
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA self-assembling peptidic platform to boost the cellular uptake and nuclear delivery of oligonucleotides†
Shabnam
Tarvirdipour
abc,
Michal
Skowicki
 a,
Cora-Ann
Schoenenberger
a,
Larisa E.
Kapinos
d,
Roderick Y. H.
Lim
d,
Yaakov
Benenson
*c and
Cornelia G.
Palivan
*ab
a,
Cora-Ann
Schoenenberger
a,
Larisa E.
Kapinos
d,
Roderick Y. H.
Lim
d,
Yaakov
Benenson
*c and
Cornelia G.
Palivan
*ab
aDepartment of Chemistry, University of Basel, Basel, CH-4058, Switzerland. E-mail: cornelia.palivan@unibas.ch
bSwiss Nanoscience Institute (SNI), University of Basel, Basel, CH-4056, Switzerland
cDepartment of Biosystems Science and Engineering, ETH Zurich, Basel, CH-4058, Switzerland. E-mail: kobi.benenson@bsse.ethz.ch
dBiozentrum, University of Basel, Basel, CH-4056 Switzerland
First published on 25th June 2022
Abstract
The design of non-viral vectors that efficiently deliver genetic materials into cells, in particular to the nucleus, remains a major challenge in gene therapy and vaccine development. To tackle the problems associated with cellular uptake and nuclear targeting, here we introduce a delivery platform based on the self-assembly of an amphiphilic peptide carrying an N-terminal KRKR sequence that functions as a nuclear localization signal (NLS). By means of a single-step self-assembly process, the amphiphilic peptides afford the generation of NLS-functionalized multicompartment micellar nanostructures that can embed various oligonucleotides between their individual compartments. Detailed physicochemical, cellular and ultrastructural analyses demonstrated that integrating an NLS in the hydrophilic domain of the peptide along with tuning its hydrophobic domain led to self-assembled DNA-loaded multicompartment micelles (MCMs) with enhanced cellular uptake and nuclear translocation. We showed that the nuclear targeting ensued via the NLS interaction with the nuclear transport receptors of the karyopherin family. Importantly, we observed that the treatment of MCF-7 cells with NLS-MCMs loaded with anti-BCL2 antisense oligonucleotides resulted in up to 86% knockdown of BCL2, an inhibitor of apoptosis that is overexpressed in more than half of all human cancers. We envision that this platform can be used to efficiently entrap and deliver diverse genetic payloads to the nucleus and find applications in basic research and biomedicine.
Introduction
Aberrant gene expression plays an essential role in numerous diseases, e.g. neurodegenerative diseases, heart dysfunction and muscular dystrophy, both at the level of transcription regulation and/or RNA processing,1–3 which make the nucleus a prominent target in gene therapy.4 In order to treat diseases associated with genetic disorders, therapeutic DNA must navigate its way to the nucleus. Being negatively charged, DNA by itself cannot easily cross cell membranes but requires a vehicle such as a non-viral vector.5 Once inside the cell, non-viral vectors have to overcome the nuclear membrane to deliver DNA to the nucleus. The gateway to and from the nucleus are nuclear pore complexes (NPCs). Generally, molecules smaller than 40–60 kDa passively diffuse through the NPC to the nucleoplasm, while larger molecules require sorting signals termed nuclear localization signals (NLS) for nuclear import.4 NLS are usually short sequences of positively charged lysines or arginines, recognized by nuclear transporters (karyopherins or kaps), which can interact with proteins of the NPCs to translocate NLS-containing proteins through the NPCs into the nucleus.6 Likewise, NLS conjugated vesicles self-assembled of poly(2-methyl-2-oxazoline)-b-poly-(dimethylsiloxane)-b-poly(2-methyl-2-oxazoline) triblock copolymers, were able to successfully traverse the NPCs into cell nuclei through interacting with karyopherins.7Non-viral delivery systems, largely short of a nuclear targeting moiety, are limited in their applications not only by poor uptake and endosomal retention, but also by inefficient nuclear delivery.8 Accordingly, substantial efforts have been directed toward tailoring non-viral gene delivery systems to better overcome plasma and nuclear membrane barriers.9,10 Almost all attempts involving NLS peptide motifs to specifically achieve nuclear targeting of DNA by non-viral vectors9 are focused on peptiplexes11,12 and lipoplexes, such as lipofectamine–nucleic acid complexes.13,14 The intrinsic limitations of these small and rather indistinct structures include in vivo instability, lack of targeting selectivity, low loading and transfection efficiency and initial burst release.15–17 While some examples of lipid-based nano-assemblies decorated with NLS motifs for DNA delivery have been reported,18 improving their stability and controlling their size remains a major hurdle for a successful passage through the nuclear pore complex.19
An efficient alternative to overcome these limitations would be the design of highly ordered peptide nanoassemblies because limitless combinations of amino acid sequences and diverse secondary structures offer many options for enhancing cellular internalization and targeting delivery.20–22 However, to make the peptide prone to self-assembly requires the rational selection of its amino acid building blocks and the precise understanding of their chemical properties that determine the interactions between peptides.
Here we introduce a structurally defined nanoassembly made purely of peptides that is tailored to efficiently entrap DNA, enter cells and ultimately deliver oligonucleotides to the nucleus. Considering hydrophobic interactions as the predominant driving force in this “bottom-up” strategy, we designed amphiphilic peptides as they can self-assemble in aqueous solution and form various nanostructures with desirable properties and functions.23 A particular advantage is that functionality can be directly integrated in the peptide sequence, as long as the effects of the respective amino acids on the self-assembly behavior are taken into account.24,25 In the design of the amphiphilic (KR)2(HR)2(WL)7W peptide, henceforth termed NLS-peptide, we considered that it should (i) comprise a nuclear targeting moiety, (ii) condense nucleic acids via electrostatic interactions, and (iii) self-assemble into multi-compartment micelles (MCMs) that exhibit cell penetration properties. In addition, we integrated four basic residues (KRKR), which are found in many NLS sequences, play a crucial role in nuclear targeting and possibly act as a monopartite NLS motif.26–28 Notably, the optimal consensus patterns required for high-affinity binding to import receptors were found to be KR(K/R)R and K(K/R)RK.29 Although several pieces of evidence indicate that KRKR suffice to function as an authentic NLS30–32 it has not been exploited for the nuclear targeting of nanoparticles so far. Tailoring supramolecular assemblies that display KRKR will allow exploring the effectiveness of this NLS in mediating nuclear import. At the same time, arginine and lysine residues based on their charge, promote interactions at the cell membrane and subsequent internalization.33
To favor self-assembly, the hydrophobic to hydrophilic weight ratio of the peptide was kept similar to our recently reported (HR)3gT nonNLS-peptide which was shown to form multi-compartment micelles (MCMs).34 Because this is the first time a purely peptidic nano-vector is functionalized with the minimal NLS via the extension of the hydrophilic domain with KRKR residues, the morphology and physicochemical properties of the self-assembly structures were systemically investigated. In particular, we examined the incorporation of three different model DNA cargoes into the nanoassemblies and the interactions of a 22nt ssDNA-loaded nanoassemblies with components of the nuclear translocation machinery by surface plasmon resonance. To resolve the nuclear targeting ability of self-assembled NLS-MCMs, ultrastructural and cellular studies with statistical analysis of DNA delivery to the nucleus were carried out and compared to corresponding MCMs lacking an NLS. Owing to the intrinsic cell penetration and nuclear targeting properties of the NLS-peptide and its propensity for self-assembly and efficient DNA entrapment, our NLS-MCMs serve as a promising DNA delivery platform that overcomes biological barriers with minimal cell toxicity.
To put this hypothesis to the test, we have used the NLS-MCM platform to deliver antisense oligonucleotides (ASOs) into the nucleus. ASOs are synthetic DNA oligomers whose potential as therapeutic agents has been extensively explored.35,36 They modulate the stability, processing, or activity of RNA by various mechanisms. In the nucleus, ASO mainly hinders mRNA maturation by inhibiting 5′-mRNA cap formation, by RNA splice modulation, and RNase H-mediated pre-RNA cleavage.37 For example, G3139 (Genasense®), an 18 nucleotide phosphorothioate ASO complementary to the first six codons of Bcl-2 mRNA, triggering RNase H dependent mRNA degradation and the inhibition of mRNA translation.38 The Bcl-2 gene product has been implicated in the growth of a variety of solid tumors including breast and lung cancer, and has been reported to promote chemo- and radiotherapy resistance.39 A ‘gapmer’ version of G3139 (G3139-GAP) containing additional 2′-methoxyethyl (2′-MOE) substitutions at the 5′ and 3′ end was even more efficient than G3139 in downregulating Bcl-2.40 Thus, we produced NLS-MCMs loaded with G3139-GAP and examined their ability to effectively downregulate BCL-2 protein levels in MCF7 cells. Our results unveil the purely peptide-based nano-vector as a promising platform for nuclear-targeted gene delivery applications.
Materials and methods
Materials
Solvents and reagents were purchased from Sigma-Aldrich unless otherwise specified. Fmoc–Trp(Boc)–OH and Rink Amide AM resin (0.71 mmol g−1) were purchased from IRIS Biotech GmbH. Ethyl cyano(hydroxyimino)acetate (Oxyma Pure) and all other Fmoc-protected amino acids were purchased from Novabiochem. Acetonitrile (ACN) and dichloromethane (DCM) were purchased from VWR chemical. Dialysis tubes for solvent exchange were purchased from Spectrum Laboratories (cellulose ester, MWCO 500–1000 Da, 3.2 cm mL−1). Atto550-labeled and unlabeled 18 nucleotide (nt) G3139-GAP, 22nt DNA, and scrambled non-specific oligonucleotide were purchased from Microsynth. All oligonucleotide sequences are provided in Fig. 1A. All purified proteins for SPR binding experiments including cysteine-tagged FG domains of human nucleoporins, Nup62, Nup214, human Kapβ1, Kapα, and wild-type Ran (RanWT) were provided by Lim Lab.41,42 Phosphate buffer saline (PBS) and fetal bovine serum (FBS) was purchased from BioConcept. Dulbecco's modified Eagle's medium (DMEM), Pen/Strep, and Opti-MEM were obtained from Gibco life technologies. Live cell imaging solution was purchased from Thermo Fisher Scientific. CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) was obtained from Promega. | ||
| Fig. 1 Self-assembled peptidic platform for oligonucleotide delivery. (A) Structure and sequence of NLS-peptide and different DNA payloads (table); schematic representation of the self-assembly process by solvent exchange: the amphiphilic NLS-peptide first forms micelles that ultimately assemble to multicompartment micelles (MCMs) with concomitant entrapment of DNA between the individual micelles. In the individual micelles, the hydrophilic domains with the N-terminal NLS (dark blue) are oriented outward and the hydrophobic domains (green) are oriented inward. (Right, middle) Cryo-TEM image of NLS-MCM reveals the multicompartment micellar structure, (B) ultrastructural morphology, and (C) size distribution of self-assembled NLS-MCMs loaded with 22nt ssDNA (left panels) and G3139-GAP (right panels). Scale bars 100 nm. | ||
Peptide synthesis and purification
(KR)2(HR)2gT (NLS-peptide) consisting of 23 amino acids (AA), H2N–[K–R]2-[H–R]2–[W–DL]7–W–NH2, and previously reported (HR)3gT (nonNLS-peptide) consisting of 19 AA, H2N–[HR]3–[W–DL]6–W–NH2, (with DL = D-Leucine) were synthesized as previously reported using Liberty Blue™ automated microwave peptide synthesizer (CEM, Kamp-Lintfort, Germany).34 Purification of crude NLS- and nonNLS-peptides was carried out by reversed phase high performance liquid chromatography (RP-HPLC) (Prominence 20A, Shimadzu, Japan) on a C18-TSE (VDSpher OptiBio PUR 300 C18-TSE, 20 × 250 mm, VDS Optilab, Germany) column. A mobile phase of water and ACN containing 0.1% TFA with a gradient of 35–50% ACN over 40 min for NLS-peptide, a gradient of 25–60% ACN over 30 min for nonNLS-peptide was used to separate peptides. Peptide purification was monitored at 280 nm. Fractions of purified peptides were collected, lyophilized and stored at −20 °C. The molecular mass of peptides was determined by PerSeptive Biosystems Voyager-DE-PRO time-of-flight mass spectrometer (MALDI-TOF-MS) in positive mode.Self-assembly of peptide nanoparticles
For self-assembly of empty and DNA-loaded peptide nanoparticles from the respective lyophilized peptide, a 1 mg mL−1 peptide stock solution was prepared in ethanol/water (50/50, v/v). Self-assembly was carried out by solvent exchange method through dialyzing the organic solvent (ethanol) against Milli-Q H2O (Merck Millipore, Milli-Q Direct 8 water purification system). To prepare DNA-loaded nanoparticles, the following synthetic DNA cargoes with and without 5′ Atto550 fluorescent label were prepared at a concentration of 100 μM in water: 22nt single-stranded DNA (22nt ssDNA), 18nt G3139-GAP ASO and scrambled non-specific sequence (sequences are provided in Fig. 1A). In order to prepare nanoparticles loaded with DNA, 100 μL of peptide stock solution were mixed with 3 μg of DNA (100 μM solution). The DNA–peptide mixture was then adjusted to a final volume of 500 μL in a final concentration of 35% ethanol and transferred to a prewashed 500–1000 MWCO dialysis tube. The self-assembly was induced by dialysis at 4 °C for approximately 20 h against two changes of 500 mL ultrapure DNase/RNase-free distilled water. To produce the control nanoparticles without DNA, peptide solutions were diluted to a concentration of 0.2 mg mL−1 in a 35% ethanol final concentration and dialyzed under the same conditions.Dynamic light scattering
The mean hydrodynamic diameter and polydispersity index (PDI) of self-assembled peptide nanoparticles were determined at 25 °C by dynamic light scattering (DLS) using a Zeta Sizer Nano ZSP (Malvern Instruments Ltd, UK) instrument at a wavelength of 633 nm with an angle detection of θ = 173°. All measurements were performed in triplicate.Zeta-potential
The zeta-potential of the self-assembled peptide nanoparticles in water was recorded by Zeta Sizer Nano ZSP (Malvern Instruments Ltd, UK) in a cuvette after each polyelectrolyte deposition. Zeta potential data represent the mean of three consecutive measurements.Nanoparticle tracking analysis
The nanoparticle tracking analysis (NTA) of the self-assembled peptide nanoparticles was performed using a NanoSight NS 300 instrument (NanoSight Ltd, Amesbury, UK). First, peptide nanoparticles were diluted 20-fold and applied to the viewing chamber using a 1 mL syringe. Nanoparticle movement was analysed by the NTA software (version 3.4, NanoSight) based on tracking each particle on a frame-by-frame basis. For each measurement, three videos of 60 s were captured at room temperature and the software provided the mean and median particle size together with the estimated concentration of nanoparticles in solution.Transmission electron microscopy
For transmission electron microscopy (TEM), aliquots of self-assembled peptide nanoparticles were deposited on a glow-discharged, carbon-coated, parlodion-(2% in n-butyl acetate) copper grid. After 2 min adsorption, excess liquid was blotted with a filter paper and grids were negatively stained with 5 μL 2% uranyl acetate for 10 s. Stained grids were washed with water and dried for 3 times. Grids were examined by a CM100 transmission electron microscope (Philips, Eindhoven, The Netherlands) at an accelerating voltage of 80 kV.Cryogenic electron microscopy
Four microliters of self-assembled peptide nanoparticles were adsorbed onto glow discharged carbon-coated grids (Lacey, Tedpella, USA). Excess sample was blotted off with grade 1 Whatman filter paper for seconds to produce a thin aqueous film which is subsequently vitrified by plunge freezing. Frozen grids were transferred at −178 °C into a Gatan 626 cryoholder and imaged by Talos electron microscope (FEI, USA). Electron micrographs were recorded at an accelerating voltage of 200 kV while keeping the sample at low temperatures. Resulting cryo-EM micrographs were recorded on a CETA camera (4096 × 4096 pixels; Thermo Fisher).Fluorescence correlation spectroscopy
Fluorescence correlation spectroscopy (FCS) experiments were performed at 20 °C on a Zeiss LSM 880 laser-scanning microscope equipped with a 40× water immersion objective (C-Apochromat 40×, NA 1.2) (Carl Zeiss, Jena, Germany) as described previously.34 In brief, for each measurement, 10 μL of sample were placed on a 22 × 50 mm glass slide and the measurements were performed using a helium/neon laser for 561 nm excitation, a 488/561/633 main beam splitter (MBS), and a pinhole size of 40 μm. Fluorescence signals were collected in a real time (5 s with 30 repetitions) and autocorrelation function was obtained by a QuickFit 3.0 software calculator.Surface plasmon resonance
Surface Plasmon Resonance (SPR) spectroscopy was used to evaluate Kapα·Kapβ1·NLS-MCMs interactions with FG Nups (Nup62, Nup214). All SPR binding assays were performed at 25 °C in HEPES (0.025 M) with 5 mM MgCl2, using a four flow cell BiacoreT200 instrument (GE Healthcare), as described previously.41,43 Briefly, HS–(CH2)11–(OCH2–CH2)3–OH (abbreviated as PUT, Nanoscience) and cysteine-terminated FG Nups were conjugated to a gold sensor chip in cell 1 (as a non-specific reference), and in cell 2 (sample). 1% (wt/vol) BSA (Sigma-Aldrich) solution was prepared in HEPES (0.025 M) with 5 mM MgCl2, pH 7.2 as a running buffer. All proteins and peptide nanoparticles were dialyzed against the same buffer before the experiment. Ten titrations of the Kapα·Kapβ1 complex were prepared by a factor 2 dilution of 400 nM Kapβ1 and 800 nM Kapα (starting maximal concentration). The NLS and non-NLS peptides were added to the Kapα·Kapβ1 complex at a ratio of peptide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Kapα:Kapβ1 of 1:4:2, terminating with a maximum concentration of 200 nM of NLS and non-NLS peptides (for 400 nM Kapβ1 and 800 nM Kapα). Before being injected, buffer solutions were filtered (0.22 μm) and degassed and all samples, protein and reagent solutions were centrifuged for 15 min at 12000 rpm to remove particles and bubbles. The binding data were analyzed using Biacore T200 Control software.
:Kapα:Kapβ1 of 1:4:2, terminating with a maximum concentration of 200 nM of NLS and non-NLS peptides (for 400 nM Kapβ1 and 800 nM Kapα). Before being injected, buffer solutions were filtered (0.22 μm) and degassed and all samples, protein and reagent solutions were centrifuged for 15 min at 12000 rpm to remove particles and bubbles. The binding data were analyzed using Biacore T200 Control software.
Thermo-responsiveness of peptide nanoparticles
To analyze the temperature response of self-assembled peptide nanoparticles, 200 μL of empty and DNA-loaded peptide nanoparticles were diluted two-fold in water and incubated at 37 °C for the times indicated. Nanoparticles size and morphology were examined by DLS and TEM before incubation and at the time points indicated.Cell culture
HeLa and Histone H2B-GFP expressing HeLa, and MCF-7 human breast carcinoma cell lines were routinely subcultured in DMEM with 1× Glutamax-I (4.5 g L−1 glucose; Gibco) supplemented with 10% fetal bovine serum (FBS; BioConcept). A549 human lung carcinoma and THP-1 human leukemia monocytic cell lines were cultured in RPMI-1640 (BioConcept) supplemented with 10% FBS and 1× GlutaMAX (Gibco). All cells were cultured at 37 °C in a humidified atmosphere with 5% CO2 up to 25 passages. For incubation with MCMs, media were supplemented with 1× penicillin–streptopmycin (Gibco).MTS cell viability assay
For cytotoxicity assessment of peptide nanoparticles, tetrazolium compound-based Cell Titer 96® AQueous One Solution Cell Proliferation Assay (MTS, Promega) was used according to the manufacturer's instructions. One day before nanoparticle treatment, HeLa cells were seeded in a 96-well plate at 3 × 103 cells in 100 μL DFBS per well. Subsequently NLS-peptide nanoparticles with and without DNA-payload were added to each well to a final peptide concentration of 50, 300, 550, 800, 1050, 1300, 1550 μg ml−1. In addition, lipofectamine–DNA complex as the gold standard for transfection was included at the highest peptide concentration (corresponding to 1550 μg peptide per ml and 96 ng DNA) to compare the cytotoxicity. Untreated cells were used as a reference value for 100% viability. After 24 h of incubation at 37 °C, 20 μl of MTS reagent was added to each well, and the plate incubated for 2 h at 37 °C. The absorbance of the plate was then measured at λ = 490 nm using a Spectramax id3 plate reader. Background absorbance of wells containing all assay components without cells was subtracted from each test well and the data was normalized to untreated control cells. All experiments were performed in triplicate wells for each condition and repeated at least twice.Cellular uptake of peptide nanoparticles
For nanoparticle uptake experiments, 3 × 104 cells per well were seeded in an eight-well glass-bottom μ-slide (Ibidi, Germany) and cultured for 24 h before equal concentrations of NLS- and nonNLS-peptide nanoparticles (determined by NTA) were added to each well, except for THP-1 which were induced to differentiate into macrophages by adding phorbol 12-myristate 13-acetate (PMA) to a final concentration of 100 nM and incubated for 48 h. Cells were imaged on a confocal laser-scanning microscope (CLSM) (Zeiss LSM 880, Carl Zeiss Meditec AG, Jena, Germany) at 5 and 24 h using the same image acquisition settings for treated and untreated cells. Moreover, to compare the relative fluorescence intensities of DNA-loaded nonNLS- and NLS-MCMs after cell uptake, CLSM images of Histone H2B-GFP expressing HeLa cells at different timepoints (0, 5, 10, 24, 36 and 48 h) were captured using C-Apochromat 40×/1.2 NA Korr FCS M27 water-immersion objective. The mean fluorescence intensity of Atto550 was measured within squares of 100 μm × 100 μm (containing at least 10 cells) using ZEN Blue software (v.3.2, Carl Zeiss Microscopy GmbH). Background fluorescence signal was subtracted from the measurement points.Protein extraction
2 × 104 cells per well were seeded in a 12-well plate and incubated overnight before addition of ASO-loaded MCMs or control MCMs for 48 h. After rinsing cells three times with ice-cold PBS, 400 μl of RIPA buffer (Merck Millipore) containing a Halt™ protease inhibitor cocktail, EDTA free (ThermoFisher) were added to each well and cells gently scraped off the surface using a rubber cell scraper. Cell lysates were then transferred to 1.5 ml Eppendorf tubes and incubated for 30 minutes at 4 °C with constant agitation. The cell lysates were spun for 20 min at 12000 rpm at 4 °C. The supernatant was gently transferred to a fresh tube and protein concentrations were determined using Pierce™ bicinchoninic acid (BCA) Protein Assay Kit (ThermoFisher). The lysates were aliquoted and stored at −80 °C.
Evaluation of BCL-2 protein expression by western blotting
Aliquots of total protein extracts normalized for concentration according to the BCA assay were denatured in Laemmli sample buffer at 95 °C for 5 minutes (ThermoFisher) and loaded onto any kD™ mini-PROTEAN® TGX™ precast protein gels (Bio-Rad). Proteins were resolved by SDS-polyacryl amide gel electrophoresis and electrophoretically transferred onto trans-blot turbo mini prewetted PVDF membranes (Bio-Rad) according to the manufacturer's instructions. The membrane was blocked with 5% milk in PBS/0.1% Tween 20 (PBST) for 2 h at room temperature with gentle agitation. The membrane was then cut horizontally at approximately 32 kDa and the lower half was incubated with 1:500 dilution monoclonal mouse anti-human Bcl-2 (ThermoFisher; Cat. no. 13-880) and the upper with 1:5000 dilution polyclonal rabbit anti-human GAPDH antibody (Bio-Rad; Cat. no. VPA00187) at 4 °C overnight with gentle agitation. After washing the blots three times for 5 minutes with PBST, the membrane containing BCL-2 proteins were probed with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody (ThermoFisher; Cat. no. G21040) at 1:5000 dilution and the membrane containing GAPDH proteins were probed with secondary HRP-conjugated goat anti-rabbit IgG (Azure, Cat. no. AC2114) at 1:10000 dilution for 2 h at room temperature. The membranes were then washed three times for 5 minutes with PBST and soaked in HRP chemiluminescence substrate (Radience Plus solution, Azure) for 30 seconds to detect the secondary HRP conjugates. western blotting images were acquired on an Azure Sapphire Biomolecular Imager. The bands were analyzed by Image Lab software from Bio-Rad.
Ultrastructural analysis of peptide nanoparticles in cells
HeLa cells (8 × 105 cells) were seeded in 100 mm × 15 mm round culture dishes (Thermo Fisher) and grown in DFBS. After 24 h, 1.28 × 107 ± 4.28 × 106 of DNA-loaded NLS- or nonNLS-peptide nanoparticles (determined by NTA) were added to each culture dish. One plate was left untreated as control. Cells were incubated for 16 h at 37 °C in 5% CO2 and then washed four times with PBS. Cells were fixed with 1:1 (v/v) medium:double strength fixative mixture (single strength fixative solution consists of 2.5% glutaraldehyde + 2% paraformaldehyde in 0.1 M PIPES buffer, pH 7) for 20 min at room temperature, harvested with cell scraper and pelleted by centrifugation at 300 rpm for 5 min at room temperature. Cell pellets were then suspended in single strength fixative for 1 h at 4 °C, then washed with 0.1 M sodium cacodylate buffer three times and embedded in low melting 2% agarose. Once solidified, agarose pellets were trimmed into 1–2 mm cubes that were post-fixed in 1% buffered osmium tetroxide for 1 h at 4 °C, rinsed in distilled water, and finally stained en bloc with aqueous 1% uranyl acetate for 1 h at 4 °C in the dark. Cubes were then dehydrated in an ethanol series (30, 50, 75, 95% and 100%). After three changes of absolute ethanol, samples were washed in acetone and finally embedded in a mixture of resin (EPON812)/acetone followed by pure Epon 812 resin. Ultra-thin sections (50–70 nm thick) were cut and mounted on grids according to the regressive EDTA staining protocol.44 In brief, the ultrathin sections were floated in a 6% aqueous uranyl acetate solution for 5 min. Thereafter, sections were rinsed with double distilled water (ddH2O) and a second floating step in 0.2 M EDTA in ddH2O, pH 7.0 was performed. The sections were rinsed again with ddH2O and stained with lead citrate for 5 min. After the final rinsing step with ddH2O, sections were imaged on a FEI Tecnai T12 Transmission Electron Microscope operating at an accelerating voltage of 120 kV. Micrographs were recorded using a TVIPS F416 CMOS digital camera (bottom mounted).
Statistical analysis of localized peptide nanoparticles under live cell conditions
Histone H2B-GFP expressing HeLa cells (3 × 104 cells per well) were seeded in DFBS medium on an eight-well glass-bottom μ-slide (Ibidi, Germany). After culturing for 24 h, 3.22 × 108 ± 6.65 × 107 of NLS or non-NLS peptide nanoparticles (determined by NTA) were added to each well. The 8-well slide was immediately mounted on a microscope stage equipped with a CO2 chamber and a heating module (37 °C, 5% CO2) for live cell confocal imaging. Samples were excited with 488 nm (green) and 561 nm (red) laser lines. Images were obtained with a C-Apochromat 40×/1.2 NA Korr FCS M27 water-immersion objective at an image acquisition resolution of 2048 × 2048 pixels. Pinhole diameters were set to 1 airy unit and at each timepoint, Z-stacks were captured the with Z-resolution of 0.26 μm. To statistically evaluate the NLS-peptide nanoparticle translocation to the nucleus, cells were imaged at 5, 10, 36, and 48 h. Image stacks were analyzed using IMARIS 9.2 software (Bitplane AG, Switzerland) on a pixel basis. Accordingly, 3D Imaris reconstructions from multiple Z-stacks were created to identify and quantify peptide nanoparticles inside and on the membrane of the nucleus. Based on the fluorescence of the objects, the Atto550-DNA-loaded peptide nanoparticles were identified as ‘spots’ and Histone H2B-GFP expressing nuclei as ‘surfaces’. Quantification of peptide nanoparticles in each image stack was performed using the SPOTS option of the IMARIS software. SPOTS option allows the detection and visualization of spherical objects in each image stack defining their X, Y and Z axial positions, size and signal intensity. Colocalization analysis of peptide nanoparticles and nuclei was performed on confocal image Z-stacks using the IMARIS distance to surface feature. Nanoparticles detected within ±0.1 μm distance to the nuclear envelope were considered associated with the nuclear membrane. Nanoparticles with a distance above 0.1 μm far away from the nuclear face of the nuclear membrane were considered to be inside the nucleus. Student's two-tailed t-test was used to demonstrate the significance of the statistical difference between NLS and non-NLS nanoparticles. A p value ≤0.05 was considered significant. The number of analyzed nuclei for 5, 10, 36 and 48 h timepoints were 38, 44, 78, 98 and 41, 41, 78, 116 for non-NLS and NLS nanoparticles, respectively. Particle counting was carried out with IMARIS 9.2 software.Results and discussion
Despite the increasing availability of non-viral delivery systems, strategies for intracellular targeting, in particular targeting the nucleus, remain in demand.9,45,46 To tackle this problem, we designed an amphiphilic peptide that is prone to self-assembly, thereby entrapping oligonucleotides, and also to target the nucleus via a short stretch of amino acids representing an NLS (Fig. 1A). In this NLS-peptide, we modified the hydrophilic and hydrophobic domain of a recently reported self-assembling (HR)3(WL)6W peptide, henceforth termed nonNLS-peptide.34 The hydrophilic domain included KRKR32 at the N-terminus, followed by two HR repeats. The hydrophobic domain comprised repeating units of tryptophan–leucine which correspond to a truncated version of gramicidin A.47 To maintain a hydrophilic to hydrophobic weight ratio conducive to multicompartment micelles assembly, the hydrophobic domain was extended by an additional tryptophan–leucine repeat compared to nonNLS-peptide.48 The resulting 23 amino acid NLS-peptide, (KR)2(HR)2(WL)7W, was synthesized using standard Fmoc-based solid phase peptide synthesis and purified by RP-HPLC. The expected molecular mass of 3455 g mol−1 was confirmed by MALDI-TOF mass spectrometry (Fig. S1, ESI†).Based on our previous findings with the nonNLS-peptide,34 we surmised that the cationic NLS-peptide would be able to entrap DNA between individual micelles of a multicompartment micelle platform via electrostatic interactions (Fig. 1B, schematic). Therefore, we induced self-assembly of the NLS-peptide by solvent exchange in the presence of three different DNAs: 22nt ssDNA, 18nt G3139-GAP (an antisense oligonucleotide gapmer against Bcl-240) or an 18nt scrambled, non-specific oligonucleotide (Fig. 1A). Ultrastructural analysis of the resulting nanoassemblies by electron microscopy revealed a spherical morphology and multi-micellar architecture of DNA-loaded NLS-MCMs similar to that observed for nonNLS-MCMs (Fig. 1B and C; Fig. S2, ESI†). NLS-MCMs have a hydrodynamic diameter, DH of 94 ± 8 nm for 22nt ssDNA and 48 ± 12 nm for G3139-GAP payloads as measured by dynamic light scattering (DLS) (Fig. 1C) with a polydispersity index (PDI) of 0.18 and 0.26, respectively. In addition, DH determined by nanoparticle tracking analysis (NTA) was consistent with DLS measurements (Fig. S3, ESI†). It is noteworthy that an average particle size below 100 nm showed maximum cellular uptake regardless of the nanoparticle core composition or surface charge.49 DNA-free NLS-MCMs had the same PDI (0.18) as their DNA-loaded counterpart but exhibited a larger hydrodynamic diameter of 145 ± 4 nm (Fig. S4, ESI†). The NLS-MCM size reduction by DNA incorporation is consistent with our previously reported observation that electrostatic interactions between DNA and peptide lead to more compact assemblies.34
Zeta potential measurements showed that the surface charge of NLS-MCMs decreased from 45 ± 7 mV to 20 ± 5 mV when negatively charged 22nt ssDNA was entrapped and to 25 ± 6 mV for entrapped G3139-GAP ASO (Fig. 2A). These data support that the majority of DNA molecules are entrapped between the individual micelles rather than being merely accumulated on the NLS-MCM surface which would yield a more negative zeta potential. The zeta potential around ±20 mV reflects the physical stability of NLS-MCMs due to the electrostatic repulsion forces between NLS-MCMs which hinder their aggregation. In addition, a zeta potential of ±20 mV was shown to be advantageous for an effective accumulation of non-viral vectors in target cells.50 Mean hydrodynamic diameters (DH), polydispersity indices (PDI) and zeta potentials for the scrambled oligo-loaded NLS-MCMs and nonNLS-MCMs loaded with different oligonucleotides are presented in Table S1.† All MCMs showed a narrow size distribution peaking below 100 nm, independent of the oligonucleotide payload.
 | ||
| Fig. 2 Quantification of NLS-MCM surface charge and DNA entrapment. (A) Zeta potential of NLS-MCMs with 22nt ssDNA (blue), with G3139-GAP (green) or without DNA (pink). (B) Normalised FCS autocorrelation curves of free Atto550-labeled 22nt ssDNA (red), free Atto550-labeled G3139-GAP (orange), Atto550-labeled 22nt ssDNA-loaded NLS-MCMs (blue), Atto550-labeled G3139-GAP-loaded NLS-MCMs (green). Dotted lines represent experimental autocorrelation curves and solid lines are fitted curves. | ||
The incorporation of DNA into NLS-MCMs was assessed by FCS using Atto550-labeled 22nt ssDNA or G3139-GAP. Diffusion times (τD) of free DNA and DNA-loaded NLS-MCMs in solution are displayed by fitting the experimental autocorrelation curves to one- and two-component fits, respectively, where τD of the free DNA was fixed as the first component (Fig. 2B).34 The increased τD of G3139-GAP-loaded NLS-MCMs (3778 ± 805 μs) compared to free G3139-GAP (138 ± 28 μs) and 22nt ssDNA-loaded NLS-MCMs (3898 ± 1028 μs) compared to free 22nt ssDNA (208 ± 12 μs) is indicative of DNA entrapment by MCMs. Moreover, the number of DNA molecules per MCM was calculated to be 17.8 ± 3.7 (G3139-GAP) and 14.3 ± 4.4 (22nt ssDNA) based on average brightness measurements and dividing the photon count per molecule of DNA-loaded NLS-MCMs to free DNA.
To evaluate if MCMs at 37 °C are able to dissociate, which conceivably facilitates cargo release, the hydrodynamic diameter and morphology of NLS-MCMs with and without DNA were examined over time (after 8, 24, 48 and 60 h). Neither DLS nor TEM analyses revealed a change in size or morphology of MCMs after 8 h at 37 °C. However, after 24 h, temperature-induced disassembly was seen for NLS-MCMs, both with and without DNA, which continued over time (Fig. S5, ESI†). DLS and TEM revealed the gradual formation of two populations, one consisting of dispersed or clustered smaller MCMs, and the other of individual micelles. Thermo-responsive disassembly (change in size and structure of MCMs) occurred faster in ‘empty’ NLS-MCMs compared to DNA-loaded NLS-MCMs (Fig. S5A and B, ESI†), suggesting that the presence of DNA affected the disassembly behaviour. It is conceivable that the electrostatic interactions between negatively charged DNA entrapped between positively charged micelles hold micelles together more tightly in DNA-loaded MCMs, resulting in a more compact MCM structure which takes longer to disassemble in response to temperature.
To evaluate the cytotoxicity of our peptidic DNA delivery platform, we measured cell proliferation/viability of MCF-7 cells treated for 24 and 48 h with different types of MCMs (Fig. 3) at a peptide concentration of 0.75 mg ml−1.
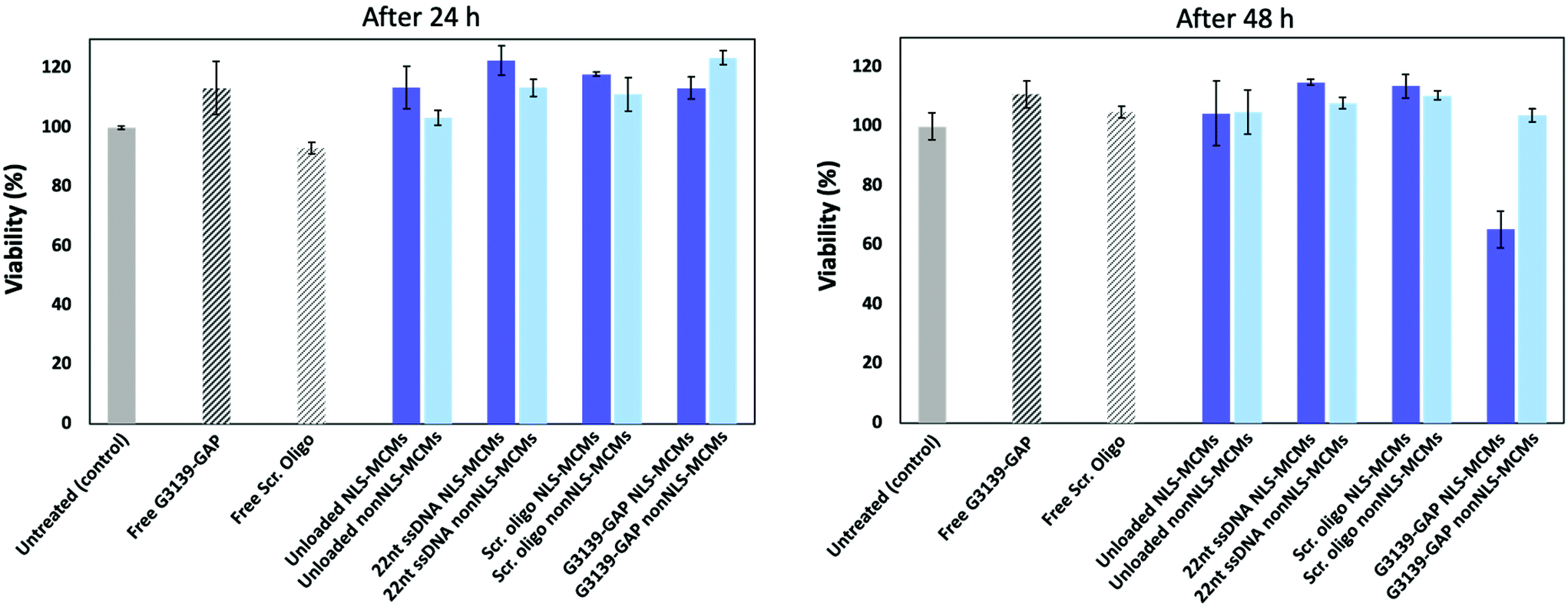 | ||
| Fig. 3 Effect of MCMs on MCF-7 cell proliferation after 24 and 48 h of exposure at 37 °C. Viability of cells treated with NLS-MCMs (dark blue bars) and nonNLS-MCMs (light blue bars) compared to untreated MCF-7 (grey). Cells incubated with free oligonucleotides are shown as shaded bars. Error bars represent standard deviation of triplicate measurements. | ||
Without DNA cargo, the NLS-MCMs and the nonNLS-MCMs did not affect cell viability after 24 h or 48 h of incubation, showing that the platform by itself is not cytotoxic, also at higher peptide concentrations (Fig. S6A, ESI†). Similarly, both NLS- and nonNLS-MCMs loaded with control 22nt ssDNA, scrambled oligonucleotide or ASO had no toxic effects on cell viability after 24 h of exposure. In contrast, lipofectamine-mediated transfection of 22nt ssDNA at the same concentration used for incorporation into NLS-MCMs (96 ng) demonstrated a 40% lower cell viability compared to non-transfected cells (Fig. S6A, ESI†). After 48 h, however, cells treated with G3139-GAP loaded NLS-MCMs demonstrated a reduced cell viability of 65 ± 6%. Conceivably, this reduction is caused by G3139-GAP delivery since G3139 ASO was shown to reduce Bcl-2 expression which was associated with apoptosis and a decrease in cell proliferation.51–54 Whereas scrambled oligo loaded NLS-MCMs had no toxic effects (Fig. 3), using lipofectamine reagent to transfect scrambled oligonucleotides revealed a 20 ± 3% decrease in cell viability after 48 h (Fig. S6B, ESI†). The apparent absence of cytotoxicity renders our peptidic platform a promising candidate for therapeutic applications.
Typically, selective transport through the NPC is facilitated by the major importin receptor Kapβ1 that binds NLS-cargoes via its adaptor, karyopherin alpha (Kapα). This is because Kapβ1 exerts multivalent interactions with intrinsically disordered phenylalanine–glycine (FG)-repeat nucleoporins (FG Nups) that form the selective NPC barrier.42 Upon reaching the nuclear interior, Ran guanosine triphosphate (RanGTP) binds Kapβ1, which results in NLS-cargo release.55 In this manner, the RanGTP gradient controls the directionality of transport by triggering the release of cargo inside the nucleus.7 Accordingly, we used surface plasmon resonance (SPR) to assess the ability of Kapα·Kapβ1 complexes to mediate the binding of NLS-MCM nanocarriers to surface tethered-FG Nup layers comprising of either Nup214 or Nup62 (Fig. 4A and Fig. S7, ESI†).56
 | ||
| Fig. 4 NLS-MCM interactions with components of the nuclear import machinery. (A) NLS-MCMs binding to FG Nups facilitated by Kaps. (B) Langmuir isotherm fits (solid lines) yielding the maximal response signal (Rmax; circles). NLS-MCM binding response measured by SPR elicited the highest FG Nup-binding response (blue). NonNLS-MCMs (green) do not bind to the Nup 214 while they demonstrate binding to Nup 62, still lower than NLS-MCMs. Standalone Kapα Kapβ1 binding is shown in red. | ||
Langmuir isotherm analyses using a two-component fit (Fig. 4B) demonstrate that there is no significant difference (within error) in the apparent equilibrium dissociation constants (KD1 and KD2) for the standalone Kapα·Kapβ1 and Kapα·Kapβ1·NLS-MCMs complexes with Nup214 or Nup62 (see Table 1). Such non-divergent binding is anticipated for multivalent species that exceed ten binding sites (e.g. Kaps).57 Obviously, no difference is expected in the presence of nonNLS-MCMs, which do not interact with the Kapα·Kapβ1 and as such do not interfere at all with the Kapα·Kapβ1 binding to FG Nups. The equilibrium dissociation constants together with the maximal SPR binding responses (Rmax1 and Rmax2 in resonance units [RU]) for the binding components with KD1 and KD2, respectively were obtained for NLS-MCMs and nonNLS-MCMs (Table 1).
| R max1 (RU) | R max2 (RU) | K D1 (nM) | K D2 (nM) | ||
|---|---|---|---|---|---|
| NLS-MCMs/Kapα·Kapβ1 | Nup214 | 3352.3 ± 373 | 7599.7 ± 1840 | 28.2 ± 4.7 | 1183.5 ± 652 |
| Nup62 | 2967.6 ± 399 | 16295 ± 1470 | 36.7 ± 7.4 | 3387.6 ± 4190 | |
| NonNLS-MCMs/Kapα·Kapβ1 | Nup214 | 2723.3 ± 415 | 5711.8 ± 1480 | 23.2 ± 5.5 | 954.8 ± 647 |
| Nup62 | 2057.2 ± 140 | 10201 ± 1260 | 20.8 ± 2.4 | 1423 ± 332 | |
| Kapα·Kapβ1 | Nup214 | 2672 ± 387 | 5711.4 ± 1850 | 19.4 ± 4.8 | 1028.1 ± 779 |
| Nup62 | 1895 ± 229 | 7495.2 ± 1420 | 22.4 ± 5.7 | 1077.3 ± 477 |
An increase of 680 and 1888 RU was detected in the Nup214 binding responses (Rmax1 and Rmax2) of NLS-MCMs/Kapα·Kapβ1 over standalone Kapα·Kapβ1. Furthermore, the binding of NLS-MCMs/Kapα·Kapβ1 to Nup62 gave a significantly larger increase of 1072 and 8800 RU in the maximal binding responses over standalone Kapα·Kapβ1. In contrast, in the presence of nonNLS-MCMs, no (in case of Nup214) or a much smaller (in case of Nup62) increase in the maximal binding response was observed for the Kapα·Kapβ1 binding to the corresponding Nup, which confirms that the observed extra response in case of NLS-MCMs/Kapα·Kapβ1 could be attributed to the additional NLS-MCMs mass in the binding complex. Possibly, the (HR) motif, which is present in nonNLS-MCMs and NLS-MCMs, could play part in the interactions with Kapα·Kapβ1 and thereby contribute to the overall binding to distinct Nups. Such interactions would explain the minor binding response of nonNLS-MCMs, and ultimately its presence in the nucleus (see below). Consistent with this notion, arginine-rich regions were shown to interact with binding sites of Kapα. For example, the arginines in NLS-STATs (signal transducers and activators of transcription) contribute to the weak interactions with minor binding sites of importin α.55,58 Alternatively, arginine residues have recently been shown to play a role in the affinity of Fused in Sarcoma (FUS) RNA-binding protein for the nuclear import receptor karyopherin β2.59,60 A systematic analysis will reveal in more detail the interactions of NLS-MCMs with the different importins. Nevertheless, our results suggest that a genuine NLS is more effective in coupling MCMs to the nuclear transport machinery. In addition, to compare the NLS functionality in self-assembled MCMs to that of free NLS-peptide, the binding of both species in the presence of Kapα·Kapβ1 complexes to FG Nups was determined (Fig. S8, ESI†). The maximal response signal (Rmax1 and Rmax2) and the equilibrium dissociation constants (KD1 and KD2) are summarized in Table S2.† NLS-MCM binding to Nup214 and Nup62 provoked a maximal binding response that was approximately ∼0.5 and 1 kRU higher than free NLS-peptide. This data corroborates that when self-assembled into MCMs, NLS-peptides, particularly at higher concentrations, more efficiently expose binding sites for karyopherin complex formation that is required for the interaction with FG Nups. Thus, the high surface to volume ratio offered by MCMs represents an attractive approach to substantially enhance the interactions of biomolecules with their microenvironment and modulated the physicochemical properties with regard to the original molecule.61,62
A critical prerequisite for an efficient DNA delivery system is cellular uptake, predominantly through different endocytosis pathways,63 followed by endosomal escape (Fig. 5A, I). Uptake of NLS-MCMs loaded with fluorescent DNA in H2B-GFP expressing HeLa cells was examined by CLSM at 5, 10, 24, 36 and 48 h (Fig. 5B and Fig. S9, ESI†). Statistical analysis of corresponding relative fluorescence intensities revealed that uptake efficiency was at least 2-fold higher for NLS- than for nonNLS-MCMs. Furthermore, to ensure that the enhanced uptake of ASO-loaded NLS-MCMs is not cell-type specific, the cellular uptake of NLS- and nonNLS-MCMs loaded with Atto550-G3139-GAP was also examined in A549, PMA-stimulated THP-1 and MCF-7 cells. For all cell lines tested, internalization of NLS-MCMs after 24 h was more efficient than nonNLS-MCMs (Fig. S10, ESI†).
 | ||
| Fig. 5 (A) Schematic representation of (I) cellular uptake and endosomal escape of NLS-MCMs, and (II) their translocation to the nucleus via NPC (created with BioRender.com). (B) CLSM merged images of H2B-GFP expressing HeLa cells recorded after 24 h incubation with DNA-loaded nonNLS- and NLS-MCMs and corresponding relative fluorescence intensities quantified after 5, 10, 24, 36 and 48 h. Scale bars, 20 μm. (C) Ultrastructural localization of DNA-loaded NLS-MCMs (left) and nonNLS-MCMs (right) in cells. Electron micrographs reveal NLS-MCMs accumulation at the nuclear envelope, associated with NPCs, and inside the nucleus. Scale bars, 1 μm. | ||
The enhanced cellular uptake can be attributed to different properties conferred upon MCMs by the NLS-peptide. The KR repeats constituting the NLS afford also cell penetrating properties and when exposed on the surface of NLS-MCMs, aid in overcoming biological barriers.64 Many studies firmly support the significance of various combinations of lysine and arginine residues in boosting the ability of nanoparticles to enter cells.25,65,66 Moreover, the higher positive surface charge and smaller size of NLS-MCMs compared to nonNLS-MCMs play a crucial role in the enhanced uptake.67 Subsequently, to finally end up in the nucleus, nanocarriers need to pass through the nuclear pore complexes (NPCs) (Fig. 5A, II).68
To reveal the fate of MCMs inside the cell at the ultrastructural level, ultrathin serial sections of cell pellets were prepared from HeLa-GFP treated with NLS-MCMs and nonNLS-MCMs, and examined by electron microscopy (Fig. 5C). Electron micrographs showed that NLS-MCMs accumulate at the cytoplasmic face of the nuclear envelope where they are often associated with NPCs (Fig. 5C). In addition, MCMs were frequently found inside the nucleus (Fig. S11B, ESI†). At first sight, the initial mean hydrodynamic diameter of 22nt-loaded NLS-MCMs (around 95 nm), in particular when increased to 110 nm by a ‘Kap corona’, appears to be incompatible with the pore size of the NPC even if the latter is more dilated in cellulo (∼70 nm-diameter).69 However, NLS- and nonNLS-MCMs are thermo-responsive and with time, disintegrate at 37 °C into smaller assemblies (Fig. S5, ESI†) that are then able to translocate through the NPC into the nucleus. Consistently, the number of particles inside the nucleus increased around 5-fold between 36 and 48 h (Fig. 6B). Moreover, entrapment of ASO further compacted NLS-MCMs which reduced their size to around 50 nm (Fig. 1C) such that nuclear translocation was evident already after 24 h (Fig. S10, ESI†). In contrast, nonNLS-MCMs were mainly distributed throughout the cytoplasm. Despite the occasional proximity of nonNLS particles to the nuclear membrane, a clear association with the NPC was not evident (Fig. 5C). This data provides further evidence that the KRKR residues at the hydrophilic end of the peptide serve as functional NLS sequence, able to mediate trafficking of micellar particles to the nucleus. Furthermore, the data are consistent with our SPR results which revealed no (in case of Nup214) or much smaller (in case of Nup62) binding response for nonNLS-MCMs compared to NLS-MCMs.
 | ||
| Fig. 6 Statistical analysis of nuclear localization of peptide nanoparticles (A) Imaris 3D reconstructions of multiple confocal sections of HeLa-GFP cells treated for 10 h with DNA-loaded NLS-MCMs and DNA-loaded nonNLS-MCMs. MCMs are represented by red dots and the nuclear boundaries as green ‘surfaces’, based upon the fluorescence of the corresponding dyes. (B) Statistical analysis of NLS- compared to nonNLS-MCMs localized inside the nucleus and at the nuclear membrane at different time-points. Bars represent number of MCMs per nucleus. Top, MCMs detected within a distance of ±0.1 μm to the nuclear membrane were considered on the membrane. Bottom, MCMs at a distance >0.1 μm away from the nuclear face of the membrane were considered inside the nucleus. The differences for each time-point were statistically significant between NLS- and nonNLS-MCMs (*** p < 0.001 and **** p < 0.0001) on both graphs. Error bars represent standard error of the mean (SEM). The number of analyzed nuclei for 5, 10, 36 and 48 h timepoints were 38, 44, 78, 98 and 41, 41, 78, 116 for nonNLS- and NLS-MCMs, respectively. | ||
For a statistical evaluation, we investigated HeLa cell expressing genetically GFP-tagged histone 2B in order delineate the cell nucleus. HeLa-GFP cells were treated with NLS-MCMs or nonNLS-MCMs loaded with fluorescently labeled DNA by confocal laser scanning microscopy. Multiple z-stacks of cells were recorded at 5, 10, 36, and 48 h of MCM incubation and used for 3D reconstructions based on which the number of MCMs associated with the nuclear membrane and inside the nuclei were determined (Fig. 6A).
Statistical analysis of MCM distribution at different time-points demonstrated that there is a significant difference in nuclear localization of DNA-loaded NLS-MCMs compared to nonNLS-MCMs (Fig. 6B). The average number of DNA-loaded NLS-MCMs inside the nucleus as well as at the nuclear membrane was at least double the number of nonNLS-MCMs (α = 0.05, P < 0.01). The statistical analysis of MCMs in individual cell nuclei confirmed that NLS-MCMs accumulate in the nucleus over time. Based on the efficient uptake and nuclear translocation, NLS-MCMs were efficiently internalized into the cytoplasm and trafficked to the nucleus with no cytotoxic effect and thus, offer themselves as suitable nanocarriers for the delivery of therapeutic antisense oligonucleotides (ASO) to the cell nucleus.
To test how effective our purely peptidic nanocarriers were in delivering therapeutics, we analyzed the effect of NLS-MCMs loaded with G3139-GAP, an RNase-H dependent ASO,70 on BCL-2 protein levels in MCF-7 cells. Western blots were carried out to quantitatively assess overall BCL-2 levels in cells incubated for 24 and 48 h with equal amounts (2.2 × 108 ± 2.96 × 107 NPs ml−1) of either NLS- or nonNLS-MCMs loaded with G3139-GAP or a scrambled oligonucleotide, with MCMs not carrying ASO, with ASO/lipofectamine complexes, and with untreated control cells. Blots representing total cell extracts were probed with antibodies against BCL-2 and GAPDH as an internal loading control (Fig. 7A), and subsequently quantified by densitometry (Fig. 7B; S12, ESI†).
 | ||
| Fig. 7 Therapeutic effects of NLS-MCMs delivering G3139-GAP ASO. (A) Western blot analysis of BCL-2 proteins in 6μg of MCF-7 total cell extract after 48 h. GAPDH was used as loading control. (B) Quantitative densitometry of the immunoblots; nonNLS-MCM-treated (light grey bars), NLS-MCM-treated (dark grey bars), untreated (black bar). (C) BCL-2 expression levels in cells transfected with G3139-GAP relative to scrambled oligo delivered with NLS- or nonNLS MCMs. Data are mean ± s.d. of the relative intensity of the bands from three independent assays (not significant (ns) p > 0.05, ** p < 0.01 and *** p < 0.001). | ||
Comparison of cells transfected with cargo-free, ‘empty’ MCMs to untransfected cells (control) after 24 and 48 h showed that the nonNLS- and NLS-MCM platforms by themselves had no significant effect on BCL-2 levels. In contrast, when NLS-MCM nanocarriers were loaded with G3139-GAP, after 24 h, a 57% reduction in BCL-2 levels relative to cells transfected with NLS-MCMs loaded with scrambled oligo was observed (Fig. 7C and S12, ESI†). More importantly, after 48 h of treatment, nuclear targeting of G3139-GAP by NLS-MCMs led to 86% downregulation of BCL-2 levels relative to cells transfected with scrambled oligo-loaded NLS-MCMs (Fig. 7C). In contrast, delivery of G3139-GAP by nonNLS-MCMs only reduced BCL-2 levels by 8 (after 24 h) and 4% (after 48 h) relative to cells transfected with scrambled oligo-loaded nonNLS-MCMs. These data show that the nuclear targeting of the nanocarriers and a corresponding release of G3139-GAP are essential for a significant decrease in BCL-2. Notably, when compared to cells transfected with lipofectamine/G3139-GAP complexes relative to lipofectamine/scrambled oligo complexes at corresponding ASO concentrations, we observed a more efficient BCL-2 reduction by G3139-GAP loaded NLS-MCMs (57% versus 45% after 24 h and 86% versus 63% after 48 h) (Fig. S13A and B, ESI†). In a recent study, cell penetrating peptide (CPP) conjugated exosomes (EXOs) were used as a platform for ASO delivery.53 Their western blot analysis revealed a decrease of 51.71% and 69.69% in BCL-2 levels by EXO-G3139 and EXO-R9-G3139 in HepG2 cells. In another example, a 70% downregulation in Bcl-2 mRNA was found in MCF-7 cells incubated with self-assembled peptide nanofibers for G3139 delivery.54 In comparison, our peptidic platform with G3139-ASO payloads displayed stronger downregulation after 48 h which suggests that NLS-MCMs are promising nanocarriers for efficient delivery of therapeutic oligonucleotides to the nucleus. Yet, there remain many challenges that need to be overcome before peptidic platforms move into the realm of clinical gene therapy. First and foremost, selective targeting of the platform to the diseased site with minimal off-target delivery needs to be addressed. As peptides can be made up of various combinations of amino acids, one could envisage directly incorporating the ligands (e.g. RGD) for specific molecular recognition sites into the peptide building block. Alternatively, peptide sequences can be designed in such a way that they bind targeting moieties that harness the platform for cell-specific delivery.
Conclusion
In this study, we devised a self-assembling cationic amphiphilic peptide specially tailored at the molecular level to maximize the potential of peptidic nanocarriers in gene delivery applications. The specific sequence of the hydrophobic domain together with the integration of a minimal NLS, (KRKR), into the hydrophilic domain of the peptide backbone to promote nuclear targeting in addition to enhancing cellular uptake, are key to the efficiency of our innovation and have not been attempted before in any non-viral vector delivery system. Importantly, integrating the NLS-targeting moiety obviates extra conjugation steps71–73 in the preparation of nanoparticles. Instead, NLS-functionalized, DNA-loaded multicompartment micellar assemblies are obtained by a single-step self-assembly process. We show that (i) these DNA-loaded NLS-MCMs enter cells more efficiently than corresponding nanocarriers lacking an NLS (non-NLS-MCMs) without adverse effects on cell proliferation, (ii) NLS-MCMs successfully ferry DNA cargo to the nucleus via NLS-mediated interactions of the nanocarrier with the nuclear translocation machinery, and (iii) ASO delivery by NLS-MCMs strongly downregulates BCL-2 protein levels in MCF-7 cancer cells and is associated with enhanced cytotoxicity. The unprecedented combination of a straightforward manner of production, non-cytotoxicity and efficient DNA delivery to the nucleus makes our purely peptidic platform superior to established nanocarriers. Together, these advantages warrant a further development of these NLS-MCMs for systemic gene therapy and vaccination applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the Swiss Nanoscience Institute (SNI), the National Centre of Competence in Research–Molecular Systems Engineering (NCCR-MSE), the University of Basel and ETH Zürich. The authors thank Dr Birthe Fahrenkrog (University of Basel) for the helpful discussions on nuclear transport, Cinzia Tiberi Schmidt (ETH Zürich), Susanne Erpel, Carola Alampi and Dr Mohamed Chami (University of Basel) for TEM measurements and Mohsen Esmailzadeh for his help with the schematic representation of multicellular compartments.References
- A. Anna and G. Monika, J. Appl. Genet., 2018, 59, 253–268 CrossRef CAS PubMed.
- P. K. Geyer, M. W. Vitalini and L. L. Wallrath, Curr. Opin. Cell Biol., 2011, 23, 354–359 CrossRef CAS PubMed.
- M. Zwerger, C. Y. Ho and J. Lammerding, Annu. Rev. Biomed. Eng., 2011, 13, 397–428 CrossRef CAS PubMed.
- A. Parodi, C. Corbo, A. Cevenini, R. Molinaro, R. Palomba, L. Pandolfi, M. Agostini, F. Salvatore and E. Tasciotti, Nanomedicine, 2015, 10, 1923–1940 CrossRef CAS PubMed.
- K. M. Wagstaff and D. A. Jans, Biochem. J., 2007, 406, 185–202 CrossRef CAS PubMed.
- D. Xu, A. Farmer and Y. M. Chook, Curr. Opin. Struct. Biol., 2010, 20, 782–790 CrossRef CAS PubMed.
- C. Zelmer, L. P. Zweifel, L. E. Kapinos, I. Craciun, Z. P. Güven, C. G. Palivan and R. Y. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2770–2778 CrossRef CAS PubMed.
- R. K. Thapa and M. O. Sullivan, Curr. Opin. Biomed. Eng., 2018, 7, 71–82 CrossRef PubMed.
- D. Dean, D. Strong and W. Zimmer, Gene Ther., 2005, 12, 881–890 CrossRef CAS PubMed.
- C. H. Jones, C.-K. Chen, A. Ravikrishnan, S. Rane and B. A. Pfeifer, Mol. Pharm., 2013, 10, 4082–4098 CrossRef CAS PubMed.
- J. M. Tomich, E. Wessel, J. Choi and L. A. Avila, in Nucleic Acid Nanotheranostics, ed. M. Filice and J. Ruiz-Cabello, Elsevier, 2019, pp. 247–276 Search PubMed.
- Y. Cheng, C. Sun, R. Liu, J. Yang, J. Dai, T. Zhai, X. Lou and F. Xia, Angew. Chem., 2019, 131, 5103–5107 CrossRef.
- W.-J. Yi, J. Yang, C. Li, H.-Y. Wang, C.-W. Liu, L. Tao, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, Bioconjugate Chem., 2012, 23, 125–134 CrossRef CAS PubMed.
- M. Bogacheva, A. Egorova, A. Slita, M. Maretina, V. Baranov and A. Kiselev, Bioorg. Med. Chem. Lett., 2017, 27, 4781–4785 CrossRef CAS PubMed.
- S. Bhattacharjee, Ther. Delivery, 2021, 12, 21–36 CrossRef CAS PubMed.
- W. C. W. Chan, Bio-Applications of Nanoparticles, Springer Science & Business Media, 2009 Search PubMed.
- R. Tenchov, R. Bird, A. E. Curtze and Q. Zhou, ACS Nano, 2021, 15, 16982–17015 CrossRef CAS PubMed.
- D. Sharma, S. Arora, J. Singh and B. Layek, Int. J. Biol. Macromol., 2021, 183, 2055–2073 CrossRef CAS PubMed.
- M. Saffari, H. R. Moghimi and C. R. Dass, Iran. J. Pharm. Res., 2016, 15, 3–17 Search PubMed.
- J. Chen and X. Zou, Bioact. Mater., 2019, 4, 120–131 CrossRef PubMed.
- S. Tarvirdipour, M. Skowicki, C.-A. Schoenenberger and C. G. Palivan, Int. J. Mol. Sci., 2021, 22, 9092 CrossRef CAS PubMed.
- M. Rad-Malekshahi, L. Lempsink, M. Amidi, W. E. Hennink and E. Mastrobattista, Bioconjugate Chem., 2016, 27, 3–18 CrossRef CAS PubMed.
- F. Qiu, Y. Chen, C. Tang and X. Zhao, Int. J. Nanomed., 2018, 13, 5003–5022 CrossRef CAS PubMed.
- T. Li, X.-M. Lu, M.-R. Zhang, K. Hu and Z. Li, Bioact. Mater., 2022, 11, 268–282 CrossRef CAS PubMed.
- S. Tarvirdipour, X. Huang, V. Mihali, C.-A. Schoenenberger and C. G. Palivan, Molecules, 2020, 25, 3482 CrossRef CAS PubMed.
- H.-J. Do, H. Song, H.-M. Yang, D.-K. Kim, N.-H. Kim, J.-H. Kim, K.-Y. Cha, H.-M. Chung and J.-H. Kim, FEBS Lett., 2006, 580, 1865–1871 CrossRef CAS PubMed.
- A. A. Beg, S. M. Ruben, R. I. Scheinman, S. Haskill, C. A. Rosen and A. S. Baldwin, Genes Dev., 1992, 6, 1899–1913 CrossRef CAS PubMed.
- R. K. Lokareddy, R. A. Hapsari, M. van Rheenen, R. A. Pumroy, A. Bhardwaj, A. Steen, L. M. Veenhoff and G. Cingolani, Structure, 2015, 23, 1305–1316 CrossRef CAS PubMed.
- S. Kosugi, M. Hasebe, N. Matsumura, H. Takashima, E. Miyamoto-Sato, M. Tomita and H. Yanagawa, J. Biol. Chem., 2009, 284, 478–485 CrossRef CAS PubMed.
- D. H. O'Day, Cells, 2019, 8, 167 CrossRef PubMed.
- X. Li and F. Zhu, J. Virol., 2009, 83, 2531–2539 CrossRef CAS PubMed.
- A. Catalano and D. H. O'Day, Histochem. Cell Biol., 2012, 138, 515–530 CrossRef CAS PubMed.
- K. Desale, K. Kuche and S. Jain, Biomater. Sci., 2021, 9, 1153–1188 RSC.
- S. Tarvirdipour, C.-A. Schoenenberger, Y. Benenson and C. G. Palivan, Soft Matter, 2020, 16, 1678–1691 RSC.
- S. Sharad, Antisense Therapy: An Overview, IntechOpen, 2019 Search PubMed.
- E. Doxakis, Med. Res. Rev., 2021, 41, 2656–2688 CrossRef CAS PubMed.
- A. Batista-Duharte, L. Sendra, M. J. Herrero, D. Téllez-Martínez, I. Z. Carlos and S. F. Aliño, Biomolecules, 2020, 10, 316 CrossRef CAS PubMed.
- A. Z. Badros, O. Goloubeva, A. P. Rapoport, B. Ratterree, N. Gahres, B. Meisenberg, N. Takebe, M. Heyman, J. Zwiebel, H. Streicher, C. D. Gocke, D. Tomic, J. A. Flaws, B. Zhang and R. G. Fenton, J. Clin. Oncol., 2005, 23, 4089–4099 CrossRef CAS PubMed.
- M. J. Morris, W. P. Tong, C. Cordon-Cardo, M. Drobnjak, W. K. Kelly, S. F. Slovin, K. L. Terry, K. Siedlecki, P. Swanson, M. Rafi, R. S. DiPaola, N. Rosen and H. I. Scher, Clin. Cancer Res., 2002, 8, 679–683 CAS.
- X. Cheng, Q. Liu, H. Li, C. Kang, Y. Liu, T. Guo, K. Shang, C. Yan, G. Cheng and R. J. Lee, Pharm. Res., 2017, 34, 310–320 CrossRef CAS PubMed.
- L. E. Kapinos, R. L. Schoch, R. S. Wagner, K. D. Schleicher and R. Y. H. Lim, Biophys. J., 2014, 106, 1751–1762 CrossRef CAS PubMed.
- L. E. Kapinos, B. Huang, C. Rencurel and R. Y. H. Lim, J. Cell Biol., 2017, 216, 3609–3624 CrossRef CAS PubMed.
- R. L. Schoch, L. E. Kapinos and R. Y. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16911–16916 CrossRef CAS PubMed.
- W. Bernhard, J. Ultrastruct. Res., 1969, 27, 250–265 CrossRef CAS PubMed.
- J. Yao, Y. Fan, Y. Li and L. Huang, J. Drug Targeting, 2013, 21, 926–939 CrossRef CAS PubMed.
- M. Durymanov and J. Reineke, Front. Pharmacol., 2018, 9, 971 CrossRef PubMed.
- T. B. Schuster, D. de B. Ouboter, E. Bordignon, G. Jeschke and W. Meier, Soft Matter, 2010, 6, 5596–5604 RSC.
- J. C. Brendel, S. Catrouillet, J. Sanchis, K. A. Jolliffe and S. Perrier, Polym. Chem., 2019, 10, 2616–2625 RSC.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef CAS PubMed.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 265–273 Search PubMed.
- E. M. Anderson, P. Miller, D. Ilsley, W. Marshall, A. Khvorova, C. A. Stein and L. Benimetskaya, Cancer Gene Ther., 2006, 13, 406–414 CrossRef CAS PubMed.
- J. Marshall, H. Chen, D. Yang, M. Figueira, K. B. Bouker, Y. Ling, M. Lippman, S. R. Frankel and D. F. Hayes, Ann. Oncol., 2004, 15, 1274–1283 CrossRef CAS PubMed.
- H. Xu, C. Liao, S. Liang and B.-C. Ye, ACS Appl. Mater. Interfaces, 2021, 13, 10760–10767 CrossRef CAS PubMed.
- S. Bulut, T. S. Erkal, S. Toksoz, A. B. Tekinay, T. Tekinay and M. O. Guler, Biomacromolecules, 2011, 12, 3007–3014 CrossRef CAS PubMed.
- M. Marfori, A. Mynott, J. J. Ellis, A. M. Mehdi, N. F. W. Saunders, P. M. Curmi, J. K. Forwood, M. Bodén and B. Kobe, Biochim. Biophys. Acta, Mol. Cell Res., 2011, 1813, 1562–1577 CrossRef CAS PubMed.
- Y. Sakiyama, A. Mazur, L. E. Kapinos and R. Y. H. Lim, Nat. Nanotechnol., 2016, 11, 719–723 CrossRef CAS PubMed.
- L. K. Davis, A. Šarić, B. W. Hoogenboom and A. Zilman, bioRxiv, 2021, 120, 1565–1577 CAS.
- R. Fagerlund, K. Melén, L. Kinnunen and I. Julkunen, J. Biol. Chem., 2002, 277, 30072–30078 CrossRef CAS PubMed.
- B. Bourgeois, S. Hutten, B. Gottschalk, M. Hofweber, G. Richter, J. Sternat, C. Abou-Ajram, C. Göbl, G. Leitinger, W. F. Graier, D. Dormann and T. Madl, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8503–8514 CrossRef CAS PubMed.
- A. Gonzalez, T. Mannen, T. Çağatay, A. Fujiwara, H. Matsumura, A. B. Niesman, C. A. Brautigam, Y. M. Chook and T. Yoshizawa, Sci. Rep., 2021, 11, 3754 CrossRef CAS PubMed.
- C. Auría-Soro, T. Nesma, P. Juanes-Velasco, A. Landeira-Viñuela, H. Fidalgo-Gomez, V. Acebes-Fernandez, R. Gongora, M. J. Almendral Parra, R. Manzano-Roman and M. Fuentes, Nanomaterials, 2019, 9, 1365 CrossRef PubMed.
- C. Huang, X. Chen, Z. Xue and T. Wang, Sci. Adv., 2020, 6, eaba1321 CrossRef PubMed.
- D. Manzanares and V. Ceña, Pharmaceutics, 2020, 12, 371 CrossRef CAS PubMed.
- Y. Zhou, S. Han, Z. Liang, M. Zhao, G. Liu and J. Wu, J. Mater. Chem. B, 2020, 8, 5564–5577 RSC.
- I. Gessner and I. Neundorf, Int. J. Mol. Sci., 2020, 21, 2536 CrossRef CAS PubMed.
- S. Silva, A. J. Almeida and N. Vale, Biomolecules, 2019, 9, 22 CrossRef PubMed.
- P. Foroozandeh and A. A. Aziz, Nanoscale Res. Lett., 2018, 13, 339 CrossRef PubMed.
- I. V. Aramburu and E. A. Lemke, Semin. Cell Dev. Biol., 2017, 68, 34–41 CrossRef CAS PubMed.
- C. E. Zimmerli, M. Allegretti, V. Rantos, S. K. Goetz, A. Obarska-Kosinska, I. Zagoriy, A. Halavatyi, G. Hummer, J. Mahamid, J. Kosinski and M. Beck, Science, 2021, 374, eabd9776 CrossRef CAS PubMed.
- X.-H. Liang, H. Sun, J. G. Nichols and S. T. Crooke, Mol. Ther., 2017, 25, 2075–2092 CrossRef CAS PubMed.
- J. Lee, Y.-E. Kwon, J. Kim, D. W. Kim, H. Guim, J. Yeon, J.-C. Kim and J. S. Choi, J. Biomater. Sci., Polym. Ed., 2021, 32, 1140–1160 CrossRef CAS PubMed.
- Q. Li, X. Hao, H. Wang, J. Guo, X. Ren, S. Xia, W. Zhang and Y. Feng, Colloids Surf., B, 2019, 184, 110510 CrossRef CAS PubMed.
- E. Park, H.-B. Cho and K. Takimoto, Cytotherapy, 2015, 17, 536–542 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00826b |
| This journal is © The Royal Society of Chemistry 2022 |