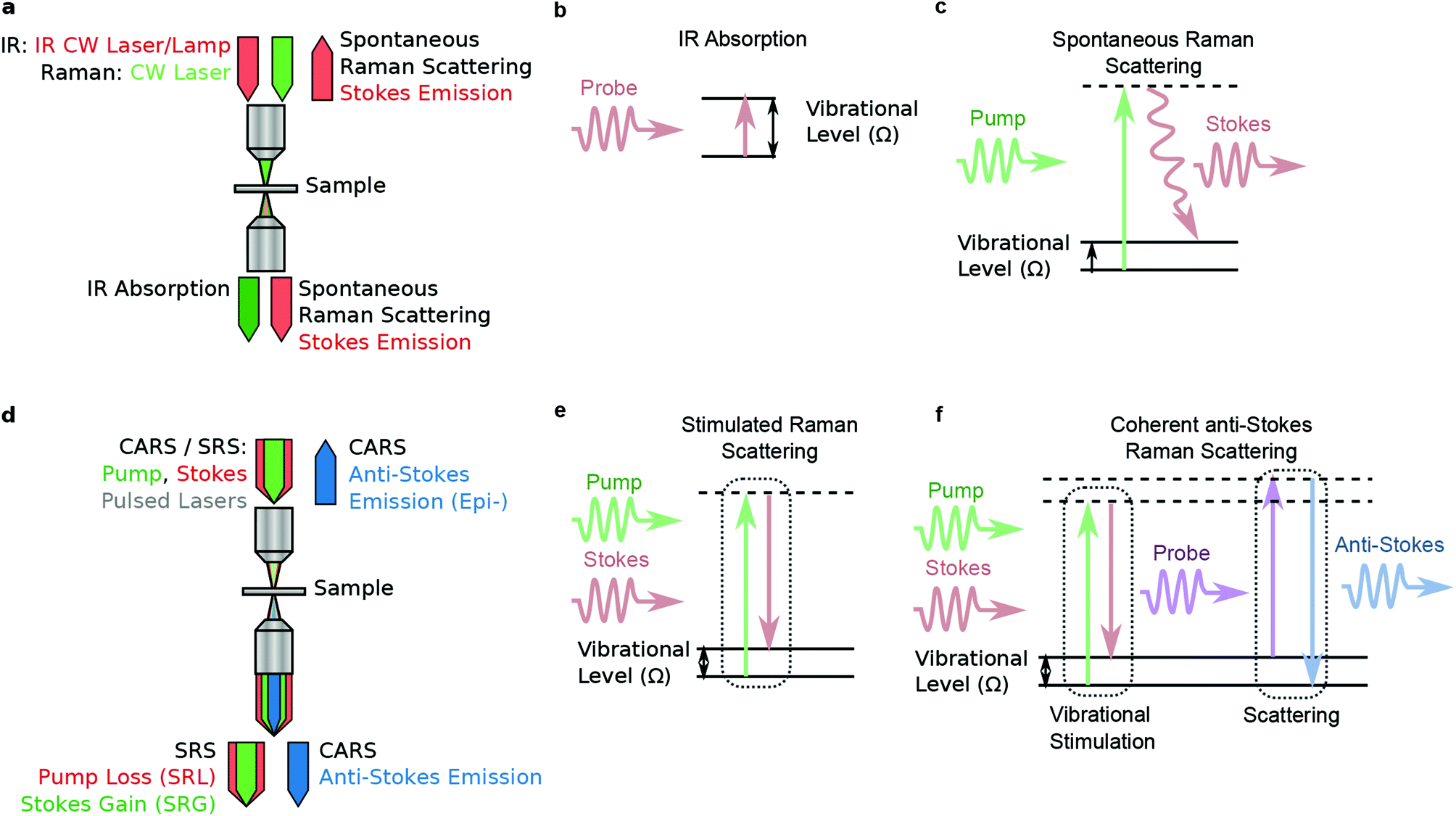 | ||
| Fig. 4 (a) Abstraction of an experimental diagram for IR absorption or spontaneous Raman imaging: a single IR or visible light source is projected through an objective. For IR, absorption is measured from the relative light intensity transmitted through the sample. In spontaneous Raman scattering, signal is emitted isotropically, equally in the forward and backwards direction, but the Raman scattered light must be separated from the excitation light. This is typically done with spectral filters or spectrometer. Frequently, IR and spontaneous Raman imaging on tissue slides will be performed in reflection mode by placing a mirrored surface after the sample. The mirror serves to increase collected signal fraction in the case of spontaneous Raman scattering. (b) and (c): Energy diagrams for IR absorption and spontaneous Raman scattering respectively. (d) Abstraction of an experimental diagram for coherent Raman imaging: typically two pulsed and synchronized laser beams are co-linearly projected into the back aperture of the microscope objective. The CARS and SRS processes occur simultaneously at the focus. All of the SRS signal and most of the CARS signal are projected forward. A small CARS signal is projected in the backwards (epi) direction. SRS imaging is frequently performed in epi-detected mode by collecting signal light that is inelastically back-scattered into the excitation objective. The SRS signal is detected as a modulation of the pump or Stokes beam, while the CARS signal is spectrally distinct from the excitation light, and can be detected after spectral separation. (e) and (f): Energy diagrams for SRS and CARS respectively. | ||
Another important difference is that CARS signal contains a nonresonant background (NRB), arising from χ(3)NR. It is termed “nonresonant” because it arises from the electronic response, and is not sensitive to vibrational resonances. Since the electrons adiabatically follow the driving field of the laser, the NRB is always in phase with the driving field, and retains a fixed phase relationship with the resonant response. The overall CARS signal has a component that is a coherent mixing between these two terms:
 | (11) |
Owing to the three terms in eqn (11), the analyte concentration dependence of the raw CARS signal depends on the ratio of resonant to nonresonant response. χ(3)NR is of similar magnitude for most biological materials but χ(3)R may vary widely owing to the density of molecular oscillators, Ñ (fraction of N0 within the focal volume) and the Raman scattering cross-section of individual vibrational modes. Typically, χ(3)NR ≤ 0.1Ñχ(3)R for CH stretch resonances and ICARS ∝ Ñ2. However, for many fingerprint peaks, χ(3)NR ≪ Ñχ(3)R and the resonant contribution to the ICARS is linear in Ñ, but NRB dominates the signal. This is why narrowband CARS is performed almost exclusively with CH stretch resonances as contrast. In fact, considering only the term |Ñχ(3)R|2, Cui et al.181 demonstrated that the resonant CARS signal amplitude is greater than spontaneous Raman only when oscillators are very concentrated, such as in bulk phase, and samples rich in lipid or structural protein.
Another important effect of the coherent interaction between resonant and non-resonant signal components arises from the Gouy phase shift, wherein a phase shift accrues with field propagation beginning at the focus of a Gaussian beam compared to propagation of a plane wave. This becomes a problem at sharp interfaces of features smaller than the Rayleigh range when resonant signal is generated only by light on one side of the focus while the non-resonant signal is generated at positions on both sides of the focus. When these mix, there is a spatial position-dependent spectral phase error182 causing spectral distortion. However, the Gouy phase is not a problem when χ(3)NR ≪ Ñχ(3)R, or when there are no sharp spatial discontinuities in resonant signal contribution.183
CH and aromatic νCH and a broad resonance between 3000 cm−1 and 3600 cm−1 due to water νOH.
The CH stretch vibrations are sensitive to their environment by direct coupling and through Fermi resonances with C–H bending modes near 1500 cm−1. Together, these influences can introduce significant shifts and broadening of the CH-stretch peaks. Spectroscopic184–187 and spectral focusing188 CARS approaches are best positioned to leverage the information contained in peak shape and position changes. Narrowband approaches19 return vibrational contrast over a narrow spectral band. In the CH stretch region, excitation is tuned to report primarily on a single resonance, but the signal generally includes lesser contributions from multiple resonances due to spectral overlap. Signal from CH2 and CH3 are frequently used to determine relative abundance of protein and lipid because the ratio of CH2 and CH3 moieties differs significantly in these chemical families. The information content in (symmetric, asymmetric) peak pairs is largely redundant, so only one peak from each pair is typically used for this purpose.
In narrowband CARS, a pair of picosecond pulses is used to excite coherence of a single selected vibrational band, and the higher frequency field is used to probe the coherence. Meyer et al.189 found that, for strong resonant signals (χ(3)R ≥ 10Nχ(3)NR), simple subtraction of NRB is sufficient to render a quantitative signal with no significant distortions to the CARS peak positions and amplitudes. Wu et al.190 have shown quantitative correspondence between νCH2,sym signals and lipid content in liver tissue. Further, large resonant signals in the CH-stretch enables rapid image acquisition, with pixel rates as high as 6 MHz,191 but more typically range from 200 kHz (ref. 192) to 1 MHz (ref. 189) for tissue samples. Repeated images can be acquired at several discrete Raman shifts to map scenes with some chemical complexity. For discriminating species that are spectrally distinct, like water and protein or lipid, a single additional scan is generally sufficient for each component.193 A larger number of spectral contrast points may be necessary to discriminate species with only subtle differences.189 Given the high pixel acquisition rates, the necessity of acquiring several scans may not itself be onerous, but latency between scans for wavelength tuning can add significant time to image acquisition for most CARS systems. Rapid spectral scan approaches have been demonstrated for narrowband CARS with only 100 μs required to scan from one frequency to another in the CH stretch region, making inter-scan latency a non-issue for such systems.194
Spectral focusing is another approach that facilitates spectroscopic scanning with narrowband CARS. In spectral focusing CARS,188 a pair of pulses that are spectrally broad and centered at distinct wavelengths are temporally broadened with nominally identical chirp. Although the absolute frequencies of the two light fields impinging on the sample changes significantly with time, variation in the difference of frequencies is minimized so a single vibrational state can be excited. Using this approach, it is feasible to collect single-frequency image pixels at 250 kHz.195 This approach to CARS can be performed with sub-picosecond pulses and spectral tuning over a limited range (typically 500 cm−1) can be accomplished by simply changing the delay of one pulse relative to the other. When spectral focusing or another CARS approach is performed at a series of Raman shifts, the effects of the NRB (distortion and nonlinear concentration response) can be separated through spectral phase retrieval approaches.196–198
Spectroscopic CARS differs from narrowband or spectral focusing CARS in that a broad swath of spectrum is obtained in each laser shot. Such an approach in the CH-stretch region has been demonstrated with picosecond/femtosecond CARS184,186,187,199 and with nonlinear interferometric vibrational imaging (NIVI).200 Histological studies focused on CH-stretch using spectroscopic CARS have been performed primarily with NIVI. From tissues, NIVI typically yields CH-stretch spectra with a bandwidth of ≈250 cm−1 at a 20 Hz spectral acquisition rate (1 kHz effective rate for a single frequency assuming 5 cm−1 resolution). These are undistorted Raman spectra, linear in N.200
Several studies have evaluated the utility of CARS in the CH stretch region for generating contrast equivalent to H&E stain. Evans et al.201 compared CARS images to H&E stained images, finding that CARS could provide similar image information when νCH2,sym and νCH3,sym or νCH3,asym were used. Similar “pseudo H&E” obtained through intravital CARS imaging202 enabled inspection of tumor environments and margins without staining, and no obvious evidence of photodamage even after 300 frames.
Quantitative tissue morphology metrics can be obtained from CARS contrast, similar to what is done with H&E-contrast images. Meyer et al.203 compare CARS with spontaneous Raman in brain, demonstrating ability to extract nucleus-to-cytoplasm ratio, cell density, nucleus size and shape. They found, however, that since nuclei lack νCH2,sym contrast, nuclear density and shape metrics may not be as reliable as H&E. On the other hand, the Wong group have used primarily νCH2,sym CARS contrast and image segmentation204–206 to determine nuclear size, cell volume, and cell–cell distance and other metrics to classify cancer subtypes in lung and breast tissue, producing encouraging results for differential diagnosis of cancer in these tissues. Uckermann et al.207 also compared metrics from CARS and H&E, finding that CARS provided additional relevant information via the signal amplitude.
In addition to conveying morphological information through imitating H&E stain, CARS νCH2 and νCH3 contrast has been used to study lipid metabolism in cancer,208 and to discriminate cell type based on lipid content. Excess lipids were found as a potential marker for circulating tumor cells,209 and Evans et al.201 found contrast in νCH2,sym to evince replacement of normal white matter with lipid deficient astrocytic glioma tissue. Chowdary et al.210 found spectral differences arising from variation in lipid to protein ratio in a rat breast cancer model, and were able to use SVD to automatically segment normal and diseased tissue regions at a 99% confidence level. Similarly, Uckermann et al.207 also found a νCH2,sym contrast gradient in the infiltrative zone around glioblastoma, with rather distinct contrast levels in the normal region. They also found that levels of νCH2,sym contrast could be used to distinguish between glioblastoma, and metasticized breast tumor in the brain. In a similar finding, Meyer et al.119 showed that CARS νCH2,sym contrast in the brain could highlight metastatic tumor regions of lung origin as the metastases retain a chemical profile similar to the originating tissue. Their finding was supported by Raman and IR imaging.
While narrowband CARS contrast from νCH2 and νCH3 alone is useful in many circumstances, it can also be combined synergistically with contrast from other methods, as exemplified in Fig. 5a and b. In other examples, Yue et al. used CARS signal from νCH3,sym to visualize mammary acini, providing guidance for acquisition of spontaneous Raman spectra. From subsequent spectral analysis, they observe changes in spatial distribution of lipids concomitant with a loss of basoapical polarity in mammary acuinus, a transformation linked to early stages of carcinogens.177 The Popp group have taken similar approaches.119,170,189,211 In comparing CARS to spontaneous Raman imaging of colon sections, Krafft et al.212 found that CARS images were consistent with expectations from spontaneous Raman scattering in the same spatial sample regions, and that with just a few CARS scans in the range 3000 to 1000 cm−1, there was sufficient information to discriminate a couple of cell types and identify regions of normal and diseased tissue. The CARS images were acquired 106 times faster than spontaneous Raman (10 μs vs. 30 s per pixel). Subsequently, they have used additional CARS peaks, in conjunction with other contrast mechanisms to characterize tissues. The same pulses that generate the CARS signal can also give rise to second harmonic generation (SHG) in collagen fibrils, and to two-photon excited fluorescence (TPEF) from intrinsic fluorophores such as NADPH, flavins, collagen, and elastin. Meyer et al.119,189 used CARS contrast at multiple Raman shifts along with intrinsic contrast from TPEF and SHG to detect white matter, the granule layer, elastin, ordered collagen, and NAD(P)H in brain, and elastic fibers, triglycerides, collagen, myelin, cellular cytoplasm, and lipid droplets in perivascular tissue.
 | ||
| Fig. 5 a & b:221 (a) Immunohistochemical stain for Sox-10 in mouse ear with haematoxylin counterstain, revealing melanocytes in red. (b) CARS image of adjacent slice to (a), showing pigmented regions, consistent with positive staining in (a). c & d:218 (c) Pseudocolour BCARS image of tumor and normal brain tissue, with nuclei highlighted in blue, lipid content in red and red blood cells in green. (d) Raman spectra retrieved from (c), with each spectrum taken from one pixel, and acquired in 3.5 ms. e & f:222 SRH image (e) is compared to a similar section of tumor imaged after formalin-fixation, paraffin-embedding and H&E staining (f). Comparison was made using a lookup table to match SRH “staining” with H&E. g & h:223 (g) Spectroscopic SRS images of pancreas and liver (left and right respectively). (h) Independent component spectra retrieved from (g) with 99% data reduction. | ||
Because the fingerprint peaks are typically much weaker than the nonresonant signal, it is not possible to obtain quantitative spectral information by simply subtracting the NRB. Because typically χ(3)R ≪ χ(3)NR for fingerprint resonances, the third (cross) term in eqn (11) dominates the resonant contribution to the CARS signal, and the overall spectral phase must be retrieved in order to extract a Raman spectrum. CARS phase retrieval is possible with an approach based on maximum entropy methods196 or one based on time-domain Kramers–Kronig.197 Both provide functionally equivalent results, but the computations for the Kramers–Kronig approach require considerably less time.213 It has recently been appreciated that the NRB shape estimate required by both of these methods can have a profound impact on extracted Raman peak amplitudes. Camp et al.198 showed that symmetry relations between the NRB and resonant signal can be used to unambiguously determine the NRB shape, and thus the relative peak amplitudes of the retrieved Raman spectrum. Additionally, the retrieved signal is linear with analyte concentration. In this way, the NRB acts as an internal calibration for spectroscopic CARS, and using this internal calibration, retrieved peak ratios are absolute.
Spectral acquisition of CARS signal is required for recovery of Raman fingerprint signals through phase retrieval, and also facilitates system noise removal via singular value decomposition.214 Spectrally focused CARS195 can be delay-scanned used to generate spectroscopic images that are amenable to phase retrieval215 Additionally, several inherently spectroscopic CARS microscopy approaches have been developed, but most are too inefficient for biological imaging. Here we will exclusively consider approaches that have been used to generate images from cells and tissues. The first of these was based on a combination of spectrally broad but uncompressed Stokes pulses, and spectrally narrow probe pulses.186,187 This approach generates broadband CARS spectra with up to 3000 cm−1 bandwidth at ≈20 Hz spectral acquisition rate (12 kHz single spectral element, assuming 5 cm−1 resolution), with strong CH stretch but quite weak fingerprint spectra.216 The quality of fingerprint spectra was improved significantly in one case by implementation of a narrow-bandwidth sub-nanosecond probe,217 and in another by using a temporally compressed Stokes source.218 In the latter work, a nearly transform-limited broadband pulse acts as both pump and Stokes, exciting vibrational coherence much more efficiently than separate pulses. This approach yielded CARS spectra in both CH and fingerprint regions with uniformly high SNR across the spectrum, and 280 Hz spectral acquisition rate (corresponding to 170 kHz single spectral element acquisition, assuming 5 cm−1 resolution).218
Perhaps because it has only recently become possible to acquire high quality fingerprint spectra in an imaging modality, there is very little work on histological samples using fingerprint CARS. With relatively weak fingerprint signal, Pohling219 was able to use principal component analysis to discriminate between gray and white matter, and identify layers of granule and Purkinje cells in mouse brain tissue. Their structural assignments agreed with those obtained from H&E staining. Also in murine brain tissue, but with significantly better SNR in the fingerprint due to impulsive coherence generation, Camp et al.198,218 were able to identify many diagnostically important species and features directly from spectral peaks, without multivariate analysis techniques (Fig. 5c and d). These included red blood cells through the 1548 cm−1 and 1565 cm−1 hemoglobin peaks (C–C stretch), as well as collagen, elastin, and nucleotides through peaks listed in Table 2 for each of these species. They were able to identify tumor margins, as there were spectral differences between normal and invasive glioblastoma image regions, such as at 785 cm−1 (nucleotide), 1004 cm−1 (phenylalanine ring breathing), and 2956 cm−1 (CH3 stretch). In both the Pohling and Camp work, a tumor-associated reduction in νCH2 was observed, as in CARS studies that focused on CH-stretch.
In the mouse glioblastoma work, Camp et al.218 obtained SHG and TPEF signals simultaneously with fingerprint CARS spectra. Although SHG is typically used to identify collagen,119,220 the multi-peak spectral identification of this protein did not demonstrate identical results. They noted that collagen generates an SHG response only when the triple-helical structure is in-tact and only when the optical polarization and fibril axes are aligned.
4.3 Stimulated Raman scattering (SRS)
In stimulated Raman scattering (SRS), energy transfers from a higher frequency (pump) field to a lower frequency (Stokes) field when the frequency differences between the fields is equal to that of a vibrational resonance in the material being probed. This effect can be monitored as stimulated Raman loss (SRL) in the pump field, or stimulated Raman gain (SRG) in the Stokes field. An expression for SRL can be given as: | (12) |
This expression differs from eqn (10) by addition of a pump field term. The pump field interacts coherently with the nonlinear polarization in the sample, generating a heterodyne term in the overall signal. Due to the relative phases of Ep and the field arising from the induced coherence, the heterodyne signal amplitude is proportional to only the imaginary part of χ(3). Furthermore, only this heterodyne component is retrieved if the SRL is detected through modulation techniques, and one directly obtains a signal proportional to the spontaneous Raman scattering cross section (see eqn (6)). Because χ(3)NR is real, the nonresonant component that is problematic for narrowband CARS is not detected in SRS. In spite of this, the theoretically achievable signal-to-noise ratio for SRS is the same as that of CARS,224 and a related value, the sensitivity limit, seems to be similar for both approaches.218 The heterodyne component of the SRL signal can be expressed as:
 | (13) |
For field strengths permissible in tissue microscopy, the relative change in pump or Stokes intensity is on the order of 10−6 to 10−8, even for the strong peaks in the CH stretch region. These small signals cannot be spectrally separated from the intense excitation light as in CARS, so must be discriminated by other means, such as lock-in or balanced detection methods. Under favorable conditions, pixel acquisition rates up to 7 MHz have been demonstrated with contrast from Raman peaks in the CH stretch region.225 However, 30–40 kHz pixel rates are more common for single spectral bands in tissue samples.222,226 Furthermore, even the strongest fingerprint peaks, such as found at 1440 & 1660 cm−1 (CH2 deformation & CC stretch) are challenging to use for image contrast with SRS. Ozeki et al.227 were unable to directly use the 1660 cm−1 peak for contrast, even when pixel dwell times were 10× longer than necessary for good SNR in the CH-stretch region.
While the NRB does not interfere with the SRS signal, other background signals can. These include two photon absorption (TPA), cross phase modulation and thermal lensing. Signals from these sources can have amplitude comparable to or greater than even the largest SRS signals. For example, in skin, the TPA signal exceeds that of CH stretch SRS by almost 100-fold.221 Not surprisingly, these background signals can easily mask weak contrast from the fingerprint region, and careful measures may be taken to suppress their effects and recover some of the stronger fingerprint signals.228,229
Because the SRS signal must be extracted directly from the laser field, lasers with very low residual intensity noise (RIN) are preferred. These are typically solid state lasers. However, fiber lasers have a number of qualities that make them desirable in a clinical setting, including being robust to physical movement, vibration, and temperature shifts, and requiring very little maintenance. A low-noise fiber source has been developed specifically for SRS230 although even for this laser, it is necessary to use both balanced detection and lock-in approaches to recover the SRS signal.
Using only contrast from the νCH3,sym band and morphological clues, Mittal et al.236 were able to identify cell membranes, nuclei, collagen and keratin in tissues containing squamous cell carcinoma. In that study, the authors found that SRS images from this single band provided similar information to that found in H&E images.
Lu et al.237 compared images from H&E staining to those generated with SRS contrast from νCH2,sym and νCH3,sym. They found that cell density counts from SRS and H&E images were indistinguishable. However, using the lipid/protein ratio from the SRS signal, and confirming this with luxol fast blue staining for the myelin sheath, they found that the two SRS bands provided a better distinction between white and gray matter than could be achieved with H&E staining. They were also able to identify red blood cells and vessels through morphological features of images with contrast at 2800 cm−1, in the nominally quiescent region. The contrast there may have arisen from TPA of heme. They also found that necrotic regions were characterized by lack of nuclei and lower protein signal, and banded regions of high protein signal appeared to be collagen.
Ji et al.238 used SRS contrast for νCH2,symm and νCH3,symm in fresh surgical brain tissues from 22 patients to create a classifier based on cellularity, axnoal density, and protein to lipid ratios, which provided tumor infiltration detection with 97.5% sensitivity and 98.5% specificity. They found that SRS images were superior to H&E images for detecting infiltrating glioma, and that SRS microscopy could detect invasive tumor cells in peri-tumor brain tissue that appears grossly normal.
Orringer et al.222 used νCH2,symm and νCH3,symm bands from SRS to recapitulate image contrast from H&E, but on unprocessed tissue, with an eye towards improving intrasurgical tissue sampling. Example images from that study are shown in Fig. 5e and f. Pathologist diagnosis based on proximally located SRS and H&E tissue samples from 30 patients was compared and found to agree 92% of the time. They also used computer algorithm-derived image attributes (based on SRS contrast) to successfully distinguish between diagnostic classes of tumors with 90% accuracy.
Cui et al.239 used SRS spectra in the region (2800 to 3000) cm−1 and multivariate curve resolution to find a correlation between cholesterol ester content in intracellular lipid droplets and Gleason scores of prostate tissue. Using acalibrated surrogate mixture for cellular lipid content, they found that the ratio between a band assigned to cholesterol at 2870 cm−1 and the νCH2,sym band was proportional to the fraction of total lipids due to cholesterol ester.
While full spectral information in the CH-stretch region is sometimes useful, a small number of peaks often provide sufficient discrimination. Otsuka et al.223 examined tissues from a tumor-graft mouse model using spectrally scanned SRS spectrally scanned over the range (2800 to 3100) cm−1. They found that through independent component analysis, they could reduce their spectral dataset by >95%, and still obtain images in which nucleus, zymogen granules, lipid droplets, and red blood cells could be visualized, and irregular glandular cell alignments detected (see Fig. 5g and h). They found that images from the reduced spectral data was qualitatively equivalent to H&E stained images. Similarly, Egawa et al.240 used SRS spectroscopy in the range (2800 to 3100) cm−1 to map spatially resolved keratinocyte differentiation, and distinguish these from Langerhans cells in the spinous layer of the epidermis. The key differences were primarily morphological, as both cell shape and nuclear prominence differ between the cell types, so the important contrast came at νCH2,symm and νCH3,symm bands.
In other studies, vibrational signal from SRS has been used in concert with other nonlinear contrast mechanisms. Freudiger et al.226 combined νCH2,sym and νCH3,sym bands with hemoglobin TPA to derive contrast of roughly equal quality to H&E-stained sections, recapitulating major histologic features in tissue from several brain regions. Galli et al.241 used νCH2 in conjunction with TPEF of an extrinsic label, and SHG from structured collagen to observe significant differences in nuclear area of normal and cancerous regions in human tumors and murine tumor models. Yue et al.242 used SRS νCH2,sym contrast for morphological imaging in liver and prostate tissue, and for guidance in identifying lipid droplets (LDs). They then used spontaneous Raman spectroscopy at LD positions to determine that the cholesterol ester content of the LDs was correlated with the ratio of 702 and 1442 cm−1 peak intensity ratios, and with the aggressiveness of the cancer.
Colon tissues from 9 patients were imaged by spectrally scanned SRS in conjunction with SHG, THG and TPEF.244 They found that an increase in protein (νCH3,sym and Amide I), and water (3250 cm−1), and a decrease in lipid (νCH2,sym, 1445 cm−1, 1745 cm−1) all correlated with presence of adenocarcinoma. They reported similarity in TPEF of NADH and 1665 cm−1 contrast images, suggesting that increased protein could be due to increased metabolic activity. They also suggested that increased water in cancerous regions could be due to increased aquaporin protein production.
5 A future role for coherent Raman imaging in histopathology
We expect that CRI methods for histopathology will be adopted to the extent that they cost-effectively meet specific needs, and fit well into the established histology work flow, possibly streamlining it. As early applications are explored, and as physicians become increasingly comfortable with the technology, it is possible that more complex and potentially disruptive adaptations of this technology may be adopted. Of course, initial adoption will be influenced by the costs of CRI instrumentation relativeto existing technology. In this section, we briefly discuss classes of problems that CRI can address, both in the near and longer terms.5.1 Immediacy of information
It is not uncommon to have 24 hours latency between tissue excision and histology analysis. While this is often acceptable, there are situations in which immediate characterization is optimal or even required, such as while sampling a suspected tumor, and during an intra-operative margin evaluation. In these cases, label-free and in situ histology modalities are desirable.Tissue involved in and surrounding a tumor is typically sampled and analyzed by histology or for biomarkers at various time points to guide therapy by establishing prognosis and following disease progression. Accordingly, inappropriate tumor sampling is thought to be a leading problem in both prognosis and evaluating treatment response.245
Tissue sampling by needle biopsy or exploratory surgery is often guided by palpitation, preparative imaging, or visual inspection, and is notoriously inefficient.245 Neoplasms may be difficult to properly sample at early stages, and the ability to categorize tissue as normal or neoplastic during the biopsy process would go a long way towards ensuring high-quality samples for subsequent analysis. Spontaneous Raman was demonstrated decades ago as an potential guidance tool for needle biopsy,246 and progress has been made toward clinical adaptation, but challenges remain with regards to low signal levels, and typically, Raman needle probes must be coupled with other detection modalities.113 Coherent Raman methods could easily provide sufficient signal levels for this application. Narrowband CRI approaches have been implemented in fiber,247–249 so could be adapted to needle biopsy, but are probably not suited for this application since disease detection would be based strictly on spectra, and thus benefit greatly from sensitivity to multiple peaks in the fingerprint spectral region. Current broadband spectroscopic CRI approaches based on impulsive excitation or spectral focusing will be difficult to implement in a fiber delivery, but other approaches may be possible in the future.
While narrowband CRI may not be appropriate for purely spectroscopic disease detection, in needle biopsy, it is effective in a microscopy modality for in vivo detection and analysis of skin carcinomas, which are present in the optically accessible epidermis layer of the skin.211,221,236,250 New approaches to signal detection may push the useful range of in vivo CRI to slightly deeper tissues,251 and to detection of fingerprint signals.229
Another widely recognized clinical problem is the need for intraoperative analysis of tissue at a resection margin. Strong correlations exist between presence of cancer cells in surgical margins and recurrence rates of cancer,252,253 and incomplete margin resection is held as a major cause for recurrence of cancers that call for tissue conservation, such as in brain and breast.
A hand-held probe is required for in situ margin analysis, and appropriate equipment has already been described for narrowband CRI247–249 although, to our knowledge, clinical studies using these probes have not yet been published. Use of a hand-held Raman probe for identification of tumor tissue in surgical margins was clinically demonstrated for brain surgery by Jermyn et al.254 and proof of principle for breast was demonstrated by Kong et al.255 As with Raman needle probes, tumor detection in these reports was based on spectroscopy alone, thus suffering somewhat from low signal and precluding imaging. This difficulty was avoided in the breast application by using autofluorescence levels to identify regions of interest, and those regions were imaged sparsely with Raman. In the brain application, the surgeon visually selected regions of interest.
Although CRI technology has not yet progressed to in situ margin analysis, it has been demonstrated as an effective tool for characterizing freshly excised, unprocessed tissue.222 Typically, margin tissue must be frozen and stained for imaging. This process can add 20 minutes to a surgery each time a margin is to be investigated. The label-free nature of CRI, and the ability to image in a backscattering mode makes it possible to obtain histology-quality images from surface regions of thick, unprocessed tissue, significantly reducing the delay between tissue removal and analysis.222
Another area in which speed of information availability would be useful is in initial evaluation of histology slides. The majority of tissue in most slides is normal, so a pathologist may spend a significant portion of their time searching for potential diseased regions. One particularly strong benefit of chemical imaging is that it can provide clear indications of tissue type and regions of normal and potentially abnormal tissue. This regional tissue typing has already been demonstrated with IR on slides from which high-resolution H&E images have been obtained, and the two images overlaid.161 CRI methods can provide both the regional tissue typing201,207,210 and the high-resolution H&E mimic without the need for staining or imaging in multiple contrast modes.201,202,222
5.2 Better quantification of morphological parameters
A survey of pathologists by the National Collation on Health Care and Best Doctors36 gave lack of sub-specialty expertise the most often suspected reason for cancer misdiagnoses from histology. In that study, physicians most frequently indicated that improved pathology resources (i.e., innovations in histopathology) is the thing most needed to improve diagnostic accuracy. One particular approach to improving pathology resources, namely CAD, aims to bridge the sub-specialty expertise gap.An important example of how CAD can bridge this gap is through providing basic quantification of image features. As mentioned previously, non-specialist pathologists were found to be much less precise in cell counts and classification than sub-specialists,88 no doubt contributing significantly to overall diagnostic uncertainty. Many of the same factors contributing to non-specialist variability, including the fact that cells are often overlaid in H&E images, with no clear delineation between nuclei,88 are also challenges to the families of morphological image analysis on which CAD is based. Further, factors such as variability in staining protocols, and in image field illumination, and issues with nonspecific staining contribute to overall image variability, adversely impacting reliability in thresholding and feature identification for CAD. These, and related issues are expected to be significant barriers to widespread adoption of CAD. In recognition of this, the US Food and Drug Administration has issued guidelines for performance of WSI instrumentation.57
CRI methods can ameliorate or resolve many of the reproducibility and reliability issues in image formation. For example, obtaining reliable cell counts will be easier because CRI has intrinsically high axial resolution, making it feasible to resolve stacked nuclei. Additionally, where imaged nuclei are spatially close, there will be a nuclear and cellular membrane signal between them, strengthening the ability of CRI-based analysis to arrive at accurate cell counts. Yang et al.206 have already demonstrated 90% accuracy and reproducibility in automated cell counting using coherent Raman imaging with only νCH2,sym, and thus poor nuclear contrast.203 This is an important near-term opportunity for coherent Raman methods.
In addition to intrinsic z-sectioning, CRI can provide a large number of contrasts through various vibrational bands. Since increased image dimensionality generally leads to higher precision segmentation and feature identification,89–91 one could expect improved performance in these aspects with these multi-contrast images. The Wong group have derived morphological parameters familiar to pathologists, such as cell shape and density, from CARS images and based on these were able to identify with 80 to 95% accuracy breast tumor types.206
Although CRI requires little or no sample preparation, it seems likely that most early implementations will include fixation and paraffin embedding of samples, simply for consistency with protocols for staining. In fact, both of these steps have potential to introduce variability in CRI signals. Paraffin has a strong CH stretch signal, and fixing agents can liberate diagnostically important molecules, particularly lipids.256 For example, compared to fresh frozen tissue, formalin-fixed brain showed modification in νCH2,assym and νCH3,assym amplitudes from CARS, but not in νCH2,sym. In the fingerprint, only choline groups of phospholipids at 717 cm−1 dropped slightly after formalin fixing. However, methanol or acetone fixation severely reduced all of these peaks and others.257
5.3 Increased chemical information
Morphological analysis of an image from a single stain such as H&E is sufficient for diagnosis in most cases. However, difficult cases often require multiple additional stains to provide needed auxiliary information, but many of these desirable stains are rare or unavailable,2 and it seems that this lack of access is a clinically significant issue.36 CRI with good fingerprint contrast has potential to make an important contribution by providing a considerable range of contrasts.Fingerprint CRI provides chemical specificity identical to that of spontaneous Raman spectroscopy,218 and therefore seems likely to be a potential substitute for most or all stains, and perhaps many IHC agents. Raman spectra can be used to recapitulate many image contrast agents used commonly in biology.258 Also, as shown by the partial listing in Table 2, fingerprint Raman has already been used to detect many diagnostic markers. With fingerprint CRI, these will be available as label-free image contrasts. An important added benefit of spectroscopic CARS is that it provides an internal reference based on NRB,198 so that spectroscopic CARS images can serve as quantitative, rather than qualitative maps of markers. This level of quantification cannot generally be achieved with stains, fluorescence, or even spontaneous Raman. The quantitative imaging and absence of non-specific staining will greatly simplify image analysis and increase confidence in image metrics and associated diagnostic decisions. A few specific examples of potential important contributions are given below.
The number density of mitotic cells plays an important role in diagnosis and grading of many cancer types,8 and is usually determined by IHC or cell morphology analysis. The best morphologically-derived automated detection of mitoses achieve F-scores (the geometric mean of precision and accuracy) between 75% and 80%.87 It seems likely that fingerprint CRI imaging could yield significantly better results. By mapping the intranuclear distribution of DNA, RNA, and chromatin, Raman spectroscopic imaging can not only positively identify whether a cell is mitotic, but which stage of mitosis it is in.163,166,167 The chemical distribution information is essentially orthogonal to morphological analysis, so can only serve to increase precision and accuracy in determining mitosis counts.
Accurate characterization of pleomorphism (atypical cell shape) is also important to proper diagnosis of many cancers.8 Pleomorphic cells of one type may be mistakenly identified as normal cells of a different type, and this was part of a 42% cell count and classification disagreement among non-specialists found by Fuchs et al.88 It is well established that Raman spectroscopy can be used to positively identify cell type.162 Applying spectral cell identification as an orthogonal metric will facilitate much better recognition of pleomorphism.
Invasion of immune cells into a tumor site, and their state of activation is an key factor in diagnosis of some cancers.8 As mentioned above, morphology-based cell identification can be difficult, and determination of immune cell activation is currently done by assessing the presence of cell surface markers, typically by IHC. For this important problem, fingerprint spectroscopic imaging will not only allow immune cell identification, but also whether they are activated.107,168
Identifying the tissue from which of CUP (cancer unknown primary) metastases originate is useful for identifying appropriate therapeutic strategies, but presently requires involved analysis.259–261 Fingerprint spectroscopic CRI may be able to positively identify metastatic tumor origins simply from chemical content of the tumor cells. Even narrowband CRI studies were able to discriminate primary (metastasized) cancer cells from cells belonging to the target tissue simply on basis of contrast at νCH2,symm and νCH3,symm.119,207
In recent years it has become clear that genomic subclasses of cancers can have distinct clinical outcomes,262 and thus require varied interventions. While spectroscopic Raman imaging has not been evaluated as a tool for identifying these subclasses, it seems reasonable that it may have power to do so in some cases. CRI will not directly detection genome changes, but it is likely to detect downstream consequences of those changes. For example, cell phenotype162 and function107,162 can be discriminated using Raman spectra. These spectral changes come not from DNA sequence or methylation modifications directly, but from the intra- and extra-cellular changes in bimolecular profiles they induce. Furthermore, microenvironment properties determined through histology have recently been found to correlate with the genomic cancer subtypes, and these microenvironment changes should be detectable with CRI in most cases.263
Broadband CRI tissue imaging using CH stretch has been demonstrated at 20 Hz for CARS185 and at 4 kHz for SRS223,240 over 300 cm−1. Fingerprint CARS spectro-microscopy has been demonstrated at 300 Hz over 3000 cm−1 in tissue.218 Overall, these range in speed from 105 to 103 times slower than WSI. This difference may be difficult to surmount. Assuming the 0.5 cm2 min−1 imaging speed is indeed an appropriate mark, it seems that present spectral CRI methods are not good candidates, although some newer spectroscopic CRI approaches may hold promise. A spectral focusing approach has been demonstrated at 77 kHz for a 200 cm−1 bandwidth,251 although not yet in tissue, and a family of approaches featuring ≈25 kHz spectroscopic CARS acquisition has been demonstrated,264,265 but with laser pulses incompatible with tissue imaging. Broadband SRS266,267 has been demonstrated with potentially fast imaging, but seem to suffer from insufficient modulation sensitivity.
Assuming that signal generation and collection efficiency of current CRI instruments is nearly optimal, and there is no way to intrinsically speed up image acquisition, there are at least two non-exclusive options for applying spectroscopic CRI to histopathology. One is to use a faster imaging modality (such as narrowband CRI) to generate a guide image from which regions of interest can be determined, and use spectroscopic CRI over ROIs covering a small fraction of the specimen. The other approach would be to use spatial multiplexing to effectively increase the rate of spectroscopic signal acquisition.
Provided sufficient excitation laser power, spatial multiplexing approaches can yield speed increases proportional to the number of detection elements. For spectroscopically detected signals, such as in spontaneous Raman scattering, 1-D (line) spatial multiplexing can realistically lead to 20×–100× imaging speed increase.164 This approach could also be used for spectroscopic CARS. 2-D multiplexing has been demonstrated with narrowband CARS268–272 though not in ways suited for clinical applications. Spectroscopic approaches using single element detectors, such as time-domain264 or spectrally scanned195 CARS could also be 2-D multiplexed, so considerable speed increases are conceivable, provided laser sources of sufficient pulse energy can be found to spread light over a larger area and still obtain signal. SRS will be more challenging to implement in spatially multiplexed modes since it requires very high sensitivity to small amplitude modulations. 2-D array detectors, such as CMOS cameras can be used in differential or lock-in mode, but these have yet to demonstrate sensitivity that would be needed for tissue imaging.266,267 Slipchenko et al.273 introduced an array of 32 LC circuits for simultaneously detecting SRS signal in as many channels, which has shown sufficient sensitivity for tissue imaging.
Finally, we note that routine use of spectroscopic CRI in histopathology could present significant data storage issues. A single-contrast slide image from a WSI instrument may occupy 1–2 Gb of storage. At a rate of 500 images a day, storing digital histopathology data is already a challenge. If all such images were acquired in full spectral mode, the data storage load would increase by 103 times – an untenable proposition. Such a scenario is neither likely or necessary; there are at least two factors that would mitigate the need for such excessive storage. One is that spectroscopic imaging would be particularly beneficial for only small areas of most tissues. The other is that, while spectral images could be stored temporarily, for processing, it quite unlikely that saving all spectral dimensions will be necessary in most cases.169 Data reduction methods such as those discussed in Sections 2.7.3 and 3.1.1 could be applied to determine the most important spectroscopic information, allowing to discard the rest and reduce the size of images for long-term storage.
6 Conclusions
We expect that coherent Raman imaging is most likely to find acceptance in the clinic as a drop-in replacement for current technologies to the extent that it is able to help resolve recognized problems related to detection, diagnosis, and treatment of disease. Such problems are likely to include fast and reliable intra-surgical margin evaluation, either through direct analysis of the surgical site or through rapid analysis of freshly excised tissue. Similarly, CRI's ability to provide label-free molecularly specific signal quickly may be useful for in situ detection of small tumors or neoplasms in exploratory surgery or needle biopsy.Intra-surgical microscopic analysis of excised tissue has already been demonstrated. In situ use will likely require a hand-held probe with fiber-delivered excitation light and signal collection. Whether in situ tissue analysis will be best done by narrowband coherent Raman imaging or by coherent Raman spectroscopic analysis is unclear at this time.
Another significant opportunity for CRI is facilitating improvements in histology-based diagnoses. The art of histopathology emerged near the end of the 19th century in nearly its present form. Significant new knowledge of disease mechanisms has improved interpretation of histological features, but, apart from the introduction of IHC in the 1940s, histopathology tools in use today are largely the same as they were in the early 20th century. Efforts to modernize histopathology practice are making headway, however, fully transforming it from a subjective art to a quantitative and objective science remains a challenge. Most of these challenges involve variability and contrast reliability in image formation. It appears that many of these issues, could be largely resolved with CRI. Furthermore, spectroscopic CRI may provide a significantly enhanced contrast space that will open new options for quickly stratifying cancer and other diseases, leading to a more personalized diagnosis.
While opportunities for CRI to improve clinical practice are clear, important barriers remain for widespread adoption in histology practice. An increase in imaging speed for narrowband CARS and a slightly larger increase for SRS would be helpful. For spectroscopic CRI, substantial imaging speed increases will be necessary if they are to be used to a significant extent in an imaging mode. Fortunately, the CRI field is still vibrant with new innovations for increased imaging speed and chemical selectivity and sensitivity. Much work remains to demonstrate the utility and reliability of these methods to the medical community. Finally, potential regulatory issues must be considered and resolved to the extent that they are found.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank C. Paul Morris, M.D., Ph.D. for helpful discussions and comments on the manuscript.References
- D. Evanko, A. Heinrichs and C. K. Rosenthal, Nat. Cell Biol., 2009, 11, S6–S22 Search PubMed.
- M. Titford, J. Histotechnol., 2009, 32, 9–19 CrossRef CAS.
- G. Hunninghake, M. B. Zimmerman, D. Schwartz, T. King, J. Lynch, R. Hegele, J. Waldron, T. Colby, N. Muller, D. Lynch, J. Galvin, B. Gross, J. Hogg, G. Toews, R. Helmers, J. A. Cooper, R. Baugman, C. Strange and M. Millard, Am. J. Respir. Crit. Care Med., 2001, 164, 193–196 CrossRef CAS PubMed.
- B. W. G. van Rhijn, G. J. L. H. van Leenders, B. C. M. Ooms, W. J. Kirkels, A. R. Zlotta, E. R. Boevé and A. C. Jöbsis, Eur. Urol., 2010, 57, 1052–1057 CrossRef PubMed.
- W. C. Allsbrook Jr., K. A. Mangold, M. H. Johnson, R. B. Lane, C. G. Lane and J. I. Epstein, Hum. Pathol., 2001, 32, 81–88 CrossRef CAS PubMed.
- M. Costantini, S. Sciallero, A. Giannini, B. Gatteschi, P. Rinaldi, G. Lanzanova, L. Bonelli, T. Casetti, E. Bertinelli, O. Giuliani, G. Castiglione, P. Mantellini, C. Naldoni and P. Bruzzi, J. Clin. Epidemiol., 2003, 56, 209–214 CrossRef PubMed.
- T. A. Longacre, M. Ennis, L. A. Quenneville, A. L. Bane, I. J. Bleiweiss, B. A. Carter, E. Catelano, M. R. Hendrickson, H. Hibshoosh, L. J. Layfield, L. Memeo, H. Wu and F. P. O'Malley, Mod. Pathol., 2005, 19, 195–207 CrossRef PubMed.
- Stanford Medicine: Surgical Pathology Criteria, http://www.surgpathcriteria.stanford.edu.
- A. H. Coons, H. J. Creech and R. N. Jones, Exp. Biol. Med., 1941, 47, 200–202 CrossRef CAS.
- C. D. Fletcher, J. J. Berman, C. Corless, F. Gorstein, J. Lasota, B. J. Longley, M. Miettinen, T. J. O'Leary, H. Remotti, B. P. Rubin, B. Shmookler, L. H. Sobin and S. W. Weiss, Hum. Pathol., 2002, 33, 459–465 CrossRef PubMed.
- M. Werner, A. Chott, A. Fabiano and H. Battifora, Am. J. Surg. Pathol., 2000, 24, 1016 CrossRef CAS PubMed.
- M. Sabah, M. Leader and E. Kay, Appl. Immunohistochem. Mol. Morphol., 2003, 11, 56–61 CAS.
- T. Kirkegaard, J. Edwards, S. Tovey, L. M. McGlynn, S. N. Krishna, R. Mukherjee, L. Tam, A. F. Munro, B. Dunne and J. M. S. Bartlett, Histopathology, 2006, 48, 787–794 CrossRef CAS PubMed.
- R. S. Weinstein, A. R. Graham, L. C. Richter, G. P. Barker, E. A. Krupinski, A. M. Lopez, K. A. Erps, A. K. Bhattacharyya, Y. Yagi and J. R. Gilbertson, Hum. Pathol., 2009, 40, 1057–1069 CrossRef PubMed.
- D. L. Woernley, Cancer Res., 1952, 12, 516–523 CAS.
- C. Kendall, N. Stone, N. Shepherd, K. Geboes, B. Warren, R. Bennett and H. Barr, J. Pathol., 2003, 200, 602–609 CrossRef PubMed.
- R. Bhargava and I. W. Levin, Appl. Spectrosc., 2004, 58, 995–1000 CrossRef CAS PubMed.
- A. S. Haka, K. E. Shafer-Peltier, M. Fitzmaurice, J. Crowe, R. R. Dasari and M. S. Feld, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12371 CrossRef CAS PubMed.
- A. Zumbusch, G. R. Holtom and X. S. Xie, Phys. Rev. Lett., 1999, 82, 4142–4145 CrossRef CAS.
- M. J. Nasse, M. J. Walsh, E. C. Mattson, R. Reininger, A. Kajdacsy-Balla, V. Macias, R. Bhargava and C. J. Hirschmugl, Nat. Methods, 2011, 8, 413–416 CrossRef CAS PubMed.
- K. Yeh, S. Kenkel, J.-N. N. Liu and R. Bhargava, Anal. Chem., 2015, 87, 485–493 CrossRef CAS PubMed.
- Protein Atlas, http://www.proteinatlas.org.
- W. H. Fridman, F. Pagès, C. Sautès-Fridman and J. Galon, Nat. Rev. Cancer, 2012, 12, 298–306 CrossRef CAS PubMed.
- W. Schubert, B. Bonnekoh, A. J. Pommer, L. Philipsen, R. Böckelmann, Y. Malykh, H. Gollnick, M. Friedenberger, M. Bode and A. W. M. Dress, Nat. Biotechnol., 2006, 24, 1270–1278 CrossRef CAS PubMed.
- P. A. Humphrey, J. Clin. Pathol., 2007, 60, 35–42 CrossRef CAS PubMed.
- J. D. Kronz, W. H. Westra and J. I. Epstein, Cancer, 1999, 86, 2426–2435 CrossRef CAS PubMed.
- S. S. Raab, R. E. Nakhleh and S. G. Ruby, Arch. Pathol. Lab. Med., 2005, 129, 459–466 Search PubMed.
- J. Ho, A. V. Parwani, D. M. Jukic, Y. Yagi, L. Anthony and J. R. Gilbertson, Hum. Pathol., 2006, 37, 322–331 CrossRef PubMed.
- A. A. Renshaw, K. M. Lezon and D. C. Wilbur, Cancer Cytopathol., 2001, 93, 106–110 CrossRef CAS.
- A. Lurkin, F. Ducimetière, D. R. Vince, A.-V. Decouvelaere, D. Cellier, F. N. Gilly, D. Salameire, P. Biron, G. de Laroche, J. Y. Blay and I. Ray-Coquard, BMC Cancer, 2010, 10, 150 CrossRef PubMed.
- J. M. Bruner, L. Inouye, G. N. Fuller and L. A. Langford, Cancer, 1997, 79, 796–803 CrossRef CAS PubMed.
- P. L. Nguyen, D. Schultz, A. A. Renshaw, R. T. Vollmer, W. R. Welch, K. Cote, A. V. DAmico and A. V. D'Amico, Urol. Oncol.: Semin. Orig. Invest., 2004, 22, 295–299 Search PubMed.
- V. L. Staradub, Ann. Surg. Oncol., 2002, 9, 982–987 CrossRef PubMed.
- T. R. Coblentz, S. E. Mills and D. Theodorescu, Cancer, 2001, 91, 1284–1290 CrossRef CAS PubMed.
- A. E. Selman, T. H. Niemann, J. M. Fowler and L. J. Copeland, Obstet. Gynecol., 1999, 94, 302–306 CAS.
- MisdiagnosisSurvey_FINALiv.pdf, http://www.bestdoctors.com/about-best-doctors/news-and-events/nchc-misdiagnosis-survey.
- S. C. Hollensead, W. B. Lockwood and R. J. Elin, J. Surg. Oncol., 2004, 88, 161–181 CrossRef PubMed.
- M. M. Mello, A. Chandra, A. A. Gawande and D. M. Studdert, Health Aff., 2010, 29, 1569–1577 CrossRef PubMed.
- ACS_2016_FactsFigures.pdf, http://www.cancer.org.
- Cost of Cancer Care, 2012.
- Z. Huang, A. McWilliams, H. Lui, D. I. McLean, S. Lam and H. Zeng, Int. J. Cancer, 2003, 107, 1047–1052 CrossRef CAS PubMed.
- N. Stone, C. Kendall, J. Smith, P. Crow and H. Barr, Faraday Discuss., 2004, 126, 141–157 RSC.
- T. C. Bakker Schut, K. Maquelin, T. van der Kwast, C. H. Bangma, D.-J. J. Kok and G. J. Puppels, Anal. Chem., 2006, 78, 7761–7769 CrossRef PubMed.
- M. Gniadecka, P. A. Philipsen, S. Sigurdsson, S. Wessel, O. F. Nielsen, D. H. Christensen, J. Hercogova, K. Rossen, H. K. Thomsen, R. Gniadecki, L. K. Hansen and H. C. Wulf, J. Invest. Dermatol., 2004, 122, 443–449 CrossRef CAS PubMed.
- M. Uhlén, E. Björling, C. Agaton, C. A.-K. Szigyarto, B. Amini, E. Andersen, A.-C. C. Andersson, P. Angelidou, A. Asplund, C. Asplund, L. Berglund, K. Bergström, H. Brumer, D. Cerjan, M. Ekström, A. Elobeid, C. Eriksson, L. Fagerberg, R. Falk, J. Fall, M. Forsberg, M. G. Björklund, K. Gumbel, A. Halimi, I. Hallin, C. Hamsten, M. Hansson, M. Hedhammar, G. Hercules, C. Kampf, K. Larsson, M. Lindskog, W. Lodewyckx, J. Lund, J. Lundeberg, K. Magnusson, E. Malm, P. Nilsson, J. Odling, P. Oksvold, I. Olsson, E. Oster, J. Ottosson, L. Paavilainen, A. Persson, R. Rimini, J. Rockberg, M. Runeson, A. Sivertsson, A. Sköllermo, J. Steen, M. Stenvall, F. Sterky, S. Strömberg, M. A. Sundberg, H. Tegel, S. Tourle, E. Wahlund, A. Waldén, J. Wan, H. Wernérus, J. Westberg, K. Wester, U. Wrethagen, L. L. Xu, S. Hober and F. Pontén, Mol. Cell. Proteomics, 2005, 4, 1920–1932 Search PubMed.
- J. B. Welsh, L. M. Sapinoso, S. G. Kern, D. A. Brown, T. Liu, A. R. Bauskin, R. L. Ward, N. J. Hawkins, D. I. Quinn, P. J. Russell, R. L. Sutherland, S. N. Breit, C. A. Moskaluk, H. F. Frierson and G. M. Hampton, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3410–3415 CrossRef CAS PubMed.
- K. Honda, T. Yamada, R. Endo, Y. Ino, M. Gotoh, H. Tsuda, Y. Yamada, H. Chiba and S. Hirohashi, J. Cell Biol., 1998, 140, 1383–1393 CrossRef CAS PubMed.
- P. Mauri, A. Scarpa, A. C. Nascimbeni, L. Benazzi, E. Parmagnani, A. Mafficini, M. D. Peruta, C. Bassi, K. Miyazaki and C. Sorio, FASEB J., 2005, 19, 1125–1127 CAS.
- P. Alfonso, A. Núñez, J. Madoz-Gurpide, L. Lombardia, L. Sánchez and J. I. Casal, Proteomics, 2005, 5, 2602–2611 CrossRef CAS PubMed.
- R. M. Levenson and J. R. Mansfield, Cytometry, Part A, 2006, 69, 748–758 CrossRef PubMed.
- P. Zrazhevskiy, L. D. True and X. Gao, Nat. Protoc., 2013, 8, 1852–1869 CrossRef CAS PubMed.
- M. J. Gerdes, C. J. Sevinsky, A. Sood, S. Adak, M. O. Bello, A. Bordwell, A. Can, A. Corwin, S. Dinn, R. J. Filkins, D. Hollman, V. Kamath, S. Kaanumalle, K. Kenny, M. Larsen, M. Lazare, Q. Li, C. Lowes, C. C. McCulloch, E. McDonough, M. C. Montalto, Z. Pang, J. Rittscher, A. Santamaria-Pang, B. D. Sarachan, M. L. Seel, A. Seppo, K. Shaikh, Y. Sui, J. Zhang and F. Ginty, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11982–11987 CrossRef CAS PubMed.
- D. P. Riordan, S. Varma, R. B. West and P. O. Brown, PLoS One, 2015, 10, e0128975 Search PubMed.
- E. Schubert, W. Gross, R. H. Siderits, L. Deckenbaugh, F. He and M. J. Becich, Semin. Diagn. Pathol., 1994, 11, 263–273 CAS.
- L. Pantanowitz, N. Farahani and A. Parwani, Pathol. Lab. Med. Int., 2015, 23 CrossRef.
- L. Pantanowitz, P. N. Valenstein, A. J. Evans, K. J. Kaplan, J. D. Pfeifer, D. C. Wilbur, L. C. Collins and T. J. Colgan, J. Pathol. Inform., 2011, 2, 36 CrossRef PubMed.
- Guidance for Industry and Food and Drug Administration Staff, http://fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM435355.pdf.
- A. J. Méndez, P. G. Tahoces, M. J. Lado, M. Souto and J. J. Vidal, Med. Phys., 1998, 25, 957–964 CrossRef PubMed.
- M. Orcutt, MIT Technology Review, 2015.
- L. He, L. R. Long, S. Antani and G. Thoma, Seq. Genome Anal., 2010, 510, 271–287 Search PubMed.
- H. Mousavi, V. Monga, G. Rao and A. Rao, J. Pathol. Inform., 2015, 6, 15–15 CrossRef PubMed.
- J. Barker, A. Hoogi, A. Depeursinge and D. L. Rubin, Med. Image Anal., 2016, 30, 60–71 CrossRef PubMed.
- E. Bengtsson, H. Danielsen, D. Treanor, M. N. Gurcan, C. MacAulay and B. Molnár, Cytometry, Part A, 2017, 91, 551–554 CrossRef PubMed.
- M. N. Gurcan, L. E. Boucheron, A. Can, A. Madabhushi, N. M. Rajpoot and B. Yener, IEEE Rev. Biomed. Eng., 2009, 2, 147–171 CrossRef PubMed.
- H. Irshad, A. Veillard, L. Roux and D. Racoceanu, IEEE Rev. Biomed. Eng., 2014, 7, 97–114 CrossRef PubMed.
- G. D. Marty, BioTechniques, 2007, 42, 716 CrossRef PubMed.
- A. C. Ruifrok and D. A. Johnston, Anal. Quant. Cytol. Histol., 2001, 23, 291–299 CAS.
- M. Macenko, M. Niethammer, J. S. Marron, D. Borland, J. T. Woosley, X. Guan, C. Schmitt and N. E. Thomas, Biomedical Imaging: From Nano to Macro, 2009. ISBI'09. IEEE International Symposium on, 2009, pp. 1107–1110.
- A. M. Khan, N. Rajpoot, D. Treanor and D. Magee, IEEE Trans. Biomed. Eng., 2014, 61, 1729–1738 CrossRef PubMed.
- N. Otsu, Automatica, 1975, 11, 23–27 Search PubMed.
- S. G. Chang, B. Yu and M. Vetterli, IEEE Trans. Image Proc., 2000, 9, 1522–1531 CrossRef CAS PubMed.
- J. B. Roerdink and A. Meijster, Fundam. Inform., 2000, 41, 187–228 Search PubMed.
- J. C. Bezdek, R. Ehrlich and W. Full, Comput. Geosci., 1984, 10, 191–203 CrossRef.
- D. L. Pham, C. Y. Xu and J. L. Prince, Ann. Rev. Biomed. Eng., 2000, 2, 315–337 CrossRef CAS PubMed.
- T. K. Moon, IEEE Signal Processing Magazine, 1996, 13, 47–60 CrossRef.
- M. Kass, A. Witkin and D. Terzopoulos, Int. J. Comput. Vis., 1988, 1, 321–331 CrossRef.
- J. S. Suri, K. Liu, S. Singh, S. N. Laxminarayan, X. Zeng and L. Reden, IEEE Trans. Inform. Technol. Biomed., 2002, 6, 8–28 CrossRef PubMed.
- S. Naik, S. Doyle, S. Agner, A. Madabhushi, M. Feldman and J. Tomaszewski, 2008 5th IEEE International Symposium on Biomedical Imaging: From Nano to Macro, 2008, pp. 284–287.
- M. Alilou, V. Kovalev and V. Taimouri, Comput. Med. Imaging. Graph., 2013, 37, 488–499 CrossRef PubMed.
- I. Jolliffe, Principal component analysis, Springer, New York, 2nd edn, 2002 Search PubMed.
- I. T. Joliffe and B. J. T. Morgan, Stat. Methods Med. Res., 1992, 1, 69–95 CrossRef CAS PubMed.
- A. Hyvärinen, J. Karhunen and E. Oja, Independent component analysis, John Wiley & Sons, New York, 2001, vol. 46 Search PubMed.
- A. M. Martínez and A. C. Kak, IEEE Trans. Pattern Anal. Mach. Intell., 2001, 23, 228–233 CrossRef.
- S. Doyle, M. Hwang, K. Shah, A. Madabhushi, M. Feldman and J. Tomaszeweski, Biomedical imaging: from nano to macro, 2007. ISBI 2007. 4th IEEE international symposium on, 2007, pp. 1284–1287.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- M. Valkonen, K. Kartasalo, K. Liimatainen, M. Nykter, L. Latonen and P. Ruusuvuori, Cytometry, Part A, 2017, 91, 555–565 CrossRef PubMed.
- D. C. Cirean, A. Giusti, L. M. Gambardella and J. Schmidhuber, Medical Image Computing and Computer-Assisted Intervention MICCAI 2013: 16th International Conference, Nagoya, Japan, September 22–26, 2013, Proceedings, Part II, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 411–418.
- T. J. Fuchs and J. M. Buhmann, Comput. Med. Imaging. Graph., 2011, 35, 515–530 CrossRef PubMed.
- D. C. Fernandez, R. Bhargava, S. M. Hewitt and I. W. Levin, Nat. Biotechnol., 2005, 23, 469–474 CrossRef CAS PubMed.
- S. S. Singh, B. Qaqish, J. L. Johnson, O. H. Ford 3rd, J. F. Foley, S. J. Maygarden and J. L. Mohler, Anal. Quant. Cytol. Histol., 2004, 26, 194–200 Search PubMed.
- D. L. Weaver, D. N. Krag, E. A. Manna, T. Ashikaga, S. P. Harlow and K. D. Bauer, Mod. Pathol., 2003, 16, 1159–1163 CrossRef PubMed.
- M. E. Brezinski, G. J. Tearney, B. E. Bouma, J. A. Izatt, M. R. Hee, E. A. Swanson, J. F. Southern and J. G. Fujimoto, Circulation, 1996, 93, 1206 CrossRef CAS PubMed.
- Z. Wang, K. Tangella, A. Balla and G. Popescu, J. Biomed. Opt., 2011, 16, 116017 Search PubMed.
- Y. L. Kim, Y. Liu, R. K. Wali, H. K. Roy, M. J. Goldberg, A. K. Kromin, K. Chen and V. Backman, IEEE J. Sel. Top. Quantum Electron., 2003, 9, 243–256 CrossRef CAS.
- H. Majeed, S. Sridharan, M. Mir, L. Ma, E. Min, J. Opt. Soc. Am. BW. Jung and G. Popescu, J. Biophotonics, 2017, 10, 177–205 CrossRef PubMed.
- N. Lue, J. Bewersdorf, M. D. Lessard, K. Badizadegan, R. R. Dasari, M. S. Feld and G. Popescu, Opt. Lett., 2007, 32, 3522–3524 CrossRef PubMed.
- P. Wang, R. Bista, R. Bhargava, R. E. Brand and Y. Liu, Opt. Lett., 2010, 35, 2840–2842 CrossRef CAS PubMed.
- V. Backman, M. B. Wallace, L. T. Perelman, J. T. Arendt, R. Gurjar, M. G. Müller, Q. Zhang, G. Zonios, E. Kline and T. McGillican, Nature, 2000, 406, 35 CrossRef CAS PubMed.
- Z. Wang, H. Ding and G. Popescu, Opt. Lett., 2011, 36, 1215–1217 CrossRef PubMed.
- R. Richards-Kortum and E. Sevick-Muraca, Annu. Rev. Phys. Chem., 1996, 47, 555–606 CrossRef CAS PubMed.
- R. Alfano, D. Tata, J. Cordero, P. Tomashefsky, F. Longo and M. Alfano, IEEE J. Quantum Electron., 1984, 20, 1507–1511 CrossRef.
- K. T. Schomacker, J. K. Frisoli, C. C. Compton, T. J. Flotte, J. M. Richter, N. S. Nishioka and T. F. Deutsch, Lasers Surg. Med., 1992, 12, 63–78 CrossRef CAS PubMed.
- S. G. Demos, R. Gandour-Edwards, R. Ramsamooj and R. d. White, J. Biomed. Opt., 2004, 9, 587–592 CrossRef PubMed.
- M. Monici, Biotechnol. Annu. Rev., 2005, 11, 227–256 CAS.
- B. Lin, S. Urayama, R. M. Saroufeem, D. L. Matthews and S. G. Demos, Opt. Express, 2010, 18, 21074–21082 CrossRef CAS PubMed.
- K. Kong, C. J. Rowlands, S. Varma, W. Perkins, I. H. Leach, A. A. Koloydenko, H. C. Williams and I. Notingher, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15189–15194 CrossRef CAS PubMed.
- M. Diem, A. Mazur, K. Lenau, J. Schubert, B. Bird, M. Miljković, C. Krafft and J. Popp, J. Biophotonics, 2013, 6, 855–886 CrossRef CAS PubMed.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. A. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protoc., 2014, 9, 1771–1791 CrossRef CAS PubMed.
- Q. Matthews, A. Brolo, J. Lum, X. Duan and A. Jirasek, Phys. Med. Biol., 2011, 56, 19–38 CrossRef CAS PubMed.
- C. Krafft, S. B. Sobottka, G. Schackert and R. Salzer, J. Raman Spectrosc., 2006, 37, 367–375 CrossRef CAS.
- L. A. Austin, S. Osseiran and C. L. Evans, Analyst, 2016, 141, 476–503 RSC.
- K. Eberhardt, C. Stiebing, C. Matthäus, M. Schmitt and J. Popp, Expert Rev. Mol. Diagn., 2015, 15, 773–787 CAS.
- O. Stevens, I. E. Iping Petterson, J. C. C. Day and N. Stone, Chem. Soc. Rev., 2016, 45, 1919–1934 RSC.
- E. Montgomery, M. P. Bronner, J. R. Goldblum, J. K. Greenson, M. M. Haber, J. Hart, L. W. Lamps, G. Y. Lauwers, A. J. Lazenby, D. N. Lewin, M. E. Robert, A. Y. Toledano, Y. Shyr and K. Washington, Hum. Pathol., 2001, 32, 368–378 CrossRef CAS PubMed.
- M. Jermyn, K. Mok, J. Desroches, J. Mercier, K. Saint-Arnaud, L. Bernstein, M.-C. C. Guiot, K. Petrecca and F. Leblond, Biomedical Optics, 2014, Miami, Florida, 2014, p. BS5A.4.
- T. Hollon, S. Lewis, C. W. Freudiger, X. Sunney Xie and D. A. Orringer, Neurosurg. Focus, 2016, 40, E9 CrossRef PubMed.
- R. Manoharan, Y. Wang and M. S. Feld, Spectrochim. Acta, Part A, 1996, 52, 215–249 CrossRef.
- C. Petibois and G. Deleris, Trends Biotechnol., 2006, 24, 455–462 CrossRef CAS PubMed.
- T. Meyer, N. Bergner, C. Bielecki, C. Krafft, D. Akimov, B. F. M. Romeike, R. Reichart, R. Kalff, B. Dietzek and J. Popp, J. Biomed. Opt., 2011, 16, 21110–21113 CrossRef PubMed.
- I. Pence and A. Mahadevan-Jansen, Chem. Soc. Rev., 2016, 45, 1958–1979 RSC.
- D. Shipp, F. Sinjab and I. Notingher, Adv. Opt. Photonics, 2017, 9, 315–428 CrossRef.
- K. Fukuda, Histochemie, 1966, 6, 127–130 CrossRef CAS.
- F. F. M. De Mul, H. Buiteveld, J. Lankester, J. Mud and J. Greve, Hum. Pathol., 1984, 15, 1062–1068 CrossRef CAS PubMed.
- N. Stone, P. Stavroulaki, C. Kendall, M. Birchall and H. Barr, Laryngoscope, 2000, 110, 1756–1763 CrossRef CAS PubMed.
- A. Beljebbar, O. Bouché, M. D. Diébold, P. J. Guillou, J. P. Palot, D. Eudes and M. Manfait, Crit. Rev. Oncol./Hematol., 2009, 72, 255–264 CrossRef CAS PubMed.
- M. Gniadecka, H. C. Wulf, O. F. Nielsen, D. H. Christensen and J. Hercogova, Photochem. Photobiol., 1997, 66, 418–423 CrossRef CAS PubMed.
- S. Koljenović, L.-P. Choo-Smith, T. C. Bakker Schut, J. M. Kros, H. J. van den Berge and G. J. Puppels, Lab. Invest., 2002, 82, 1265–1277 CrossRef.
- S. Rehman, Z. Movasaghi, A. T. Tucker, S. P. Joel, J. A. Darr, A. V. Ruban and I. U. Rehman, J. Raman Spectrosc., 2007, 38, 1345–1351 CrossRef CAS.
- P.-H. H. Chen, R. Shimada, S. Yabumoto, H. Okajima, M. Ando, C.-T. T. Chang, L.-T. T. Lee, Y.-K. K. Wong, A. Chiou and H.-o. O. Hamaguchi, Sci. Rep., 2016, 6, 20097 CrossRef CAS PubMed.
- S. Kaminaka, H. Yamazaki, T. Ito, E. Kohda and H.-o. O. Hamaguchi, J. Raman Spectrosc., 2001, 32, 139–141 CrossRef CAS.
- A. Nijssen, K. Maquelin, L. F. Santos, P. J. Caspers, T. C. Bakker Schut, J. C. den Hollander, M. H. A. Neumann and G. J. Puppels, J. Biomed. Opt., 2007, 12, 034004 CrossRef PubMed.
- A. Mizuno, H. Kitajima, K. Kawauchi, S. Muraishi and Y. Ozaki, J. Raman Spectrosc., 1994, 25, 25–29 CrossRef CAS.
- J. T. Kwak, A. Kajdacsy-Balla, V. Macias, M. Walsh, S. Sinha and R. Bhargava, Sci. Rep., 2015, 5, 8758 CrossRef CAS PubMed.
- R. Manoharan, K. Shafer, L. Perelman, J. Wu, K. Chen, G. Deinum, M. Fitzmaurice, J. Myles, J. Crowe, R. R. Dasarl and M. S. Feld, Photochem. Photobiol., 1998, 67, 15–22 CrossRef CAS PubMed.
- C. H. Liu, B. B. Das, W. L. S. Glassman, G. C. Tang, K. M. Yoo, H. R. Zhu, D. L. Akins, S. S. Lubicz, J. Cleary, R. Prudente, E. Celmer, A. Caron and R. R. Alfano, J. Photochem. Photobiol., B, 1992, 16, 187–209 CrossRef CAS.
- U. Utzinger, D. L. Heintzelman, A. Mahadevan-Jansen, A. Malpica, M. Follen and R. Richards-Kortum, Appl. Spectrosc., 2001, 55, 955–959 CrossRef CAS.
- S. You, H. Tu, Y. Zhao, Y. Liu, E. J. Chaney, M. Marjanovic and S. A. Boppart, Sci. Rep., 2016, 6, 32922 CrossRef CAS PubMed.
- R. E. Kast, G. K. Serhatkulu, A. Cao, A. K. Pandya, H. Dai, J. S. Thakur, V. M. Naik, R. Naik, M. D. Klein, G. W. Auner and R. Rabah, Biopolymers, 2008, 89, 235–241 CrossRef CAS PubMed.
- M. Miljković, T. Chernenko, M. J. Romeo, B. Bird, C. Matthäus and M. Diem, Analyst, 2010, 135, 2002–2013 RSC.
- B. Vajna, G. Patyi, Z. Nagy, A. Bódis, A. Farkas and G. Marosi, J. Raman Spectrosc., 2011, 42, 1977–1986 CrossRef CAS.
- H. J. Butler, L. Ashton, B. Bird, G. Cinque, K. Curtis, K. Esmonde-white, N. J. Fullwood, B. Gardner, P. L. Martin, M. J. Walsh, M. R. Mcainsh, N. Stone, F. L. Martin, H. J. Butler and P. L. Martin-hirsch, Nat. Protoc., 2016, 11, 1–47 Search PubMed.
- F. Masia, A. Glen, P. Stephens, P. Borri and W. Langbein, Anal. Chem., 2013, 85, 10820–10828 CrossRef CAS PubMed.
- J. J. Andrew and T. M. Hancewicz, Appl. Spectrosc., 1998, 52, 797–807 CrossRef CAS.
- I. I. Patel, J. Trevisan, G. Evans, V. Llabjani, P. L. Martin-Hirsch, H. F. Stringfellow and F. L. Martin, Analyst, 2011, 136, 4950–4959 RSC.
- D. Zhang, P. Wang, M. N. Slipchenko, D. Ben-Amotz, A. M. Weiner and J. X. Cheng, Anal. Chem., 2013, 85, 98–106 CrossRef CAS PubMed.
- A. Alfonso-García, J. Paugh, M. Farid, S. Garg, J. Jester and E. Potma, J. Raman Spectrosc., 2017, 803–812 CrossRef PubMed.
- A. J. Berger, T. W. Koo, I. Itzkan, G. Horowitz and M. S. Feld, Appl. Opt., 1999, 38, 2916–2926 CrossRef CAS PubMed.
- M. S. Bergholt, W. Zheng, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Yan So, A. Shabbir and Z. Huang, Gastroenterology, 2014, 146, 27–32 CrossRef PubMed.
- E. Widjaja, W. Zheng and Z. Huang, Int. J. Oncol., 2008, 32, 653–662 CAS.
- P. Rösch, M. Harz, M. Schmitt, O. Ronneberger, H. Burkhardt, M. Lankers, S. Hofer, H. Thiele, P. Ro, K.-d. D. Peschke and H.-w. W. Motzkus, Appl. Environ. Microbiol., 2005, 71, 1626–1637 CrossRef PubMed.
- S. K. Teh, W. Zheng, D. P. Lau and Z. Huang, Analyst, 2009, 134, 1232 RSC.
- A. Kallenbach-Thieltges, F. Großerüschkamp, A. Mosig, M. Diem, A. Tannapfel and K. Gerwert, J. Biophotonics, 2013, 6, 88–100 CrossRef PubMed.
- O. J. Old, G. R. Lloyd, J. Nallala, M. Isabelle, L. M. Almond, N. A. Shepherd, C. A. Kendall, A. C. Shore, H. Barr and N. Stone, Analyst, 2016, 1227–1234 Search PubMed.
- A. J. Berger, T.-w. Koo, I. Itzkan and M. S. Feld, Anal. Chem., 1998, 70, 623–627 CrossRef CAS PubMed.
- G. R. Lloyd, L. M. Almond, N. Stone, N. Shepherd, S. Sanders, J. Hutchings, H. Barr and C. Kendall, Analyst, 2014, 139, 381–388 RSC.
- Y. LeCun, B. Yoshua and H. Geoffrey, Nature, 2015, 521, 436–444 CrossRef CAS PubMed.
- M. Gniadecka, H. C. Wulf, N. N. Mortensen, O. F. Nielsen and D. H. Christensen, J. Raman Spectrosc., 1997, 28, 125–129 CrossRef CAS.
- R. Goodacre, E. M. Timmins, R. Burton, N. Kaderbhai, A. M. Woodward, D. B. Kell and P. J. Rooney, Microbiology, 1998, 144, 1157–1170 CrossRef CAS PubMed.
- P. Manescu, Y. Jong Lee, C. Camp, M. Cicerone, M. Brady and P. Bajcsy, Med. Image Anal., 2017, 37, 37–45 CrossRef PubMed.
- M. J. Pilling, A. Henderson, J. H. Shanks, M. D. Brown, N. W. Clarke and P. Gardner, Analyst, 2016, 1258–1268 Search PubMed.
- J. T. Kwak, S. M. Hewitt, S. Sinha and R. Bhargava, BMC Cancer, 2011, 11, 62 CrossRef PubMed.
- J. W. Chan, D. K. Lieu, T. Huser and R. A. Li, Anal. Chem., 2009, 81, 1324–1331 CrossRef CAS PubMed.
- C. Matthäus, S. Boydston-White, M. Miljković, M. Romeo and M. Diem, Appl. Spectrosc., 2006, 60, 1–8 CrossRef PubMed.
- M. Okada, N. I. Smith, A. F. Palonpon, H. Endo, S. Kawata, M. Sodeoka and K. Fujita, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 28–32 CrossRef CAS PubMed.
- H. G. Schulze, S. O. Konorov, J. M. Piret, M. W. Blades and R. F. Turner, Analyst, 2013, 138, 3416–3423 RSC.
- S. Boydston-White, T. Chernenko, A. Regina, M. S. Miljković, C. Matthäus and M. Diem, Vib. Spectrosc., 2005, 38, 169–177 CrossRef CAS.
- S. Boydston-White, T. Gopen, S. Houser, J. Bargonetti and M. Diem, Biospectroscopy, 1999, 5, 219–227 CrossRef CAS PubMed.
- J. W. Chan, D. S. Taylor, S. M. Lane, T. Zwerdling, J. Tuscano and T. Huser, Anal. Chem., 2008, 80, 2180–2187 CrossRef CAS PubMed.
- S. Tiwari, J. Raman, V. Reddy, A. Ghetler, R. P. Tella, Y. Han, C. R. Moon, C. D. Hoke and R. Bhargava, Anal. Chem., 2016, 88, 10183–10190 CrossRef CAS PubMed.
- C. Krafft, M. Schmitt, I. W. Schie, D. Cialla-May, C. Matthäus, T. Bocklitz and J. Popp, Angew. Chem., Int. Ed., 2016, 4392–4430 Search PubMed.
- J. A. Armstrong, N. Bloembergen, J. Ducuing and P. S. Pershan, Phys. Rev., 1962, 127, 1918–1939 CrossRef CAS.
- P. D. Maker and R. W. Terhune, Phys. Rev., 1965, 137, A801–A818 CrossRef.
- M. D. Duncan, J. Reintjes and T. J. Manuccia, Opt. Lett., 1982, 7, 350–352 CrossRef CAS PubMed.
- A. Volkmer, J. Phys. D: Appl. Phys., 2005, 38, R59 CrossRef CAS.
- C. L. Evans and X. S. Xie, Annu. Rev. Anal. Chem., 2008, 1, 883–909 CrossRef CAS PubMed.
- J. P. Pezacki, J. A. Blake, D. C. Danielson, D. C. Kennedy, R. K. Lyn and R. Singaravelu, Nat. Chem. Biol., 2011, 7, 137–145 CrossRef CAS PubMed.
- S. Yue, J. Cárdenas-Mora, L. Chaboub, S. Lelièvre and J.-X. X. Cheng, Biophys. J., 2012, 102, 1215–1223 CrossRef CAS PubMed.
- C. H. Camp Jr. and M. T. Cicerone, Nat. Photonics, 2015, 9, 295–305 CrossRef.
- E. O. Potma and S. Mukamel, in Theory of Coherent Raman Scattering, CRC Press, Boca Raton, FL, 2013 Search PubMed.
- W. M. Tolles, J. W. Nibler, J. R. McDonald and A. B. Harvey, Appl. Spectrosc., 1977, 31, 253–271 CrossRef CAS.
- M. Cui, B. R. Bachler and J. P. Ogilvie, Opt. Lett., 2009, 34, 773–775 CrossRef PubMed.
- K. I. Popov, A. F. Pegoraro, A. Stolow and L. Ramunno, Opt. Express, 2011, 19, 5902–5911 CrossRef CAS PubMed.
- A. M. Barlow, K. Popov, M. Andreana, D. J. Moffatt, A. Ridsdale, A. D. Slepkov, J. L. Harden, L. Ramunno and A. Stolow, Opt. Express, 2013, 21, 15298–15307 CrossRef CAS PubMed.
- J. X. Chen, A. Volkmer, L. D. Book and X. S. Xie, J. Phys. Chem. B, 2002, 106, 8493–8498 CrossRef.
- D. L. Marks and S. A. Boppart, Phys. Rev. Lett., 2004, 92, 123905 CrossRef PubMed.
- T. W. Kee and M. T. Cicerone, Opt. Lett., 2004, 29, 2701–2703 CrossRef CAS PubMed.
- H. Kano and H. Hamaguchi, Appl. Phys. Lett., 2004, 85, 4298–4300 CrossRef CAS.
- T. Hellerer, A. M. Enejder and A. Zumbusch, Appl. Phys. Lett., 2004, 85, 25–27 CrossRef CAS.
- T. Meyer, M. Chemnitz, M. Baumgartl, T. Gottschall, T. Pascher, C. Matthäus, B. F. M. Romeike, B. R. Brehm, J. Limpert, A. Tünnermann, M. Schmitt, B. Dietzek and J. Popp, Anal. Chem., 2013, 85, 6703–6715 CrossRef CAS PubMed.
- Y.-M. M. Wu, H.-C. C. Chen, W.-T. T. Chang, J.-W. W. Jhan, H.-L. L. Lin and I. Liau, Anal. Chem., 2009, 81, 1496–1504 CrossRef CAS PubMed.
- C. L. Evans, E. O. Potma, M. Puoris’ haag, D. Côté, C. P. Lin and X. S. Xie, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16807–16812 CrossRef CAS PubMed.
- I. Veilleux, J. A. Spencer, D. P. Biss, D. Cote and C. P. Lin, IEEE J. Sel. Top. Quantum Electron., 2008, 14, 10–18 CrossRef CAS.
- E. Y. Lin, J. G. Jones, P. Li, L. Zhu, K. D. Whitney, W. J. Muller and J. W. Pollard, Am. J. Pathol., 2003, 163, 2113–2126 CrossRef PubMed.
- S. Bégin, B. Burgoyne, V. Mercier, A. Villeneuve, R. Vallée and D. Côté, Biomed. Opt. Express, 2011, 2, 1296–1306 CrossRef PubMed.
- A. F. Pegoraro, A. Ridsdale, D. J. Moffatt, J. P. Pezacki, B. K. Thomas, L. B. Fu, L. Dong, M. E. Fermann and A. Stolow, Opt. Express, 2009, 17, 20700–20706 CAS.
- E. M. Vartiainen, J. Opt. Soc. Am. B, 1992, 9, 1209–1214 CrossRef CAS.
- Y. X. Liu, Y. J. Lee and M. T. Cicerone, Opt. Lett., 2009, 34, 1363–1365 CrossRef PubMed.
- C. H. Camp, Y. J. Lee and M. T. Cicerone, J. Raman Spectrosc., 2016, 47, 408–415 CrossRef CAS PubMed.
- M. Muller and J. M. Schins, J. Phys. Chem. B, 2002, 106, 3715–3723 CrossRef.
- W. A. Benalcazar, P. D. Chowdary, Z. Jiang, D. L. Marks, E. J. Chaney, M. Gruebele and S. A. Boppart, IEEE J. Sel. Top. Quant., 2010, 16, 824–832 CrossRef CAS PubMed.
- C. L. Evans, X. Xu, S. Kesari, X. S. Xie, S. T. Wong and G. S. Young, Opt. Express, 2007, 15, 12076–12087 CAS.
- M. Lee, A. Downes, Y.-Y. Y. Chau, B. Serrels, N. Hastie, A. Elfick, V. Brunton, M. Frame and A. Serrels, IntraVital, 2015, e1055430 CrossRef PubMed.
- T. Meyer, N. Bergner, A. Medyukhina, B. Dietzek, C. Krafft, B. F. M. Romeike, R. Reichart, R. Kalff and J. Popp, J. Biophotonics, 2012, 5, 729–733 CrossRef CAS PubMed.
- L. Gao, F. Li, M. J. Thrall, Y. Yang, J. Xing, A. A. Hammoudi, H. Zhao, Y. Massoud, P. T. Cagle and Y. Fan, J. Biomed. Opt., 2011, 16, 096004–096004 CrossRef PubMed.
- L. Gao, Z. Wang, F. Li, A. A. Hammoudi, M. J. Thrall, P. T. Cagle and S. T. Wong, Arch. Pathol. Lab. Med., 2012, 136, 1502–1510 CrossRef CAS PubMed.
- Y. Yang, F. Li, L. Gao, Z. Wang, M. J. Thrall, S. S. Shen, K. K. Wong and S. T. Wong, Biomed. Opt. Express, 2011, 2, 2160–2174 CrossRef PubMed.
- O. Uckermann, R. Galli, S. Tamosaityte, E. Leipnitz, K. D. Geiger, G. Schackert, E. Koch, G. Steiner and M. Kirsch, PLoS One, 2014, 9, e107115 Search PubMed.
- T. T. Le, T. B. Huff and J. X. Cheng, BMC Cancer, 2009, 9, 42 CrossRef PubMed.
- R. Mitra, O. Chao, Y. Urasaki, O. B. Goodman and T. T. Le, BMC Cancer, 2012, 12, 540 CrossRef CAS PubMed.
- P. D. Chowdary, Z. Jiang, E. J. Chaney, W. A. Benalcazar, D. L. Marks, M. Gruebele and S. A. Boppart, Cancer Res., 2010, 70, 9562–9569 CrossRef CAS PubMed.
- S. Heuke, N. Vogler, T. Meyer, D. Akimov, F. Kluschke, H.-J. J. Röwert-Huber, J. Lademann, B. Dietzek and J. Popp, Br. J. Dermatol., 2013, 169, 794–803 CrossRef CAS PubMed.
- C. Krafft, A. A. Ramoji, C. Bielecki, N. Vogler, T. Meyer, D. Akimov, P. Rösch, M. Schmitt, B. Dietzek, I. Petersen, A. Stallmach and J. Popp, J. Biophoton., 2009, 2, 303–312 CrossRef PubMed.
- M. T. Cicerone, K. A. Aamer, Y. J. Lee and E. Vartiainen, J. Raman Spectrosc., 2012, 43, 637–643 CrossRef CAS.
- A. Rajwade, A. Rangarajan and A. Banerjee, IEEE Trans. Pattern Anal. Mach. Intell., 2013, 35, 849–862 CrossRef PubMed.
- A. F. Pegoraro, A. D. Slepkov, A. Ridsdale, D. J. Moffatt and A. Stolow, J. Biophotonics, 2012, 49–58 Search PubMed.
- S. H. Parekh, Y. J. Lee, K. A. Aamer and M. T. Cicerone, Biophys. J., 2010, 99, 2695–2704 CrossRef CAS PubMed.
- M. Okuno, H. Kano, P. Leproux, V. Couderc, J. P. R. Day, M. Bonn and H.-o. O. Hamaguchi, Angew. Chem., Int. Ed., 2010, 49, 6773–6777 CrossRef CAS PubMed.
- C. H. Camp Jr., Y. J. Lee, J. M. Heddleston, C. M. Hartshorn, W. R. Hight, J. N. Rich, J. D. Lathia and M. T. Cicerone, Nat. Photonics, 2014, 8, 627–634 CrossRef CAS PubMed.
- C. Pohling, T. Buckup, A. Pagenstecher and M. Motzkus, Biomed. Opt. Express, 2011, 2, 2110–2116 CrossRef PubMed.
- Y. J. Lee, Y. Liu and M. T. Cicerone, Opt. Lett., 2007, 32, 3370–3372 CrossRef CAS PubMed.
- H. Wang, S. Osseiran, V. Igras, A. J. Nichols, E. M. Roider, J. Pruessner, H. Tsao, D. E. Fisher and C. L. Evans, Sci. Rep., 2016, 6, 37986 CrossRef CAS PubMed.
- D. A. Orringer, B. Pandian, Y. S. Niknafs, T. C. Hollon, J. Boyle, S. Lewis, M. Garrard, S. L. Hervey-Jumper, H. J. L. Garton, C. O. Maher, J. A. Heth, O. Sagher, D. A. Wilkinson, M. Snuderl, S. Venneti, S. H. Ramkissoon, K. A. McFadden, A. Fisher-Hubbard, A. P. Lieberman, T. D. Johnson, X. S. Xie, J. K. Trautman, C. W. Freudiger and S. Camelo-Piragua, Nat. Biomed. Eng., 2017, 1, 0027 CrossRef PubMed.
- Y. Otsuka, S. Satoh, M. Kyogaku, H. Hashimoto, K. Itoh and Y. Ozeki, SPIE BiOS, 2014, pp. 89470C–89470C.
- Y. Ozeki, F. Dake, S. Kajiyama, K. Fukui and K. Itoh, Opt. Express, 2009, 17, 3651–3658 CrossRef CAS PubMed.
- B. G. Saar, C. W. Freudiger, J. Reichman, C. M. Stanley, G. R. Holtom and X. S. Xie, Science, 2010, 330, 1368–1370 CrossRef CAS PubMed.
- C. W. Freudiger, R. Pfannl, D. A. Orringer, B. G. Saar, M. Ji, Q. Zeng, L. Ottoboni, W. Ying, C. Waeber, J. R. Sims, P. L. De Jager, O. Sagher, M. A. Philbert, X. Xu, S. Kesari, X. S. Xie and G. S. Young, Lab. Invest., 2012, 92, 1492–1502 CrossRef CAS PubMed.
- Y. Ozeki, W. Umemura, Y. Otsuka, S. Satoh, H. Hashimoto, K. Sumimura, N. Nishizawa, K. Fukui and K. Itoh, Nat. Photonics, 2012, 6, 845–851 CrossRef CAS.
- P. Berto, E. R. Andresen and H. Rigneault, Phys. Rev. Lett., 2014, 112, 053905 CrossRef PubMed.
- D. Fu, W. Yang and X. S. Xie, J. Am. Chem. Soc., 2017, 139, 583–586 CrossRef CAS PubMed.
- C. W. Freudiger, W. Yang, G. R. Holtom, N. Peyghambarian, X. S. Xie and K. Q. Kieu, Nat. Photonics, 2014, 8, 153–159 CrossRef CAS PubMed.
- P. Nandakumar, A. Kovalev and A. Volkmer, New J. Phys., 2009, 11, 033026 CrossRef.
- C. W. Freudiger, W. Min, B. G. Saar, S. Lu, G. R. Holtom, C. W. He, J. C. Tsai, J. X. Kang and X. S. Xie, Science, 2008, 322, 1857–1861 CrossRef CAS PubMed.
- D. Fu, G. Holtom, C. Freudiger, X. Zhang and X. S. Xie, J. Phys. Chem. B, 2013, 117, 4634–4640 CrossRef CAS PubMed.
- M. S. Alshaykh, C.-S. S. Liao, O. E. Sandoval, G. Gitzinger, N. Forget, D. E. Leaird, J.-X. X. Cheng and A. M. Weiner, Opt. Lett., 2017, 42, 1548–1551 Search PubMed.
- C.-S. S. Liao, M. N. Slipchenko, P. Wang, J. Li, S.-Y. Y. Lee, R. A. Oglesbee and J.-X. X. Cheng, Light: Sci. Appl., 2015, 4, e265 CrossRef CAS PubMed.
- R. Mittal, M. Balu, T. Krasieva, E. O. Potma, L. Elkeeb, C. B. Zachary and P. WilderSmith, Lasers Surg. Med., 2013, 45, 496–502 Search PubMed.
- F.-K. K. Lu, D. Calligaris, O. I. Olubiyi, I. Norton, W. Yang, S. Santagata, X. S. Xie, A. J. Golby and N. Y. R. Agar, Cancer Res., 2016, 3451–3462 CrossRef CAS PubMed.
- M. Ji, S. Lewis, S. Camelo-Piragua, S. H. Ramkissoon, M. Snuderl, S. Venneti, A. Fisher-Hubbard, M. Garrard, D. Fu, A. C. Wang, J. A. Heth, C. O. Maher, N. Sanai, T. D. Johnson, C. W. Freudiger, O. Sagher, X. S. Xie and D. A. Orringer, Sci. Transl. Med., 2015, 7, 309ra163 CrossRef PubMed.
- S. Cui, P. Wang and S. Yue, Proc. SPIE, 2017, 100690M-1 Search PubMed.
- M. Egawa, K. Tokunaga, J. Hosoi, S. Iwanaga and Y. Ozeki, J. Biomed. Opt., 2016, 21, 86017 Search PubMed.
- R. Galli, O. Uckermann, A. Temme, E. Leipnitz, M. Meinhardt, E. Koch, G. Schackert, G. Steiner and M. Kirsch, J. Biophotonics, 2016, 404–414 Search PubMed.
- S. Yue, J. Li, S.-Y. Y. Lee, H. Lee, T. Shao, B. Song, L. Cheng, T. Masterson, X. Liu, T. Ratliff and J.-X. X. Cheng, Cell Metab., 2014, 19, 393–406 CrossRef CAS PubMed.
- X. Zhang, M. B. J. Roeffaers, S. Basu, J. R. Daniele, D. Fu, C. W. Freudiger, G. R. Holtom and X. S. Xie, ChemPhysChem, 2012, 13, 1054–1059 CrossRef CAS PubMed.
- Z. Wang, W. Zheng, J. Lin and Z. Huang, Proc. SPIE, 2016, 9712, 97120G Search PubMed.
- A. L. Tam, H. J. Lim, I. I. Wistuba, A. Tamrazi, M. D. Kuo, E. Ziv, S. Wong, A. J. Shih, R. J. Webster, G. S. Fischer, S. Nagrath, S. E. Davis, S. B. White and K. Ahrar, J. Vasc. Interv. Radiol., 2015, 27, 8–19 CrossRef PubMed.
- C. J. Frank, R. L. McCreery and D. C. Redd, Anal. Chem., 1995, 67, 777–783 CrossRef CAS PubMed.
- M. Balu, G. Liu, Z. Chen, B. J. Tromberg and E. O. Potma, Opt. Express, 2010, 18, 2380–2388 CrossRef CAS PubMed.
- B. G. Saar, R. S. Johnston, C. W. Freudiger, X. S. Xie and E. J. Seibel, Opt. Lett., 2011, 36, 2396–2398 CrossRef PubMed.
- A. A. Hammoudi, K. K. Wong, L. Gao, P. Luo, S. T. C. Wong, Y. Yang and Z. Wang, Opt. Express, 2011, 19, 7960–7970 CrossRef PubMed.
- S. Heuke, N. Vogler, T. Meyer, D. Akimov, F. Kluschke, H.-J. J. Röwert-Huber, J. Lademann, B. Dietzek and J. Popp, Healthcare, 2013, 1, 64–83 CrossRef PubMed.
- C.-S. Liao and J.-X. Cheng, Sci. Adv., 2015, E1500738 Search PubMed.
- B. Spivack, M. M. Khanna, L. Tafra, G. Juillard and A. E. Giuliano, Arch. Surg., 1994, 129, 952–957 CrossRef CAS PubMed.
- M. Lacroix, D. Abi-Said, D. R. Fourney, Z. L. Gokaslan, W. Shi, F. DeMonte, F. F. Lang, I. E. McCutcheon, S. J. Hassenbusch, E. Holland, K. Hess, C. Michael, D. Miller and R. Sawaya, J. Neurosurg., 2001, 95, 190–198 CrossRef CAS PubMed.
- M. Jermyn, K. Mok, J. Mercier, J. Desroches, J. Pichette, K. Saint-Arnaud, L. Bernstein, M.-C. C. Guiot, K. Petrecca and F. Leblond, Sci. Transl. Med., 2015, 7, 274ra19 CrossRef CAS PubMed.
- K. Kong, F. Zaabar, E. Rakha, I. Ellis, A. Koloydenko and I. Notingher, Phys. Med. Biol., 2014, 59, 6141 CrossRef PubMed.
- M. G. Shim and B. C. Wilson, Photochem. Photobiol., 1996, 63, 662–671 CrossRef CAS PubMed.
- R. Galli, O. Uckermann, E. Koch, G. Schackert, M. Kirsch and G. Steiner, J. Biomed. Opt., 2013, 19, 071402 CrossRef PubMed.
- K. Klein, A. Gigler, T. Aschenbrenner, R. Monetti, W. Bunk, F. Jamitzky, G. Morfill, R. Stark and J. Schlegel, Biophys. J., 2012, 102, 360–368 CrossRef CAS PubMed.
- S. Gröschel, M. Bommer, B. Hutter, J. Budczies, D. Bonekamp, C. Heining, P. Horak, M. Fröhlich, S. Uhrig, D. Hübschmann, C. Geörg, D. Richter, N. Pfarr, K. Pfütze, S. Wolf, P. Schirmacher, D. Jäger, C. von Kalle, B. Brors, H. Glimm, W. Weichert, A. Stenzinger and S. Fröhling, Cold Spring Harbor Mol. Case Stud., 2016, 2, a001180 CrossRef PubMed.
- G. Mountzios, M.-A. A. Dimopoulos, J.-C. C. Soria, D. Sanoudou and C. A. Papadimitriou, Crit. Rev. Oncol./Hematol., 2010, 75, 94–109 CrossRef PubMed.
- N. Navin, A. Krasnitz, L. Rodgers, K. Cook, J. Meth, J. Kendall, M. Riggs, Y. Eberling, J. Troge and V. Grubor, Genome Res., 2010, 20, 68–80 CrossRef CAS PubMed.
- C. Curtis, S. P. Shah, S.-F. F. Chin, G. Turashvili, O. M. Rueda, M. J. Dunning, D. Speed, A. G. Lynch, S. Samarajiwa, Y. Yuan, S. Gräf, G. Ha, G. Haffari, A. Bashashati, R. Russell, S. McKinney, M. Group, A. Langerød, A. Green, E. Provenzano, G. Wishart, S. Pinder, P. Watson, F. Markowetz, L. Murphy, I. Ellis, A. Purushotham, A.-L. L. Børresen-Dale, J. D. Brenton, S. Tavaré, C. Caldas and S. Aparicio, Nature, 2012, 486, 346 CAS.
- R. Natrajan, H. Sailem, F. K. Mardakheh, M. A. Garcia, C. J. Tape, M. Dowsett, C. Bakal and Y. Yuan, PLoS Med., 2016, 13, e1001961 Search PubMed.
- T. Ideguchi, S. Holzner, B. Bernhardt, G. Guelachvili, N. Picque and T. W. Hansch, Nature, 2013, 502, 355–358 CrossRef CAS PubMed.
- K. Hashimoto, M. Takahashi, T. Ideguchi and K. Goda, Sci. Rep., 2016, 6, 21036 CrossRef CAS PubMed.
- E. Ploetz, S. Laimgruber, S. Berner, W. Zinth and P. Gilch, Appl. Phys. B: Lasers Opt., 2007, 87, 389–393 CrossRef CAS.
- W. Rock, M. Bonn and S. H. Parekh, Opt. Express, 2013, 21, 15113 CrossRef CAS PubMed.
- K. Shi, H. Li, Q. Xu, D. Psaltis and Z. Liu, Phys. Rev. Lett., 2010, 104, 093902 CrossRef PubMed.
- C. Heinrich, A. Hofer, A. Ritsch, C. Ciardi, S. Bernet and M. Ritsch-Marte, Opt. Express, 2008, 16, 2699–2708 CrossRef CAS PubMed.
- I. Toytman, K. Cohn, T. Smith, D. Simanovskii and D. Palanker, Opt. Lett., 2007, 32, 1941–1943 CrossRef CAS PubMed.
- M. Lei, M. Winterhalder, R. Selm and A. Zumbusch, J. Biomed. Opt., 2011, 16, 021102 Search PubMed.
- P. Berto, D. Gachet, P. Bon, S. Monneret and H. Rigneault, Phys. Rev. Lett., 2012, 109, 093902 CrossRef PubMed.
- M. N. Slipchenko, R. A. Oglesbee, D. Zhang, W. Wu and J.-X. X. Cheng, J. Biophotonics, 2012, 801–807 CrossRef PubMed.
Footnote |
| † Official contribution of the National Institute of Standards and Technology; not subject to copyright in the United States. |
| This journal is © The Royal Society of Chemistry 2018 |