
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of sulfidation of small zinc oxide nanoparticles
Progna Banerjee a and
Prashant K. Jain*abcd
a and
Prashant K. Jain*abcd
aDepartment of Physics, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: jain@illinois.edu
bDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
cMaterials Research Laboratory, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
dBeckman Institute of Advanced Science and Technology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
First published on 8th October 2018
Abstract
ZnO has industrial utility as a solid sorbent for the removal of polluting sulfur compounds from petroleum-based fuels. Small ZnO nanoparticles may be more effective in terms of sorption capacity and ease of sulfidation as compared to bulk ZnO. Motivated by this promise, here, we study the sulfidation of ZnO NPs and uncover the solid-state mechanism of the process by crystallographic and optical absorbance characterization. The wurtzite-structure ZnO NPs undergo complete sulfidation to yield ZnS NPs with a drastically different zincblende structure. However, in the early stages, the ZnO NP lattice undergoes only substitutional doping by sulfur, while retaining its wurtzite structure. Above a threshold sulfur-doping level of 30 mol%, separate zincblende ZnS grains nucleate, which grow at the expense of the ZnO NPs, finally yielding ZnS NPs. Thus, the full oxide to sulfide transformation cannot be viewed simply as a topotactic place-exchange of anions. The product ZnS NPs formed by nucleation-growth share neither the crystallographic structure nor the size of the initial ZnO NPs. The reaction mechanism may inform the future design of nanostructured ZnO sorbents.
Zinc oxide (ZnO) nanoparticles (NPs), due to their cost-effectiveness and biodegradability, have a multitude of applications1–3 including coatings4–8 and pigments,9,10 catalysis,11,12 energy storage,13,14 and environmental remediation.15–22 ZnO NPs have particular appeal as sorbents for scavenging polluting sulfur compounds such as mercaptans and hydrogen sulfide (H2S) from petroleum-based fuels:23–27 ZnO + H2S → ZnS + H2O. Lattice O2− in the ZnO is replaced with S2− scavenged from the pollutant. Bulk powders of ZnO have already been used for adsorptive removal of H2S,28,29 but NPs have specific advantages. With smaller grain sizes, mass transport limitations are lifted.23 Whereas sulfidation is limited to the surface of bulk ZnO, with NPs, the entire mass of ZnO can undergo sulfidation, enabling high sorbent capacity.23 Volume and morphology changes resulting from restructuring of the solid can also be more easily accommodated with NPs,23 allowing regenerable use of the sorbent. Finally, the high specific surface area of NPs allows more enhanced kinetics of the sulfidation reaction, potentially facilitating much lower desulfurization temperatures as compared to the conventional operating temperatures of 650–800 °C.23,29
In this context, small few-nm size ZnO NPs can be expected to be particularly promising, but it is important to understand the manner in which these NPs undergo sulfidation. The structural mechanism of the sulfidation process30 may have critical differences compared to bulk ZnO powders or even larger NPs of tens of nm in size24 and may therefore influence sorbent design. In a seminal study, Park et al.30 studied the sulfidation of hexagonal-shaped 14 nm ZnO nanocrystals (NCs) at high temperature (235 °C) using hexamethyldisilathiane. The reaction was found to involve the anion exchange of O2− with S2− in the NC lattice. The overall shape and crystallography of ZnS NCs was templated by the initial ZnO NCs. However, due to the faster outward diffusion of Zn2+ as compared to the inward diffusion of S2−, the exchange reaction was accompanied by a nanoscale Kirkendall phenomenon, as a result of which the ZnS NCs formed were hollow.
Here, we track the step-wise sulfidation of smaller (ca. 5 nm) ZnO NPs using optical spectroscopy and X-ray crystallography. Prior to the onset of sulfidation, O2− in wurtzite ZnO NPs undergoes substitutional doping with S2− without any major change in its structure. Upon reaching a critical concentration of sulfur doping, separate zincblende ZnS grains form and grow into ZnS NPs. Thus, the sulfidation of these small ZnO NPs studied here is not simply a topotactic or templated place-exchange of anions; rather the nucleation and growth of a separate ZnS crystallite is involved in the latter stages.
Experimental methods
Synthesis of ZnO NPs
The synthesis of 4 nm ZnO NPs was based on a procedure from Kamat and coworkers31 with some changes. Briefly, 0.05 M of zinc acetate dihydrate (1.095 g) was added to 50 mL of ethanol in a 100 mL three-necked round-bottom flask. The flask was put under Ar, heated to 80 °C, and refluxed for 2 h followed by gradual cooling down to room-temperature over the course of 1.5 h. The zinc precursor solution (0.05 M) was stored in a desiccator and was used for a period of up to three months from the preparation date. Whenever needed, a batch of ZnO NP colloid was prepared by the hydrolysis of the zinc precursor using lithium hydroxide (LiOH) powder. Typically, 20 mg of LiOH was added directly to 3 mL of the precursor solution diluted with 12 mL of ethanol. The resulting reaction mixture was sonicated for 20 min without heating. The resulting colloid solution was washed by adding an equal volume of acetone and centrifuging at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min. The precipitated colloid was redispersed in 25 mL of ethanol. The colloid was allowed to stand for 5 h or more to allow NP growth to stabilize before sulfidation studies or other measurements. The ZnO colloid was typically used up to a week from its preparation.
000 rpm for 20 min. The precipitated colloid was redispersed in 25 mL of ethanol. The colloid was allowed to stand for 5 h or more to allow NP growth to stabilize before sulfidation studies or other measurements. The ZnO colloid was typically used up to a week from its preparation.
Sulfidation of ZnO NPs
The conversion from ZnO to ZnS NPs was carried out at a temperature of 45 °C. We started with a colloid of ZnO NPs dispersed in ethanol (at a concentration which yields an absorbance of ∼1 at the ZnO excitonic peak maximum). The colloid was transferred to a 20 mL glass vial immersed in a water bath set at 45 °C. To this colloid, a solution of sodium sulfide (Na2S) in ethanol (3.1 mg mL−1) was added in aliquots of 10 μL with vigorous stirring. After each addition, the reaction was allowed to approach completion by waiting a duration of 40 min, after which ∼3.5 mL of the colloid was transferred to a quartz spectrophotometry cuvette and a UV-vis extinction spectrum in the range of 220–600 nm was measured. After the measurement, the colloid was returned to the reaction vial. The reaction appeared to reach saturation upon the addition of 11.9 μmol of S2−, at which point no further change in the excitonic features in the absorbance spectrum were observed.At various stages of sulfidation, the NPs were subject to other characterization described below. For this, the colloid subject was subject to sulfidation in steps, as described above, up to a specific stage identified by the total molar amount of S2− added. The colloid was then purified from excess reagents and salts by washing with acetone and centrifugation at 10000 rpm for 20 min. The precipitated colloid was redispersed in ethanol for further characterization.
pXRD
Powder X-ray diffraction (pXRD) patterns were collected for NPs at multiple stages of sulfidation. The diffraction measurements were conducted on a Rigaku Miniflex 600 powder X-ray diffractometer operated at full power (40 kV to 15 mA) with Cu Kα radiation wavelength (1.54 Å). Data was collected in reflection mode in the 2θ range of 20°–75° using a step size of 0.04°, with scan times lasting for 2–3 h. pXRD samples were prepared by drop-casting NPs from a colloid dispersed in ethanol into a thick film on a zero-background quartz substrate. Before each run, the quartz substrate was cleaned by rinsing in aqua regia and DI water. Diffraction patterns were plotted in Origin Lab software and manual baseline subtraction was performed for each of the patterns.Elemental analysis
The elemental composition of the NPs, specifically the molar ratio of S:Zn, was determined at three stages: the initial ZnO NPs before sulfidation, the intermediate stage obtained by the addition of 1.99 μmol of S2−, and the final ZnS NPs achieved by the addition of 11.9 μmol of S2− to the ZnO NPs. These samples were each purified of reagents and salts twice by washing with acetone and centrifugation. The washed precipitate was subsequently dried in quartz digestion tubes by flowing N2 gas. Samples were subject to inductively-coupled plasma atomic emission spectroscopy (ICP-AES) measurements, the results of which are presented in Table 1.
| Stage | S/Zn, molar ratio |
|---|---|
| Initial (ZnO) | 0 |
| Intermediate (1.99 μmol of S2− added) | 0.27 |
| Final (ZnS) | 1.06 |
Electron microscopy
The initial ZnO NPs and final ZnS NPs were subject to TEM imaging. For ZnO NPs, 6 mL of ZnO colloid prepared as described above was treated with 0.3 mL of oleic acid and washed with 6 mL of acetone followed by centrifugation at 10000 rpm for 20 min. Then the precipitated colloid was dispersed in ethanol. About 50 μL of the diluted ZnO NP colloid was drop cast onto an ultrathin carbon grid, which was then allowed to dry in a vacuum desiccator. ZnS NPs were prepared by complete sulfidation of 6 mL of ZnO colloid accomplished, as described above, by addition of S2−. The ZnS NP colloid was treated with 0.3 mL of oleic acid and washed with 6 mL of acetone followed by centrifugation at 10000 rpm for 20 min. Then the precipitated colloid was dispersed in ethanol. The TEM grid, held by tweezers was dipped into the colloid once and then allowed to dry in a vacuum desiccator. TEM imaging was performed on a JEOL 2010-EFFEG instrument operating at 200 kV. NP size analysis was performed using the Fiji software. The diameter of each NP identified in a representative image was determined manually along three arbitrary directions across the NP and averaged.
Structure factor simulations
For the initial ZnO and the early-stage sulfur-doped ZnO NCs, pXRD patterns were simulated using the program PowderCell. The program performs a structure factor calculation using crystal symmetry, lattice parameters, atomic positions, site occupancies and Debye–Scherer broadening as input parameters. Input parameters for each simulated pattern are presented in Table 2. The X-ray wavelength was set to 1.54 Å corresponding to Cu Kα1 radiation. The starting model was ZnO in a wurtzite lattice, characterized by the space group P63mmc. The O and the Zn atoms were placed in the 2b Wycoff positions. The lattice parameters were set to 3.247 Å and 5.179 Å along the a and the c crystallographic directions, respectively, which provided the closest fit to the peak positions of the experimentally measured pXRD reflections of the wurtzite ZnO NPs. These lattice parameters, while close to the known parameters of bulk ZnO (a = 3.250 Å and c = 5.204 Å),32 are marginally smaller, which may be explained by a small degree of compressive strain in the NPs. A peak-dependent Debye–Scherer broadening (FWHM) of 0.79° was applied at a 2θ of 20°, along with a Lorentzian peak profile. The pXRD pattern simulated with these parameters matched the experimental pattern of the initial ZnO NPs, especially in terms of the {002}:{101} peak intensity ratio. For sulfur-doped ZnO NCs, the structure was modeled by substitution of a fraction of the O2− by S2− in the 2b Wycoff positions. Four different S/Zn molar ratios of 0.1, 0.11, 0.24, and 0.29 were tested, simulated pXRD patterns of which matched experimental patterns (in terms of the {002}:{101} peak intensity ratio) of the NPs at added S2− amount of 0.4, 0.79, 1.99, and 2.78 μmol, respectively.| S/Zn molar ratio | Element | Wycoff position | x | y | z | Site occupancy factor (SOF) | {002}/{101} intensity |
|---|---|---|---|---|---|---|---|
| 0.00 | O | 2b | 0.3333 | 0.6667 | 0.6600 | 1.00 | 0.72 |
| Zn | 2b | 0.3333 | 0.6667 | 0.2200 | 1.00 | ||
| 0.10 | O | 2b | 0.3333 | 0.6667 | 0.6600 | 0.90 | 0.77 |
| S | 2b | 0.3333 | 0.6667 | 0.6600 | 0.10 | ||
| Zn | 2b | 0.3333 | 0.6667 | 0.2200 | 1.00 | ||
| 0.11 | O | 2b | 0.3333 | 0.6667 | 0.6600 | 0.89 | 0.77 |
| S | 2b | 0.3333 | 0.6667 | 0.6600 | 0.11 | ||
| Zn | 2b | 0.3333 | 0.6667 | 0.2200 | 1.00 | ||
| 0.19 | O | 2b | 0.3333 | 0.6667 | 0.6600 | 0.81 | 0.81 |
| S | 2b | 0.3333 | 0.6667 | 0.6600 | 0.19 | ||
| Zn | 2b | 0.3333 | 0.6667 | 0.2200 | 1.00 | ||
| 0.24 | O | 2b | 0.3333 | 0.6667 | 0.6600 | 0.76 | 0.84 |
| S | 2b | 0.3333 | 0.6667 | 0.6600 | 0.24 | ||
| Zn | 2b | 0.3333 | 0.6667 | 0.2200 | 1.00 | ||
| 0.29 | O | 2b | 0.3333 | 0.6667 | 0.6600 | 0.71 | 0.86 |
| S | 2b | 0.3333 | 0.6667 | 0.6600 | 0.29 | ||
| Zn | 2b | 0.3333 | 0.6667 | 0.2200 | 1.00 |
Results and discusions
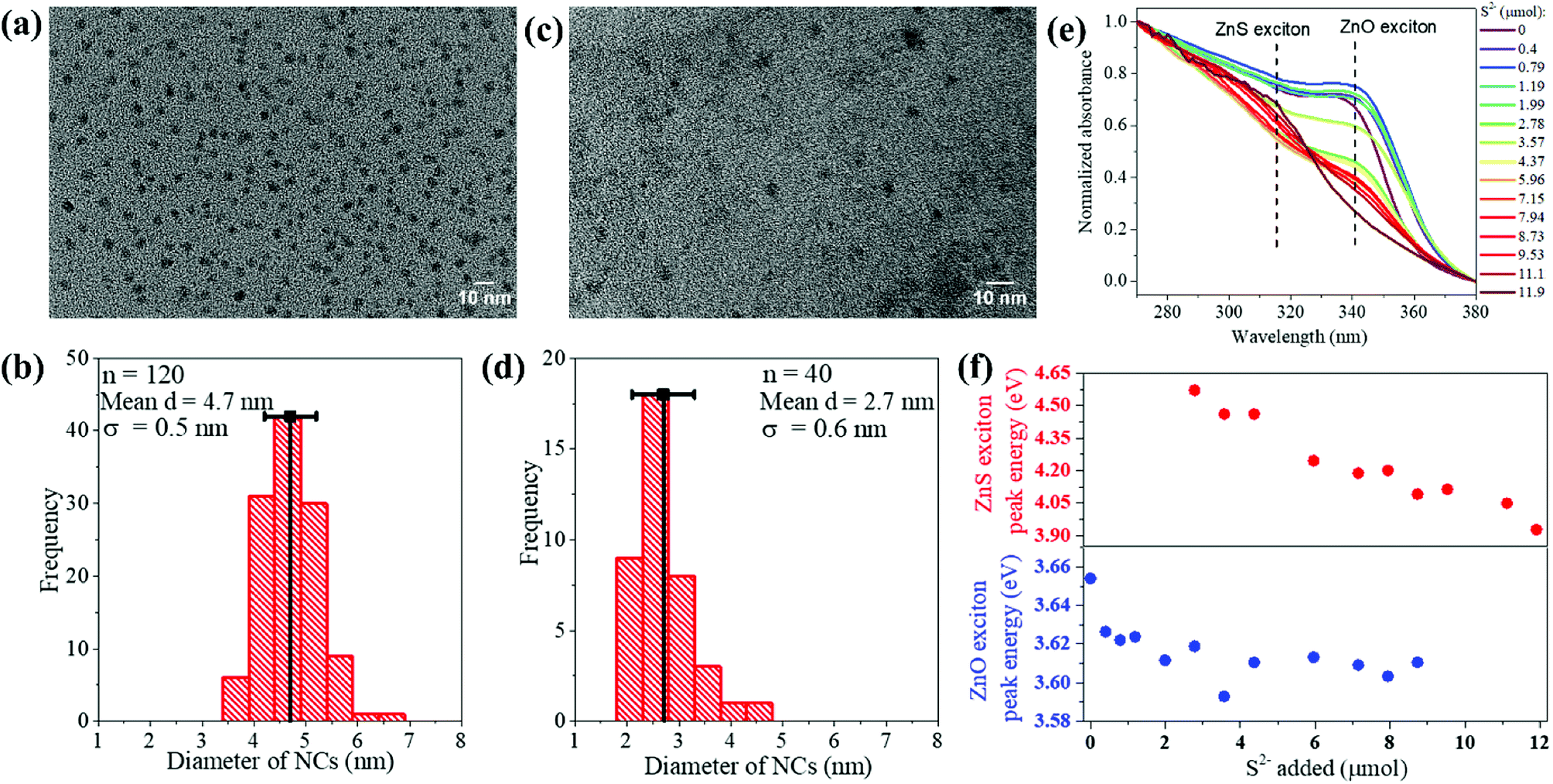
The ZnO NPs, synthesized by a known procedure,31 had an average size of 4.7 nm (Fig. 1a and b) as found from TEM and a wurtzite structure as shown by their pXRD pattern (Fig. 2a, dark purple curve). The NPs were subject to sulfidation with Na2S in ethanol at 45 °C. The sulfidation was performed as a titration, wherein molar aliquots of Na2S were added in a step-wise manner. Upon cumulative addition of 11.9 μmol of Na2S or S2−, the ZnO NPs underwent complete sulfidation. This is evidenced by the complete loss of diffraction peaks of wurtzite ZnO and the appearance of the {111} and {220} reflections of zincblende ZnS in the pXRD pattern (Fig. 2a, brown curve). In the elemental analysis (Table 1) of the final-stage NPs, a S/Zn molar of ca. 1 was measured, which supports the complete sulfidation of ZnO NPs to form ZnS. TEM showed ZnS NPs with an average size of 2.7 nm, significantly smaller than the size of the starting ZnO NPs. The smaller grain size of the ZnS is also reflected in the greater Debye–Scherer broadening of the pXRD peaks of the final-stage ZnS NPs (Fig. 2a, brown curve) as compared to those of the starting ZnO NPs (Fig. 2a, dark purple curve). From the Debye–Scherer broadening of the {111} peak (2θ = 29°, FWHM = 3.79°) of the final ZnS NPs, we estimated the NP size D:to be 22.6 Å or 2.3 nm. λ is the incident X-ray wavelength of 1.54 Å. The size estimated from pXRD is close to the one estimated by TEM (Fig. 1d).
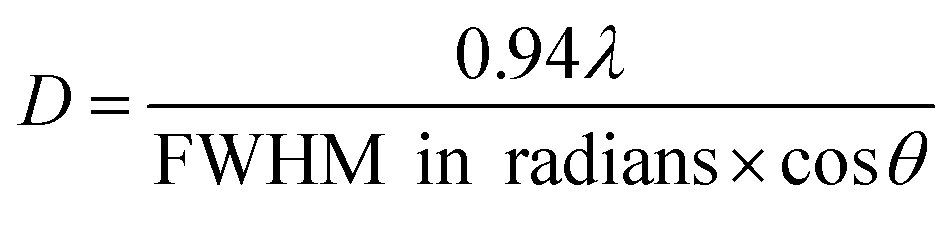
 | ||
| Fig. 1 (a) Representative TEM image of the synthesized ZnO NPs, (b) histogram of NP diameter for n = 120 NPs showing an average of 4.7 nm with a standard deviation (σ) of 0.5 nm. (c) Representative TEM image of the synthesized ZnS NPs, (d) histogram of NP diameter for n = 40 NPs showing an average of 2.7 nm with a standard deviation (σ) of 0.6 nm. (e) Absorbance spectra (normalized from 0 to 1 in the 270–380 nm range) of the colloid at various stages of ZnO NP sulfidation, in terms of the moles of Na2S added. The exciton peak positions of the initial ZnO NPs and the final ZnS NPs are marked by the vertical dotted lines. (f) The ZnO NP and the ZnS NP exciton peak energies in eV plotted as a function of the moles of Na2S added. The exciton peak energies were extracted from the absorbance spectra in (e), which were first subject to manual baseline subtraction, without which it is difficult to discern from the plotted spectra the broad and weak ZnS exciton peak at the early stages of sulfidation. | ||
 | ||
| Fig. 2 (a) Experimental pXRD patterns (baseline subtracted) are shown for NPs at various stages of the sulfidation transformation from wurtzite ZnO to zincblende ZnS, indicated by the moles of Na2S added. Peaks are assigned to major reflections, as labeled by the {hkl} Miller plane notation, with wurtzite ZnO reflections shown by purple sticks and zincblende ZnS ones by brown sticks. (b) The {002}:{101} peak intensity ratio from experimental patterns (black data points) and from simulations (red data points) plotted at the early stages of sulfidation. Lines are meant to guide the eye. (c) A magnified view of the experimental pXRD patterns plotted to highlight the peak reflection changes occurring in the early stages of sulfidation: 0, 0.4, 0.79, 1.99, and 2.78 μmol of S2−. The color legend is the same as that of (a). (d) Simulated pXRD patterns for NCs at early stages of sulfidation, plotted with the same magnified view as that in (c). The legend specifies the molar ratio of S/Zn used in the pXRD simulation. The crystallographic parameters used for the pXRD simulations at each stage are tabulated in Table 2. All patterns are shown vertically stacked and major reflections are labeled by the {hkl} Miller plane notation. | ||
At each step of the titration, the NP colloid was characterized by UV-vis extinction spectrophotometry (Fig. 1e) and pXRD (Fig. 2a), which shed light on the manner of sulfidation. The starting ZnO NPs exhibited an extinction spectrum with an exciton peak around 3.66 eV. This excitonic peak reflects the band-gap of these NPs, which is smaller than the bulk value33 of 3.37 eV due to the effect of quantum confinement.
With the sequential addition of S2− in the range of 0–1.99 μmol, the ZnO exitonic peak shifted to lower energies (Fig. 1f). This shift cannot be attributed to the formation of ZnS. ZnS has a higher band-gap than ZnO,34 so ZnS formation would have resulted in a shift to higher energies. Moreover, pXRD showed that, in the 0–1.99 μmol range of added S2−, the NPs maintain their wurtzite ZnO structure (Fig. 2a) and no diffraction peaks related to ZnS are observed. However, elemental analysis of the NPs obtained by treatment with 1.99 μmol of S2− revealed a S/Zn molar ratio of 0.27 (Table 1). Thus, the ZnO NPs appear to be significantly sulfur-doped. Such sulfur doping can explain the red-shift of the exciton peak. Zinc oxysulfides, ZnO1−xSx, with x = 0–0.3 are known to have a lower band gap than ZnO due to the effect of band-bowing.34 If the sulfur-doping were to be limited to an outer shell of each NP, the resulting intermediate-stage NPs would consist of a shrunken ZnO core surrounded by a ZnS shell. Such NPs would exhibit a blue-shifted ZnO exciton peak as compared to the exciton peak of the initial ZnO NPs. On the other hand, we observe a red-shift of the ZnO exciton peak at the early stages in the sulfidation (Fig. 1f, bottom panel), which is consistent with the attainment of an oxysulfide composition.34 In other words, the sulfur-doping extends across the NP lattice.
Further confirmation of and insight into the sulfur-doping of the ZnO NPs at the early stages of the sulfidation (0–2.78 μmol of S2−) is obtained from a closer analysis of the pXRD findings. We performed pXRD simulations (Fig. 2d) of sulfur-doped ZnO lattices, where a fraction of the O2− anions were replaced by S2−, without any other modification of the wurtzite structure. The simulated pXRD patterns of sulfur-doped ZnO resemble the simulated pattern for wurtzite ZnO (Fig. 2d). However, there is one difference: the ratio of the {002}:{111} peak intensity is higher for the sulfur-doped ZnO NPs as compared to the undoped ZnO NPs. Larger the S/Zn molar ratio (Fig. 2d), greater is the ratio of the {002}:{111} peak intensity. This computed trend matches with the trend observed in the experimental pXRD, as shown in Fig. 2b. Thus, the pXRD analysis confirms that at the early stages of the sulfidation, the ZnO NPs undergo substitutional doping by sulfur. Simulations predict that NPs subject to 1.99 μmol S2− have a S/Zn molar ratio of 0.24, which is close to the elemental analysis findings (Table 1).
At the point of 2.78 μmol added S2−, a broad absorption peak emerged at higher energies, i.e., 4.6 eV. This peak can be attributed to newly formed ZnS NPs. After all, ZnS is known to have a higher band-gap than ZnO.34 Upon further addition of S2−, the newly emerged excitonic peak increased in absorbance and shifted to lower energies (Fig. 1e and f). Alongside, the ZnO exciton peak decreased in strength until it was fully extinguished. These spectral trends can be explained by the following manner of sulfidation.
A small ZnS domain nucleates from the heavily sulfur-doped ZnO NPs. The considerably higher excitonic peak energy of 4.6 eV relative to the bulk ZnO band-gap35 of 3.72 eV suggests that the nucleated domain is ultrasmall. As more S2− becomes available, the ZnS domains grow further, which is manifested in the red-shift of the ZnS excitonic peak and increase in its absorbance (Fig. 1e and f). Concomitantly, the ZnO domains, which serve as the source of Zn, shrink in volume, which is reflected in a small blue-shift of the ZnO excitonic peak along with a decrease in its absorbance beyond 3.57 μmol added S2−. Eventually, the ZnO excitonic peak was fully extinguished. The final ZnS NPs exhibited a spectrum with a single excitonic peak at 3.94 eV, corresponding to small NPs.
The pXRD results provide further support to the above-described mechanism of sulfidation in the post-doping stages. At 4.37 μmol of added S2−, a broad peak appeared at the position corresponding to the major {111} reflection of zincblende ZnS, which serves as a structural indicator of ZnS nucleation. This diffraction peak grew in relative intensity and also narrowed with increasing addition of S2−, which would be consistent with a growth in ZnS domains. Alongside, with increasing addition of S2−, the wurtzite ZnO diffraction peaks became gradually weaker in relative intensity, until they were fully extinguished. The final pXRD pattern is indicative of NPs in a single ZnS phase, as also confirmed by the elemental analysis (Table 1). The final ZnS NPs appear to have a broader size dispersion (Fig. 1d, standard deviation of 22%) as compared to that of the initial ZnO NPs (Fig. 1b, standard deviation of 11%). The final NPs are formed by sulfur-doping of ZnO NPs followed by nucleation and growth of separate ZnS domains, a process expected to be replete with inherent heterogeneities.
Conclusion
The scheme in Fig. 3 summarizes the mechanism of sulfidation that is drawn from the results taken as a whole. The sulfidation of ZnO NPs is preceded by a stage where the NPs undergo substitutional doping by S2−. The wurtzite ZnO lattice remains otherwise unmodified in its structure. Above a concentration of ca. 30 mol% S2− dopants, ZnS nucleates from the ZnO. The threshold concentration probably corresponds to the solubility limit of S2− in ZnO. As more S2− becomes available, ZnS domains grow by supply of Zn2+ from ZnO. The O2− diffuses into solution. Thus, the complete phase transformation of the NPs from ZnO to ZnS cannot be considered simply as a topotactic exchange of anions in analogy to cation exchange reactions.36–47 Rather the post-doping stage sulfidation is more akin to a dissolution–reprecipitation process known in mineralogy,48 although the reprecipitation (nucleation-growth of ZnS) may occur in interfacial contact with the sulfur-doped ZnO NPs. The ZnS NPs formed from the nucleation-growth process share neither the crystallographic structure nor the morphology or size of the initial ZnO NPs. In topotactic sulfidation, the differences between the equilibrium unit cells of ZnO and ZnS would result in lattice strain and stress; in sulfidation occurring via the reprecipitation-like mechanism found here, such stresses and any resulting thermodynamic or kinetic bottlenecks in the solid-state transformation would be relieved. This mechanism of sulfidation must be taken into consideration when developing small ZnO NP-based sorbents for uptake of sulfur compounds from fuels. The major restructuring involved in going from the oxide to the sulfide can have major implications for the engineering design and operation of NP-based sorbent columns.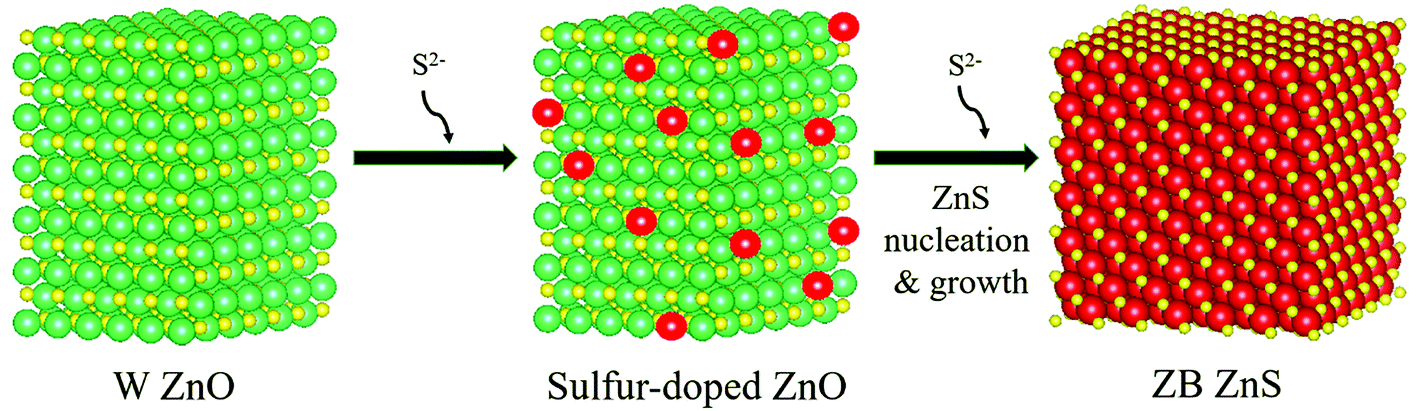 | ||
| Fig. 3 Pictorial depiction of the atomistic mechanism of sulfidation. Zn atoms are shown in yellow, O in green, and S in red. The structures were generated in Vesta software. The ZnO crystallite was based on a wurtzite (W) P63mmc unit cell containing 2592 atoms. The sulfur-doped crystallite was made by replacing some of the O atoms by S atoms. The ZnS crystallite is based on zincblende (ZB) F43m structure containing 1963 atoms. The morphologies and shapes of the final ZnS NPs formed by the nucleation-growth mechanism do not bear a topotaxial relationship with the initial ZnO NPs. | ||
Conflicts of interest
P. K. J. is an inventor on two patents on nanostructured ZnO sorbents and has served in the past as a scientific consultant on nanostructured ZnO for desulfurization technologies.Acknowledgements
P. B. conducted materials synthesis and characterization, performed simulations and analysis, and co-wrote the manuscript. P. K. J. conceived the project, designed experiments, performed supporting simulations and analysis, and wrote the manuscript. We acknowledge the donors of the American Chemical Society Petroleum Research Fund for support of this research. Ryan Sanders and Cody Tripp are acknowledged for their preliminary studies. This work was conducted in part at the Frederick Seitz Materials Research Laboratory.Notes and references
- A. Kołodziejczak-Radzimska and T. Jesionowski, Zinc Oxide—From Synthesis to Application: A Review, Materials, 2014, 7(4), 2833–2881 CrossRef PubMed.
- A. Moezzi, A. M. McDonagh and M. B. Cortie, Zinc Oxide Particles: Synthesis, Properties and Applications, Chem. Eng. J., 2012, 185–186, 1–22 CrossRef CAS.
- X. Tang, E. S. G. Choo, L. Li, J. Ding and J. Xue, Synthesis of ZnO Nanoparticles with Tunable Emission Colors and Their Cell Labeling Applications, Chem. Mater., 2010, 22(11), 3383–3388 CrossRef CAS.
- K. Ghule, A. V. Ghule, B.-J. Chen and Y.-C. Ling, Preparation and Characterization of ZnO Nanoparticles Coated Paper and Its Antibacterial Activity Study, Green Chem., 2006, 8(12), 1034–1041 RSC.
- R. Tankhiwale and S. K. Bajpai, Preparation, Characterization and Antibacterial Applications of ZnO-Nanoparticles Coated Polyethylene Films for Food Packaging, Colloids Surf., B, 2012, 90, 16–20 CrossRef CAS PubMed.
- B. Stieberova, M. Zilka, M. Ticha, F. Freiberg, P. Caramazana-González, J. McKechnie and E. Lester, Application of ZnO Nanoparticles in a Self-Cleaning Coating on a Metal Panel: An Assessment of Environmental Benefits, ACS Sustainable Chem. Eng., 2017, 5(3), 2493–2500 CrossRef CAS.
- A. M. El Saeed, M. A. El-Fattah and A. M. Azzam, Synthesis of ZnO Nanoparticles and Studying Its Influence on the Antimicrobial, Anticorrosion and Mechanical Behavior of Polyurethane Composite for Surface Coating, Dyes Pigm., 2015, 121, 282–289 CrossRef CAS.
- J. Ma, W. Zhu, Y. Tian and Z. Wang, Preparation of Zinc Oxide-Starch Nanocomposite and Its Application on Coating, Nanoscale Res. Lett., 2016, 11(1), 200 CrossRef PubMed.
- D. Álvarez, A. Collazo, X. R. Nóvoa and C. Pérez, Assessment of ZnO Nanoparticles as Anticorrosive Pigment in Hybrid Sol–Gel Films, Prog. Org. Coat., 2016, 96, 3–12 CrossRef.
- T. G. Smijs and S. Pavel, Titanium Dioxide and Zinc Oxide Nanoparticles in Sunscreens: Focus on Their Safety and Effectiveness, Nanotechnol., Sci. Appl., 2011, 4, 95–112 CrossRef CAS PubMed.
- J. Yin, Q. Lu, Z. Yu, J. Wang, H. Pang and F. Gao, Hierarchical ZnO Nanorod-Assembled Hollow Superstructures for Catalytic and Photoluminescence Applications, Cryst. Growth Des., 2010, 10(1), 40–43 CrossRef CAS.
- J. Zhang, Y. Wang, S. Zheng, H. Xue and H. Pang, N-Doped Mesoporous ZnO with Oxygen Vacancies for Stable Hydrazine Electrocatalysis, ChemNanoMat, 2018, 4 DOI:10.1002/cnma.201800363.
- H. Pang, Y. Ma, G. Li, J. Chen, J. Zhang, H. Zheng and W. Du, Facile Synthesis of Porous ZnO–NiO Composite Micropolyhedrons and Their Application for High Power Supercapacitor Electrode Materials, Dalton Trans., 2012, 41(43), 13284–13291 RSC.
- J. Zhang, P. Gu, J. Xu, H. Xue and H. Pang, High Performance of Electrochemical Lithium Storage Batteries: ZnO-Based Nanomaterials for Lithium-Ion and Lithium–Sulfur Batteries, Nanoscale, 2016, 8(44), 18578–18595 RSC.
- A. A. Keller, S. McFerran, A. Lazareva and S. Suh, Global Life Cycle Releases of Engineered Nanomaterials, J. Nanopart. Res., 2013, 15(6), 1692 CrossRef.
- V. Kandavelu, H. Kastien and K. R. Thampi, Photocatalytic Degradation of Isothiazolin-3-Ones in Water and Emulsion Paints Containing Nanocrystalline TiO2 and ZnO Catalysts, Appl. Catal., B, 2004, 48(2), 101–111 CrossRef CAS.
- B. Nowack and T. D. Bucheli, Occurrence, Behavior and Effects of Nanoparticles in the Environment, Environ. Pollut., 2007, 150(1), 5–22 CrossRef CAS PubMed.
- R. B. Reed, D. A. Ladner, C. P. Higgins, P. Westerhoff and J. F. Ranville, Solubility of Nano-Zinc Oxide in Environmentally and Biologically Important Matrices, Environ. Toxicol. Chem., 2012, 31(1), 93–99 CrossRef CAS PubMed.
- L. J. Gimbert, R. E. Hamon, P. S. Casey and P. J. Worsfold, Partitioning and Stability of Engineered ZnO Nanoparticles in Soil Suspensions Using Flow Field-Flow Fractionation, Environ. Chem., 2007, 4(1), 8–10 CrossRef CAS.
- O. Bondarenko, K. Juganson, A. Ivask, K. Kasemets, M. Mortimer and A. Kahru, Toxicity of Ag, CuO and ZnO Nanoparticles to Selected Environmentally Relevant Test Organisms and Mammalian Cells in vitro: A Critical Review, Arch. Toxicol., 2013, 87(7), 1181–1200 CrossRef CAS PubMed.
- Y. Ju-Nam and J. R. Lead, Manufactured Nanoparticles: An Overview of Their Chemistry, Interactions and Potential Environmental Implications, Sci. Total Environ., 2008, 400(1), 396–414 CrossRef CAS PubMed.
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Modeled Environmental Concentrations of Engineered Nanomaterials (TiO2, ZnO, Ag, CNT, Fullerenes) for Different Regions, Environ. Sci. Technol., 2009, 43(24), 9216–9222 CrossRef CAS PubMed.
- M. Behl, J. Yeom, Q. Lineberry, P. K. Jain and M. A. Shannon, A Regenerable Oxide-Based H2S Adsorbent with Nanofibrous Morphology, Nat. Nanotechnol., 2012, 7(12), 810–815 CrossRef CAS PubMed.
- M. Behl and P. K. Jain, Catalytic Activation of a Solid Oxide in Electronic Contact With Gold Nanoparticles, Angew. Chem. Int. Ed., 2015, 54(3), 992–997 CrossRef CAS PubMed.
- M. A. Sayyadnejad, H. R. Ghaffarian and M. Saeidi, Removal of Hydrogen Sulfide by Zinc Oxide Nanoparticles in Drilling Fluid, Int. J. Environ. Sci. Technol., 2008, 5(4), 565–569 CrossRef CAS.
- X. Wang, T. Sun, J. Yang, L. Zhao and J. Jia, Low-Temperature H2S Removal from Gas Streams with SBA-15 Supported ZnO Nanoparticles, Chem. Eng. J., 2008, 142(1), 48–55 CrossRef CAS.
- J. Skrzypski, I. Bezverkhyy, O. Heintz and J.-P. Bellat, Low Temperature H2S Removal with Metal-Doped Nanostructure ZnO Sorbents: Study of the Origin of Enhanced Reactivity in Cu-Containing Materials, Ind. Eng. Chem. Res., 2011, 50(9), 5714–5722 CrossRef CAS.
- I. Rosso, C. Galletti, M. Bizzi, G. Saracco and V. Specchia, Zinc Oxide Sorbents for the Removal of Hydrogen Sulfide from Syngas, Ind. Eng. Chem. Res., 2003, 42(8), 1688–1697 CrossRef CAS.
- S. Lew, K. Jothimurugesan and M. Flytzani-Stephanopoulos, High-Temperature Hydrogen Sulfide Removal from Fuel Gases by Regenerable Zinc Oxide-Titanium Dioxide Sorbents, Ind. Eng. Chem. Res., 1989, 28(5), 535–541 CrossRef CAS.
- J. Park, H. Zheng, Y. Jun and A. P. Alivisatos, Hetero-Epitaxial Anion Exchange Yields Single-Crystalline Hollow Nanoparticles, J. Am. Chem. Soc., 2009, 131(39), 13943–13945 CrossRef CAS PubMed.
- V. Subramanian, E. E. Wolf and P. V. Kamat, Green Emission to Probe Photoinduced Charging Events in ZnO–Au Nanoparticles. Charge Distribution and Fermi-Level Equilibration, J. Phys. Chem. B, 2003, 107(30), 7479–7485 CrossRef CAS.
- H. Karzel, W. Potzel, M. Köfferlein, W. Schiessl, M. Steiner, U. Hiller, G. M. Kalvius, D. W. Mitchell, T. P. Das and P. Blaha, et al., Lattice Dynamics and Hyperfine Interactions in ZnO and ZnSe at High External Pressures, Phys. Rev. B, 1996, 53(17), 11425–11438 CrossRef CAS.
- H. Morkoç and Ü. Özgür, General Properties of ZnO, in Zinc Oxide, Wiley-VCH Verlag GmbH & Co. KGaA, 2009; pp. 1–76 Search PubMed.
- B. K. Meyer, A. Polity, B. Farangis, Y. He, D. Hasselkamp, T. Krämer and C. Wang, Structural Properties and Bandgap Bowing of ZnO1−xSx Thin Films Deposited by Reactive Sputtering, Appl. Phys. Lett., 2004, 85(21), 4929–4931 CrossRef CAS.
- X. Fang, T. Zhai, U. K. Gautam, L. Li, L. Wu, Y. Bando and D. Golberg, ZnS Nanostructures: From Synthesis to Applications, Prog. Mater. Sci., 2011, 56(2), 175–287 CrossRef CAS.
- D. H. Son, S. M. Hughes, Y. Yin and A. Paul Alivisatos, Cation Exchange Reactions in Ionic Nanocrystals, Science, 2004, 306(5698), 1009–1012 CrossRef CAS PubMed.
- J. B. Rivest and P. K. Jain, Cation Exchange on the Nanoscale: An Emerging Technique for New Material Synthesis, Device Fabrication, and Chemical Sensing, Chem. Soc. Rev., 2013, 42(1), 89–96 RSC.
- B. J. Beberwyck and A. P. Alivisatos, Ion Exchange Synthesis of III–V Nanocrystals, J. Am. Chem. Soc., 2012, 134(49), 19977–19980 CrossRef CAS PubMed.
- S. L. White, P. Banerjee, I. Chakraborty and P. K. Jain, Ion Exchange Transformation of Magic-Sized Clusters, Chem. Mater., 2016, 28(22), 8391–8398 CrossRef CAS.
- P. K. Jain, L. Amirav, S. Aloni and A. P. Alivisatos, Nanoheterostructure Cation Exchange: Anionic Framework Conservation, J. Am. Chem. Soc., 2010, 132(29), 9997–9999 CrossRef CAS PubMed.
- R. D. Robinson, B. Sadtler, D. O. Demchenko, C. K. Erdonmez, L.-W. Wang and A. P. Alivisatos, Spontaneous Superlattice Formation in Nanorods Through Partial Cation Exchange, Science, 2007, 317(5836), 355–358 CrossRef CAS PubMed.
- P. K. Jain, K. Manthiram, J. H. Engel, S. L. White, J. A. Faucheaux and A. P. Alivisatos, Doped Nanocrystals as Plasmonic Probes of Redox Chemistry, Angew. Chem., Int. Ed., 2013, 52(51), 13671–13675 CrossRef CAS PubMed.
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss and D. J. Milliron, et al., Prospects of Nanoscience with Nanocrystals, ACS Nano, 2015, 9(2), 1012–1057 CrossRef CAS PubMed.
- H. Li, M. Zanella, A. Genovese, M. Povia, A. Falqui, C. Giannini and L. Manna, Sequential Cation Exchange in Nanocrystals: Preservation of Crystal Phase and Formation of Metastable Phases, Nano Lett., 2011, 11(11), 4964–4970 CrossRef CAS PubMed.
- D. Dumett Torres, P. Banerjee, S. Pamidighantam and P. K. Jain, A Non-Natural Wurtzite Polymorph of HgSe: A Potential 3D Topological Insulator, Chem. Mater., 2017, 29(15), 6356–6366 CrossRef CAS.
- P. Banerjee and P. K. Jain, Lithiation of Copper Selenide Nanocrystals, Angew. Chem. Int. Ed., 2018, 57(30), 9315–9319 CrossRef CAS PubMed.
- S. L. White, P. Banerjee and P. K. Jain, Liquid-like Cationic Sub-Lattice in Copper Selenide Clusters, Nat. Commun., 2017, 8, 14514 CrossRef CAS PubMed.
- A. Putnis, Mineral Replacement Reactions: From Macroscopic Observations to Microscopic Mechanisms, Mineral. Mag., 2002, 66(5), 689–708 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |