DOI:
10.1039/C8RA05693E
(Paper)
RSC Adv., 2018,
8, 29924-29927
Electron paramagnetic resonance spectroscopic studies of the electron transfer reaction of Hantzsch ester and a pyrylium salt†
Received
3rd July 2018
, Accepted 13th August 2018
First published on 23rd August 2018
Abstract
The oxidation of Hantzsch ester by a pyrylium cation takes place via electron–proton–electron transfer. The reaction was investigated with EPR spectroscopy using TEMPO and DMPO for inhibition and spin trapping, respectively, of the radicals appearing during the reaction. The present in-depth EPR study of the radical reactions of a NADH analogue indicate a complex electron transfer mechanism in the title reaction.
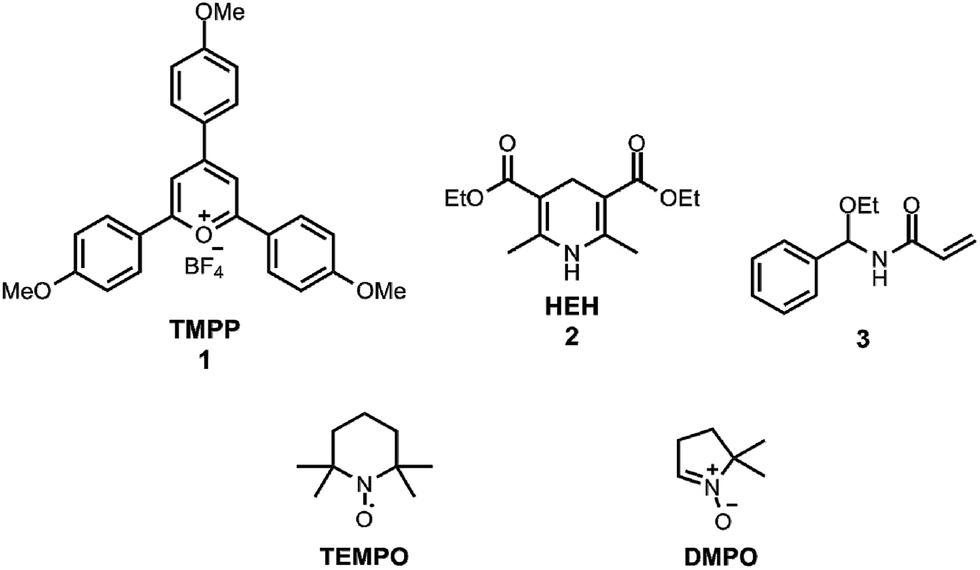
NADH and its synthetic analogues with a dihydropyridine structure serve as the sources of two electrons and one proton, which can be transferred via three different reaction pathways: direct hydride transfer,1,2 electron transfer followed by hydrogen atom transfer3 and electron–proton–electron transfer.4 This oxidation mechanism was already investigated sixty years ago,5 but despite considerable experimental and thermodynamic calculation efforts the characterisation of the free radical reactions has not been completed yet.6 In this work we applied the 2,4,6-tris(4-methoxyphenyl)pyryliumtetrafluoroborate7 cation (TMPP, Scheme 1) for the oxidation of the NADH analogue8 Hantzsch ester (diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate, HEH, Scheme 1) under an air and argon atmosphere. Here we present our empirical findings uncovered in this model reaction. The stable radical 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO, Scheme 1) and a spin trap 5,5-dimethyl-1-pyrroline-N-oxide (DMPO, Scheme 1) were applied for the electron paramagnetic resonance (EPR) spectroscopic measurements of free radicals produced during the reactions9 (see ESI† for experimental methods).
 |
| | Scheme 1 | |
In the course of our ongoing investigations related to the reactivity of N-acyl-N,O-acetals10 (compound 3 as a representative is depicted in Scheme 1), we have discovered that the reaction between 1 and 2 should proceed via a radical pathway since the EPR signal of TEMPO monitored during their reaction was decreased with time. This signal decay was monitored over several days both in the presence and absence of 3 and at three different concentrations (see ESI†). Analysis of the kinetics revealed a decay that could be fitted with two exponentials indicating two parallel possible reactions that neutralize the TEMPO radical. Additionally, a control experiment, where TMPP was replaced by a Brønsted acid11 catalyst, resulted in no TEMPO consumption indicating the pivotal role of pyrylium salt in the generation of transient radical species.12
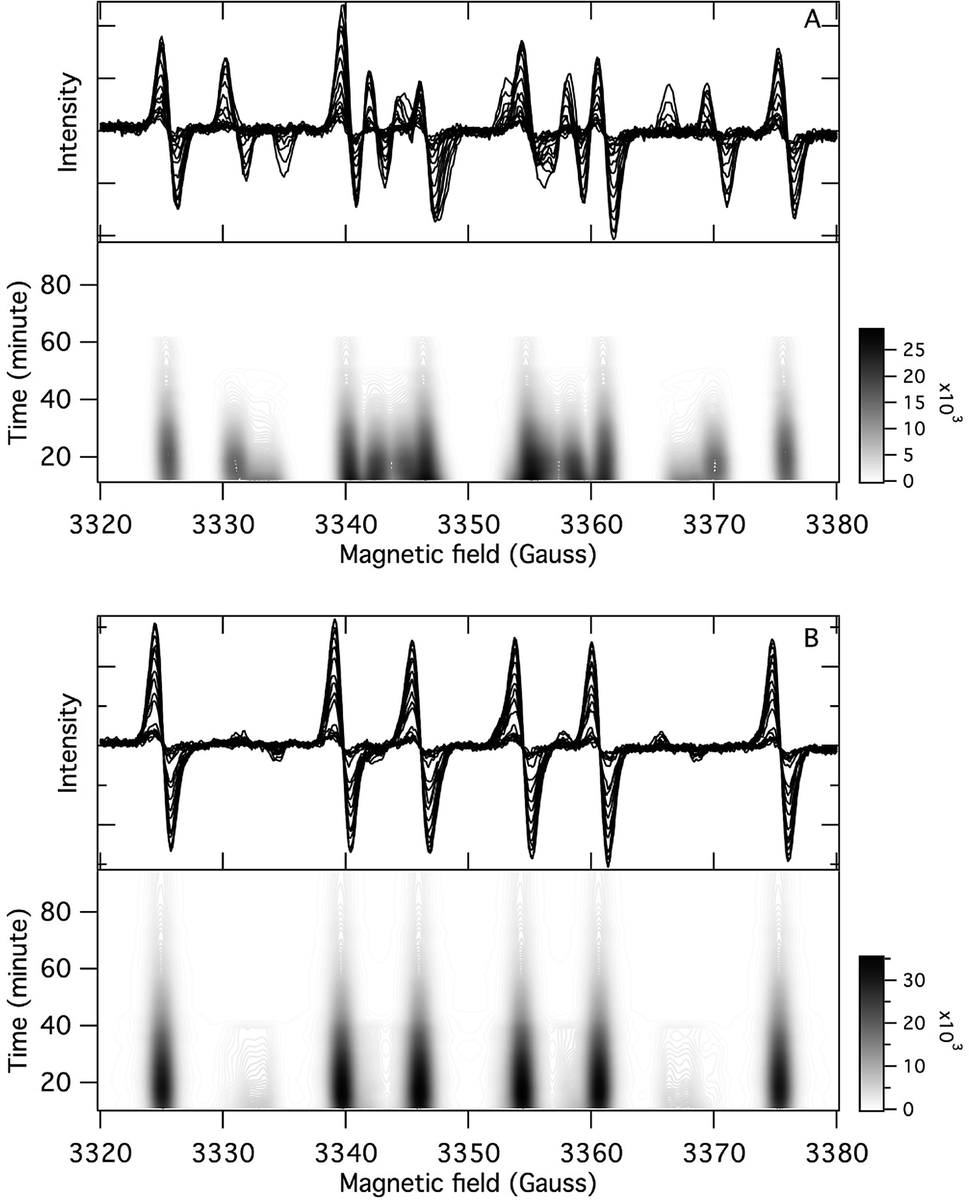
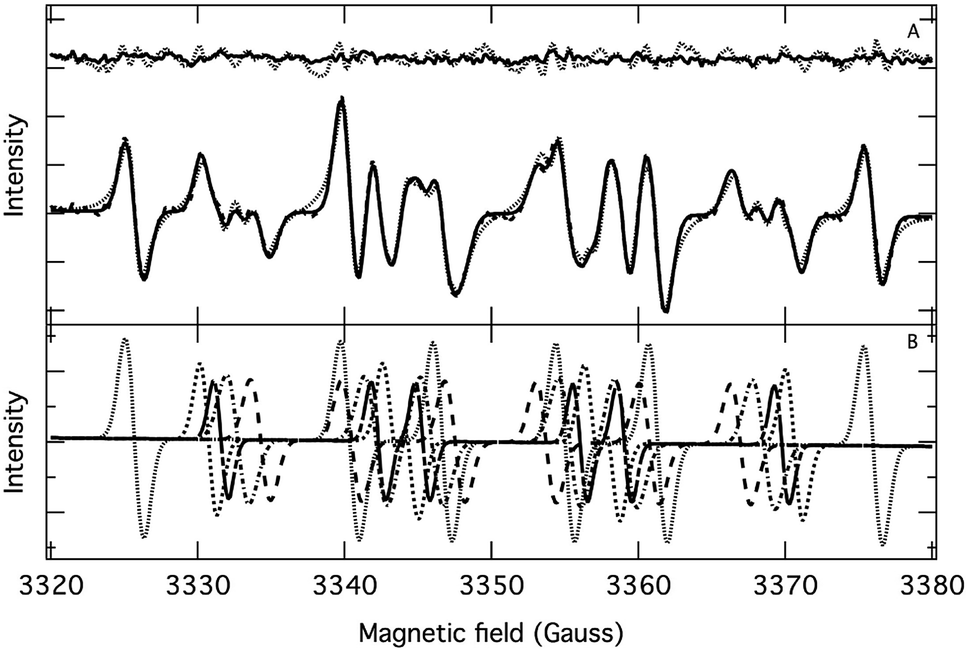
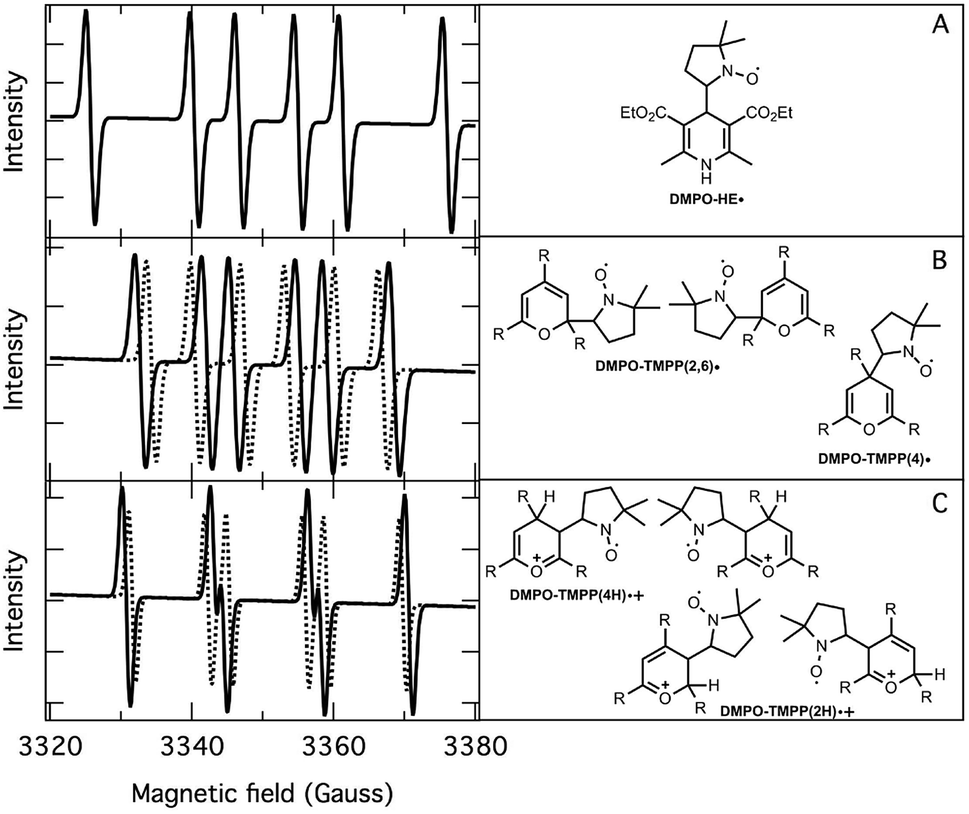
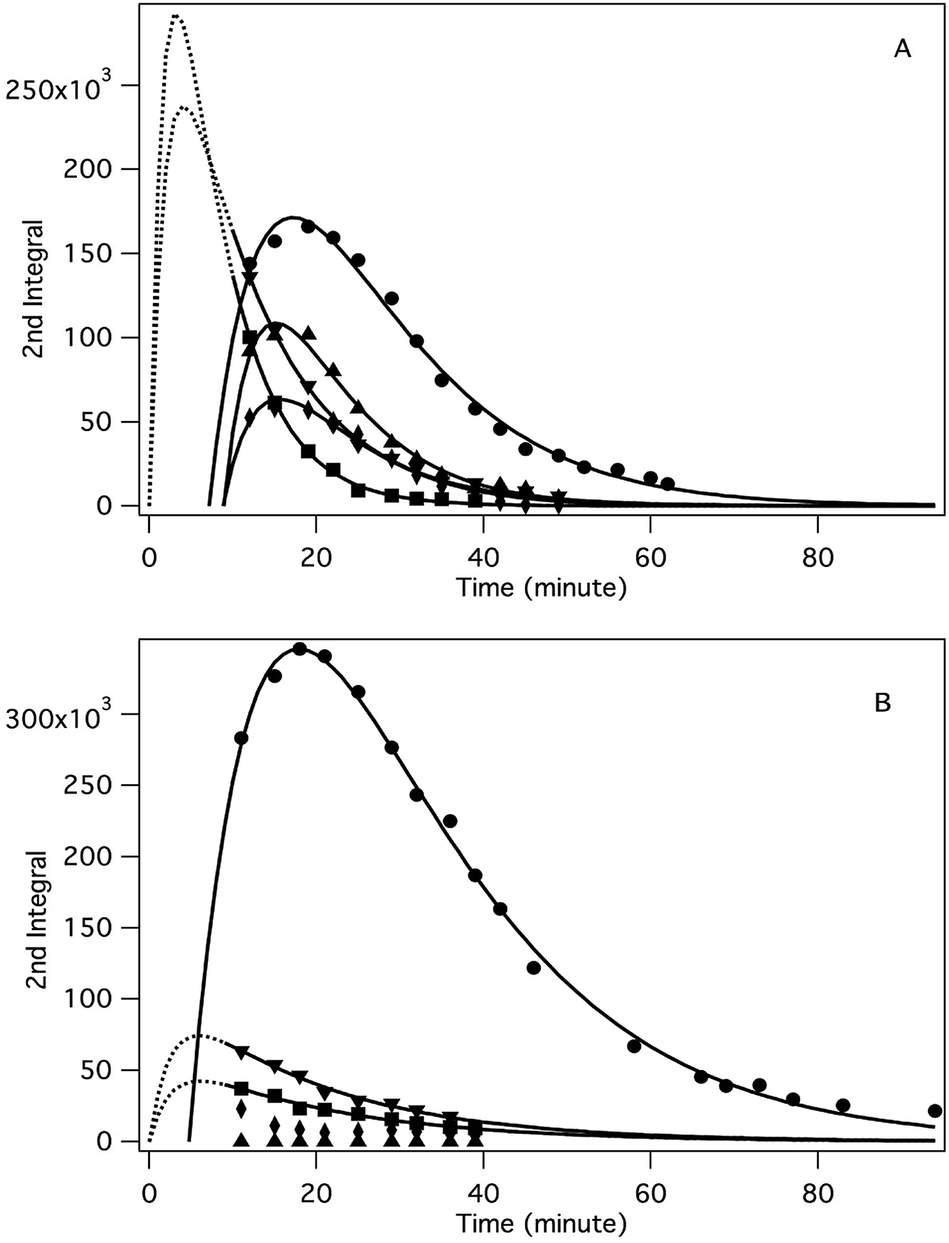
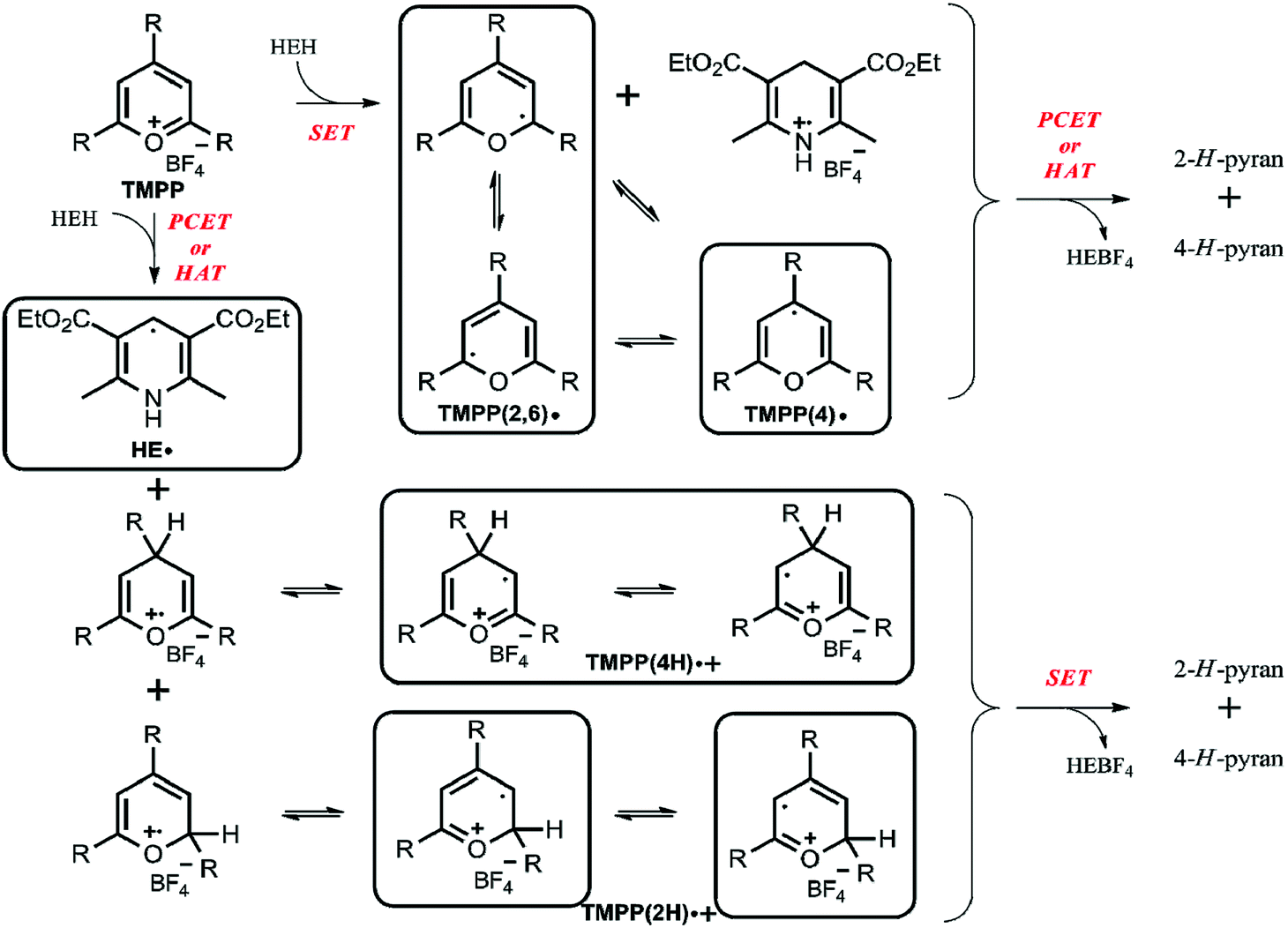
The application of the spin trap DMPO, instead of TEMPO, and its competitive reactions made it possible to detect radicals trapped as DMPO-adducts over a period of 1–1.5 hours. Fig. 1 (top traces of A and B) represents the EPR spectra of trapped radicals during the oxidation of HEH in the absence (A) and presence (B) of 3. The contour plots (Fig. 1, bottom traces of A and B) show the time dependence of absorption spectra, which are the corresponding first integrals. The original spectra and their first integrals show clearly that the EPR spectra were less complex and the reaction time was ∼1.5 times longer in the presence (panel B) than in the absence (panel A) of compound 3. Computer simulation and deconvolution of EPR spectra provided detailed information on the radicals trapped by DMPO.6j,13 A total of twenty-eight independent parameters were needed for accurate description of the spectra by fitting them with a superposition of first derivatives of either Lorentzian (L′(B)) or Voigt (V′(B))14 line shapes. The spectral parameters, used as fitting parameters were the following: the hyperfine splitting from nitrogen and hydrogen nucleus (aN and aH, respectively), the inhomogeneous Gaussian line width, the amplitude and the magnetic field position of the component spectra of each DMPO-adduct, two parameters of a linear background (used only for baseline correction) and a global line shape parameter (see Fig. 2 and ESI†). The individual EPR spectra of the different DMPO-adducts are presented in Fig. 3 together with our proposed structures (also supported by literature data3) corresponding to each spectrum. Table 1 summarizes the fitted hyperfine splitting constants for the five detected components. (The full table with all the parameters is given in the ESI.†) The time dependence of the concentration of each detected component was also studied. The strategy for the assignment of component spectra to specific chemical structures was as follows. Possible candidate radicals were first identified based on literature data.7d,15 It was then considered with how many equivalent and non-equivalent N and H nuclei the unpaired electron can interact in each corresponding DMPO adduct. A comparison of the possible theoretical hyperfine splitting patterns with the observed spectra and also taking into account the lines showing the same time dependence, hence belonging to the same radical, led to the assignment of the spectra to specific DMPO adducts and free radicals. The consistency between the proposed structures and the component spectra and their time dependence gives confidence of the assignment. Fig. 4 represents the concentration distribution of the DMPO-adducts in the absence (A) and presence (B) of 3. Data points for each adduct are the second integrals of each corresponding deconvoluted EPR spectrum. The time dependences were fitted with two exponentials corresponding to the formation and decay of each radical adduct (for the rate constants of formation and consumption see ESI†). Based on the above data, we propose that three different basic types of DMPO-adducts were formed in this reaction. One type of adduct was formed with the Hantzsch ester radical, one with the pyranyl radical and one with the pyranyl radical cation. The latter two adducts are consisting of their corresponding electromers and tautomers, in agreement with the observed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 spectrum ratios. The simultaneous presence of both pyranyl radical and radical cation, together with the TEMPO decay curves indicating two simultaneous reactions neutralizing the radical signal, strongly suggests a complex reaction mechanism involving an electron–proton–electron transfer process. This sequence can be sub-grouped as an initial single electron transfer (SET) followed by a proton-coupled electron transfer (PCET) or hydrogen atom transfer (HAT)16 and vice versa.
:2 spectrum ratios. The simultaneous presence of both pyranyl radical and radical cation, together with the TEMPO decay curves indicating two simultaneous reactions neutralizing the radical signal, strongly suggests a complex reaction mechanism involving an electron–proton–electron transfer process. This sequence can be sub-grouped as an initial single electron transfer (SET) followed by a proton-coupled electron transfer (PCET) or hydrogen atom transfer (HAT)16 and vice versa.
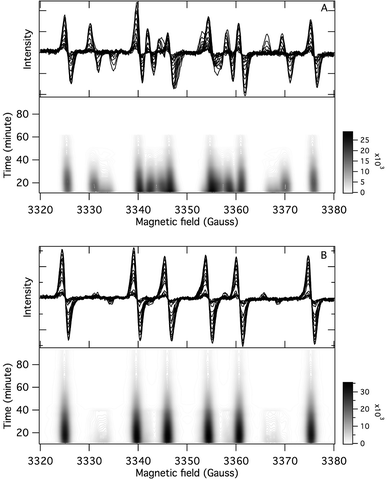 |
| | Fig. 1 Time dependence of the EPR spectra of DMPO adducts (top) and contour plots of the absorption spectra (bottom). Conditions: [HEH]0 = 0.04 M, [TMPP]0 = 0.04 M, [DMPO]0 = 0.045 M, DMSO, under argon, room temperature, (A): [3]0 = 0 M, (B): [3]0 = 0.2 M. | |
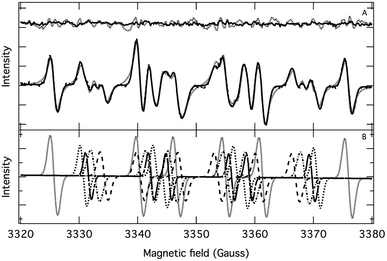 |
| | Fig. 2 (A) Fitting of an experimental EPR spectrum (dashed line) with a superposition of first derivative of Lorentzian (dotted line) and Voigt (solid line) functions. Top trace: residuals. (B) Deconvoluted individual EPR spectra. Experimental conditions are the same as for Fig. 1 (A, in the absence of compound 3) and the spectrum was recorded 12 minutes after the onset of HEH oxidation. | |
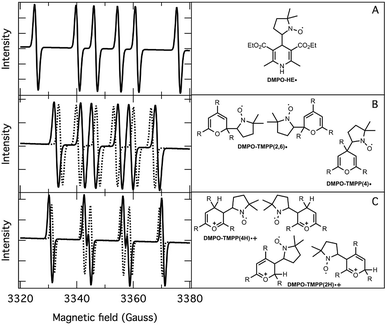 |
| | Fig. 3 Deconvoluted EPR spectra (left) and corresponding structure of DMPO-adducts (right). (A): DMPO–HE˙; (B): DMPO–TMPP(4)˙ (dotted line), DMPO–TMPP(2,6)˙ (solid line); (C): DMPO–TMPP(4H)˙+ (solid line), DMPO–TMPP(2H)˙+ (dotted line). | |
Table 1 The hyperfine splitting constants of DMPO-adducts
| Adduct |
aN/G |
aH/G |
| DMPO–HE˙ |
14.66 ± 0.01 |
20.98 ± 0.02 |
| DMPO–TMPP(4)˙ |
13.30 ± 0.06 |
5.97 ± 0.09 |
| DMPO–TMPP(2,6)˙ |
13.05 ± 0.16 |
9.48 ± 0.32 |
| DMPO–TMPP(4H)˙+ |
14.04 ± 0.45 |
10.60 ± 0.25 |
| DMPO–TMPP(2H)˙+ |
13.75 ± 0.03 |
12.33 ± 0.28 |
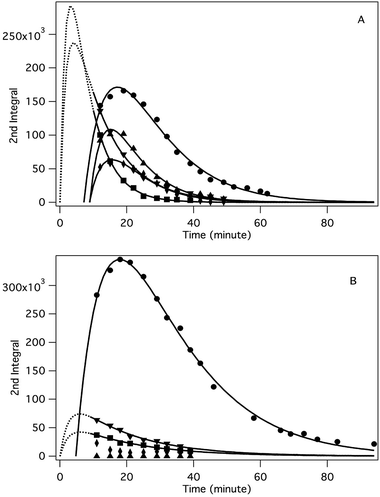 |
| | Fig. 4 Time dependence of the concentration of the different spin-trapped radical adducts, measured as the integrated intensity of the corresponding EPR absorption spectra. Conditions: [HEH]0 = 0.04 M, [TMPP]0 = 0.04 M, [DMPO]0 = 0.045 M, DMSO, under argon, room temperature, (A): [3]0 = 0 M, (B): [3]0 = 0.2 M. Symbols: DMPO–HE˙ (●), DMPO–TMPPH˙(4) (■), DMPO–TMPP˙(2,6) (▼), DMPO–TMPP(4H)˙+ (♦), DMPO–TMPP(2H)˙+ (▲). | |
Based on our results, in particular the observed radicals and their time dependence, we propose a mechanism on Scheme 2, where the framed compounds do form DMPO-adducts. If TMPP reacts by an initial single electron transfer from HEH (top pathway), neutral radicals TMPP(2,6) and (4) are formed together with a Hantzsch ester radical cation. A consecutive PCET or HAT from the Hantzsch ester radical cation produces 2-H- and 4-H-pyrans as primary products, though these species are not stable enough to be characterized from the reaction mixture. Carbon-centered TMPP(2,6) and (4) radicals can, while the Hantzsch ester radical cation cannot form a stable adduct with DMPO. On the other hand, when TMPP reacts by an initial PCET or HAT with the Hantzsch ester (bottom pathway), the DMPO-trappable carbon-centered Hantzsch ester neutral radical is formed besides primary O-centered TMPPH radical cations. While O-centered radical cations cannot form stable DMPO-adducts, stabilization of these TMPP(4H) and (2H) species as their electromers results DMPO-trappable carbon-centered radicals. Eventually, a single electron transfer from the Hantzsch ester neutral radical yields identical primary products 2-H- and 4-H-pyrans as outlined in the other reaction pathway above. Both pathways provide oxidized Hantzsch ester tetrafluoroborate salt as a by-product. The role of compound 3 in rendering the observed EPR spectrum less complex (Fig. 1) arise most likely from its Brønsted basic character. Literature data shows that N,O-acetals react with Brønsted acids readily.17 Therefore, we propose that compound 3 scavenges radical cation type intermediates (TMPP(4H)˙+ and (2H)˙+) by deprotonation, thus inhibiting facile quenching of HE˙ and consequently prolonging the lifetime of this radical together with its DMPO adduct. This explanation is further supported by the large excess (5 fold) of compound 3.
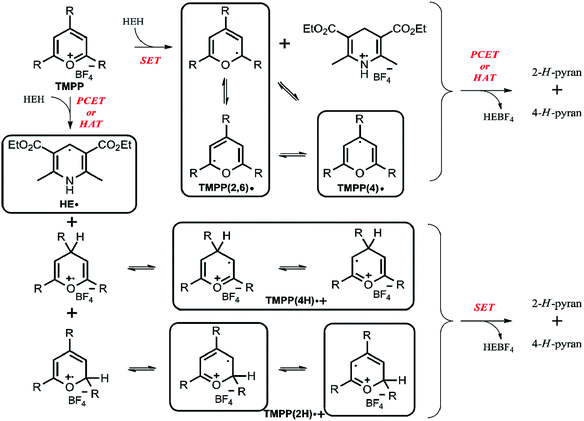 |
| | Scheme 2 The proposed reaction mechanism (R = 4-methoxyphenyl; HEBF4 = oxidized Hantzsch ester tetrafluoroborate salt). | |
In conclusion, we have carried out an EPR study with a very detailed analysis of the radical reaction of Hantzsch ester and a pyrylium salt. The complex electron–proton–electron mechanism of NADH and its analogues show great variation depending on the substrates (see, e.g., ref. 3 and 4). However, such schemes have not been supported by spin-trapping experiments yet, and such complex reactivity of a pyrylium salt in a single chemical reaction is unprecedented so far. Based on our empirical findings, we propose six DMPO-adducts, and overall eight transient radical species, in this reaction, which strongly support a complex electron transfer mechanism. We believe that such mathematical deconvolution of complex EPR spectra could become a useful tool for the identification of transient chemical entities in distinct complex radical reactions as well.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported in part by the Hungarian National Research Development and Innovation Fund (K112716) and also by GINOP-2.3.2-15-2016-00001 programme.
Notes and references
- X.-Q. Zhu, Y.-C. Liu and J.-P. Cheng, J. Org. Chem., 1999, 64, 8980–8981 CrossRef
 .
. - B. Zhao, X. Zhu, Y. Lu, C.-Z. Xia and J.-P. Cheng, Tetrahedron Lett., 2000, 41, 257–260 CrossRef .
- J.-P. Cheng and Y. Lu, J. Phys. Org. Chem., 1997, 10, 577–584 CrossRef .
- X.-Q. Zhu, H.-R. Li, Q. Li, T. Ai, J.-Y. Lu, Y. Yang and J.-P. Cheng, Chem.–Eur. J., 2003, 9, 871–880 CrossRef PubMed .
- D. Mauzerall and F. H. Westheimer, J. Am. Chem. Soc., 1955, 77, 2261–2264 CrossRef .
-
(a) S. Fukuzumi, S. Koumitsu, K. Hironaka and T. Tanaka, J. Am. Chem. Soc., 1987, 109, 305–316 CrossRef ;
(b) J. Zielonka, A. Marcinek, J. Adamus and J. Gębicki, J. Phys. Chem. A, 2003, 107, 9860–9864 CrossRef ;
(c) S. Fukuzumi, H. Kotani, Y. M. Lee and W. Nam, J. Am. Chem. Soc., 2008, 130, 15134–15142 CrossRef PubMed ;
(d) J. A. Birrell and J. Hirst, Biochemistry, 2013, 52, 4048–4055 CrossRef PubMed ;
(e) J.-P. Cheng, Y. Lu, X. Zhu and L. Mu, J. Org. Chem., 1998, 63, 6108–6114 CrossRef PubMed ;
(f) S. Fukuzumi, K. Ohkubo, Y. Tokuda and T. Suenobu, J. Am. Chem. Soc., 2000, 122, 4286–4294 CrossRef ;
(g) J.-P. Cheng, Y. Lu, X.-Q. Zhu, Y. Sun, F. Bi and J. He, J. Org. Chem., 2000, 65, 3853–3857 CrossRef PubMed ;
(h) T. Matsuo and J. M. Mayer, Inorg. Chem., 2005, 44, 2150–2158 CrossRef PubMed ;
(i) J. Yuasa and S. Fukuzumi, J. Am. Chem. Soc., 2006, 128, 14281–14292 CrossRef PubMed ;
(j) G. M. Rosen and E. J. Rauckman, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 7346–7349 CrossRef PubMed .
- Representative examples for the application of 2,4,6-tris(4-methyoxphenyl)pyrylium tetrafluoroborate as an electron transfer catalyst:
(a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef PubMed ;
(b) K. Wang, L.-G. Meng and L. Wang, Org. Lett., 2017, 19, 1958–1961 CrossRef PubMed ;
(c) E. Alfonzo, F. S. Alfonso and A. B. Beeler, Org. Lett., 2017, 19, 2989–2992 CrossRef PubMed ;
(d) M. A. Miranda and H. Garcia, Chem. Rev., 1994, 94, 1063–1089 CrossRef .
- Representative examples for the radical reactivity of 1,4-dihydropyridines:
(a) Á. Gutiérrez-Bonet, J. C. Tellis, J. K. Matsui, B. A. Vara and G. A. Molander, ACS Catal., 2016, 6, 8004–8008 CrossRef PubMed ;
(b) W. Chen, Z. Liu, J. Tian, J. Li, J. Ma, X. Cheng and G. Li, J. Am. Chem. Soc., 2016, 138, 12312–12315 CrossRef PubMed ;
(c) X. Cheng and W. Huang, Synlett, 2017, 28, 148–158 CrossRef ;
(d) S. Sumino, M. Uno, T. Fukuyama, I. Ryu, M. Matsuura, A. Yamamoto and Y. Kishikawa, J. Org. Chem., 2017, 82, 5469–5474 CrossRef PubMed .
- S. Niizuma, N. Sato, H. Kawata and Y. Suzuki, Bull. Chem. Soc. Jpn., 1985, 58, 2600–2607 CrossRef .
- These results will be published in full detail in due course..
- Brønsted-acid mediated Hantzsch ester activation is well known in the literature, Enantioselective Organocatalyzed Reactions I: Enantioselective Oxidation, Reduction, Functionalization and Desymmetrization, ed. R. Mahrwald, 2011, DOI:10.1007/978-90-481-3865-4_2 .
- Small effect (<5%) of oxygen was observed on k1 (1.10e−5 ± 8.0e−7 s−1 (air), 1.15e−5 ± 2.0e−7 s−1 (argon)), while ∼30% on k2 (7.98e−5 ± 2.7e−6 s−1 (air), 5.76e−5 ± 1.8e−7 s−1 (argon)). To exclude the reactions generated by oxygen and eliminate its effect on any oxygen-sensitive reactions, further experiments were carried out under argon..
-
(a) Q. Guo, S. Y. Qian and R. P. Mason, J. Am. Soc. Mass Spectrom., 2003, 14, 862–871 CrossRef PubMed ;
(b) D. H. Yoo, S. K. Han, M. J. Lee and J. W. Kang, J. Ind. Eng. Chem., 2005, 11, 215–221 Search PubMed ;
(c) F. Li, L. Xiao and L. Liu, Sci. Rep., 2016, 6, 22876, DOI:10.1038/srep22876 .
- U. Platt and J. Stutz in Differential Optical Absorption Spectroscopy – Principles and Applications. Springer-Verlag Berlin Heidelberg, 2008, Ch. 3 Search PubMed .
- T.-S. Balaban and A. T. Balaban, Tetrahedron Lett., 1987, 28, 1341–1344 CrossRef .
-
(a) A. Sirjoosingh and S. Hammes-Schiffer, J. Phys. Chem. A, 2011, 115, 2367–2377 CrossRef PubMed ;
(b) A. K. Harshan, T. Yu, A. V. Soudackov and S. Hammes-Schiffer, J. Am. Chem. Soc., 2015, 137, 13545–13555 CrossRef PubMed .
- Y.-Y. Huang, C. Chai, X. Yang, Z.-C. Lv and U. Schneider, ACS Catal., 2016, 6, 5747–5763 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05693e |
|
| This journal is © The Royal Society of Chemistry 2018 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
T. Páli
*b and
T. Páli