
Synergistic effects of particulate matter (PM2.5) and sulfur dioxide (SO2) on neurodegeneration via the microRNA-mediated regulation of tau phosphorylation†
Tingting
Ku‡
,
Minjun
Chen‡
,
Ben
Li
,
Yang
Yun
,
Guangke
Li
and
Nan
Sang
*
College of Environment and Resource, Research Center of Environment and Health, Shanxi University, Taiyuan, Shanxi, 030006 PR China. E-mail: sangnan@sxu.edu.cn; Fax: +86-351-7011932; Tel: +86-351-7011932
First published on 9th November 2016
Abstract
Because air pollution is a complex mixture of pollutants consisting of both particulate and gaseous components, understanding the health risks from these pollutants requires an evaluation of their combined effects rather than predictions based on the toxicities of single chemicals alone. Particulate matter (PM2.5) and sulfur dioxide (SO2) commonly co-exist in the atmospheric environment, and epidemiological studies have linked air pollution to the development of neurodegenerative disorders, in addition to increased morbidity from cardiopulmonary diseases. However, few studies have examined the potential effects from combinations of these pollutants on neurodegeneration, especially at NOEC doses. In the present study, we first found that PM2.5 and SO2 co-exposure leads to neurodegeneration at low doses, including neuronal apoptosis, the reduction of synaptic structural protein postsynaptic density (PSD-95) and synaptic functional protein N-methyl-D-aspartate (NMDA) receptor subunits (NR2B), and the elevation of tau phosphorylation in vitro and in vivo, which did not induce clear effects when the compounds were tested separately. Furthermore, we clarified that the microRNA (miRNA) miR-337-5p, which is homologous to a human miRNA that targets tau, was involved in the combined effect and contributed to synergistic neurodegeneration. This work implies the potential risk of neuronal dysfunction from the co-existence of PM2.5 and SO2 in coal-burning areas and provides new insights into the molecular markers for the relevant diseases.
Introduction
Ambient air pollution is a complex mixture of pollutants that includes both particulate and gaseous components. These components are primarily derived from sources related to the combustion of fuel by motor vehicles, diesel-powered transport and local industrial processes.1 Multiple pollutants vary spatially and temporally in their characteristics and the toxicity of the mixture could differ from the toxicity of the individual pollutant.2 Populations are seldom exposed to single air pollutants, and the experimental investigations that have focused on the harmful effects induced by individual pollutants may not represent the actual exposure risks. In particular, it has recently been proposed that air quality management must evolve from a single pollutant perspective towards the regulation of emissions in a multi-pollutant manner.3 Recent studies have investigated the combined toxicity of simultaneously co-existing chemicals,4 but the communication of potentially synergistic toxicity and possible mechanisms to the public is still a significant problem.Coal consumption covers more than 70% of China's total energy demands,5 and particulate matter (PM) and sulfur dioxide (SO2) are the major anthropogenic contributors to air pollution from burning of coal. Although coal combustion in large cities has been well-controlled in recent decades, the extensive use of coal combustion without effective control technologies continues to occur in rural areas by residential stoves for cooking and heating.6 Several studies have reported that PM and SO2 were the predominant atmospheric pollutants in 31 provincial capital cities of China.7 Importantly, PM2.5 is a complex mixture of small particles and liquid droplets that are suspended in the atmosphere, and they can be emitted directly as primary PM or formed through the gas-to-particle partitioning of semi-volatile products as secondary PM.8 The PM constituents include sulfates, nitrates, carbon, ammonium, hydrogen ions, lipopolysaccharides (LPS), metals, and water,9 and most of the sulfates and nitrates are formed by the oxidation of the precursor gases SO2 and NOX.10 Therefore, the major source of fine PM aerosols in the mega-cities was the secondary formation from coal/biomass combustion products.11 The dominant secondary inorganic aerosol species account for 30–60% of PM2.5 mass concentrations.12
Epidemiological studies have indicated that air pollutants not only cause increased morbidity through cardiopulmonary diseases,13,14 but they are also associated with the development and progression of neurodegenerative disorders,15 which are characterized by the age-dependent deposition of aggregated and misfolded proteins, cognitive disturbance and neuronal loss.16 Several studies have reported that PM2.5 has the ability to reach the brain, and it might be associated with neurodegeneration in vivo and in vitro.17,18 Furthermore, experimental evidence shows that PM2.5 exposure can cause changes in the immune-related transcription factor, nuclear factor-κB (NF-κB), and activator protein-1 (AP-1) and in two pro-inflammatory cytokines, namely tumor necrosis factor-α (TNF-α) and interleukin-1α (IL-1α). Exposure to PM2.5 can also cause brain inflammation,19 which has been shown to exacerbate the progression of neurodegeneration.20 Generally, the lowest concentrations at which significant neuronal loss occurred were 3 mg kg−1 and 25–50 μg mL−1.21,22 Additionally, other studies have shown that neurological disorders occurred after environmental SO2 exposure.22 In particular, chronic SO2 inhalation at levels above the environmental standard led to impaired neuronal behavior, repressed glutamate receptor gene expression and memory-related kinase activation via neuroinflammation in rats, and thus tended to cause neurodegeneration.23,24
Thus far, although PM2.5 and SO2 commonly co-exist in the atmospheric environment, few studies have examined their potential joint effects on neurodegeneration. In the present study, we assessed the combined effects of PM2.5 and SO2 on the induction of neurodegeneration, and we clarified the underlying regulatory mechanisms. Here, we first found that co-exposure to PM2.5 and SO2 produced neurodegeneration at low doses that did not induce obvious effects on their own. After that, we determined that miR-337-5p, which is homologous with human miRNA that targets tau, was involved in the combined effect and contributed to synergistic neurodegeneration.
Materials and methods
PM2.5 sample collection
PM2.5 samples were collected during the winter in Taiyuan (112°21-34′E longitude, 37°47-48′N latitude), Shanxi Province. The samples were collected onto quartz filters with PM middle volume air samplers (TH-150CIII, Wuhan, China). 69 effective samples were collected during the winter except for the rainfall period, equipment failure, power outages and student holidays. For the bioassays, PM extracts were prepared by soaking the filters in ultrapure water, drying them under nitrogen, and reconstituting the samples to 10 mg mL−1. The final extracts were stored at −80 °C.Primary cortical neuron culture and treatment
Cortical tissue that was dissected from a C57BL/6 mouse pup (postnatal day 0, P0) was incubated in oxygenated trypsin for 10 min at 37 °C and then mechanically triturated. The cells were then spun down and resuspended in Neurobasal/B27 medium (Invitrogen, USA) supplemented with 1.25 μM L-glutamine, penicillin/streptomycin, and 25 μM glutamate. The cells (1 × 106) were plated into poly-D-lysine-coated 24-well plates or 35 mm culture dishes for viability and immunoblot analyses. The cells (5 × 104) were plated onto poly-D-lysine-coated cover slips for immunofluorescence analysis. The cultures were used between 13 and 15 days in vitro (DIV). The control group was incubated in Neurobasal/B27 medium with the vehicle solution, and the different treatment groups were treated with the PM2.5 extract (1, 3, 10, 30, 100 and 300 μg mL−1) and SO2 derivatives (bisulfite and sulfite, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 M/M, 0.1, 0.3, 1, 3, 10 and 30 μg mL−1) either individually or in binary combinations at an isobologram and fixed mixture ratios.
:3 M/M, 0.1, 0.3, 1, 3, 10 and 30 μg mL−1) either individually or in binary combinations at an isobologram and fixed mixture ratios.
Animals and exposure
Healthy male C57BL/6 mice (6–8 weeks old) were purchased from the Academy of Military Medical Sciences (Beijing, China). They were randomly divided into four groups, each of which contained six mice. The mice in the treatment groups were exposed to SO2 dynamic inhalation (6 h d−1, 28 d) and PM2.5 intranasal instillation (every other day, 28 d). And the mice in the control group were exposed to fresh air (6 h d−1, 28 d) and received an intranasal instillation of saline (every other day, 28 d) using the same protocol. Before intranasal instillations, the mice were anesthetized with isoflurane (Yi Pin Pharmaceutical Co., Ltd, Hebei, China). The mice were sacrificed 18 h after their last exposure. Their cortices were removed, quickly frozen in liquid nitrogen and stored at −80 °C until further analysis. The details of the process are shown in the ESI.† All animal experiment procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the Chinese Animal Welfare Committee. The protocol was approved by the Shanxi University's Institutional Animal Care and Use Committee.Neuronal viability determination
Cortical neurons were plated into 24-well plates at a concentration of 5 × 105 cells per well, and the cultures were treated with PM2.5 (1, 3, 10, 30, 100 or 300 μg mL−1), SO2 derivatives (0.1, 0.3, 1, 3, 10 or 30 μg mL−1) or PM2.5 & SO2 derivatives (fixed mixture ratios, 10:1) on DIV14. After the treatment, neuronal viability was determined by the MTT assay.
Concentration-response analysis
Using the “best fit” approach,25 we analyzed the concentration–response relationship with suitable nonlinear regression models. Additionally, we calculated the 95% confidence intervals of the estimated mean effects and the NOECs of the individual chemicals and mixtures with a Dunnett test.Calculation of predicted mixture effects
The cortical neurons were treated with equitoxic mixtures at a fixed ratio of 10:1. Based on the individual effects of the PM2.5 and SO2, the predicted combination effects were calculated with the following CA model, assuming that the combination effect of a mixture with n components is concentration-additive.| EC xmix = [∑pi/EC xi]−1 | (1) |
In eqn (1), the EC xi terms were the concentrations of individual component i that generated X% effect; the EC xmix was the concentration of a mixture that jointly generated X% effect; and pi indicates the percentage of the individual component i concentration within the total mixture concentration.
Immunocytochemistry
The in vitro neuronal apoptosis was assessed with a KeyGen Biotech TUNEL Apoptosis Detection Kit (KeyGen, China). The cells plated on cover slips had been pretreated with poly-D-lysine, then incubated for 13 days. The supernatant was aspirated, and the cover slips were fixed with 4% paraformaldehyde. After washing with PBS, the cover slips were permeabilized with 1% Triton X-100 and washed again. The cover slips were then incubated with TUNEL reagent in a humidified chamber, washed, and stained with 4′,6-diamidino-2-phenylindole (DAPI) in the dark. The images were then visualized and analyzed under a fluorescence microscope (Olympus, Japan) with Image-Pro Express 6.0 software (Media Cybernetics, USA) at 200× magnification.Immunoblot analyses
Proteins were extracted from the primary cultured cortical neurons or cortical tissues and quantified.26 In brief, 20–60 μg of the total protein was resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose (NC) membrane. The membrane was blocked with 3% bovine serum albumin and then incubated with rabbit antibodies to caspase-3, cleaved caspase-3, β-actin (1:1000, Cell Signaling Technology, USA), PSD95, NMDAR2B, tau or phospho-tau (1:200, Bioss, China) at 4 °C overnight. The membranes were then washed with PBS and incubated with an IR Dye 800CW-conjugated secondary antibody (1:5000, LiCor Biosciences, USA), scanned and detected with a LI-COR Odyssey Infrared Fluorescent System.
MicroRNA-gene target prediction
MicroRNA-gene target prediction was performed by using Targetscan (http://www.targetscan.org/), RNA22 (http://cbcsrv.watson.ibm.com/rna22.html), RNAhybrid (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/) and miRMap (http://mirmap.ezlab.org/) to search for the target gene. Microtubule-associated protein tau (Mapt) is a potential target of miR-337-5p, which has been included in the above databases.Real-time quantitative reverse transcription-PCR analysis
To determine the expression of mmu-miR-337-5p, total RNA was extracted using the TRIzol Reagent (Invitrogen, USA), and first-strand complementary DNA (cDNA) was synthesized with a reverse transcription kit (Qiagen Biotechnology, Germany). The primer sequences are shown in the ESI.† The relative quantification of the gene expression was determined on a qTOWER 2.2 Real-Time PCR (Analytik Jena AG, Jena, Germany), with mmu-miR-202 as an internal control. Detailed procedures are presented in the ESI.†Luciferase reporter assay
Two psiCHECK-2 vectors (Promega) were constructed to carry a Mapt 3′UTR DNA fragment containing the indicated miR-337-5p binding site. In addition, two matched mutants were constructed to disrupt the putative miR-337-5p binding sites in the 3′-UTR of MAPT by using KOD Plus neo DNA polymerase (ToYoBo). Both the wild-type and mutated plasmid were cloned into the site of the psiCHECK-2 vector downstream of the luciferase gene with PCR. A mimic or inhibitor was transfected into HEK293T cells with the above vectors according to the instruction. All experiments were performed in triplicate. The detailed procedures are shown in the ESI.†Statistical analysis
A one-way analysis of variance (ANOVA) was used to analyze the mean differences among the groups relative to the vehicle control by Origin 8.0. A two-tailed Student t-test was used to analyze the experimental data between pairs of groups. The data are presented as the means ± SE. Values with p < 0.05 were considered statistically significant.Results and discussion
Concentration-effect analysis for PM2.5- and SO2-induced neuronal injuries
To evaluate the potential combined effect of PM2.5 and SO2, we first clarified the concentration–cytotoxic response relationship for each component on primary cultured cortical neurons by determining the neuronal viability by the MTT assay. After a 24 h treatment, both the PM2.5 and SO2 derivatives decreased the cell survival ratios in a concentration-dependent manner (Fig. 1A and B). Based on the data and the “best fit” approach, we conducted a nonlinear regression and found the concentration-neuronal survival data were fit well by the Langmuir function. Following this exploration, the NOECs of PM2.5 and SO2 derivatives were calculated to be 10 and 1 μg mL−1 (see Table S1, ESI†), respectively. Increasing epidemiological and experimental evidence has shown that air pollutants can pass the blood–brain barrier, damage the central nervous system and cause neurological dysfunction, such as neuroinflammation, oxidative stress and neurodegenerative pathology.27 PM2.5 and SO2 are two major pollutants that coexist in the vicinity of burning coal. However, most experimental studies have focused on the adverse health effects of single air pollutants, ignoring the combined toxicity of these pollutants. Air pollution consists of gaseous pollutants and PM, which can jointly induce neurotoxicity at low concentrations. Thus, some measures have been taken to control the emission of multi-pollutants. The “Twelfth Five-Year Plan for National Environment Protection” has resulted in reduction in PM and SO2 emissions from the 2010 levels to 10% and 8%, respectively, in 2015.28 It is vital to define whether the combined effects occur at lower concentrations below their NOECs.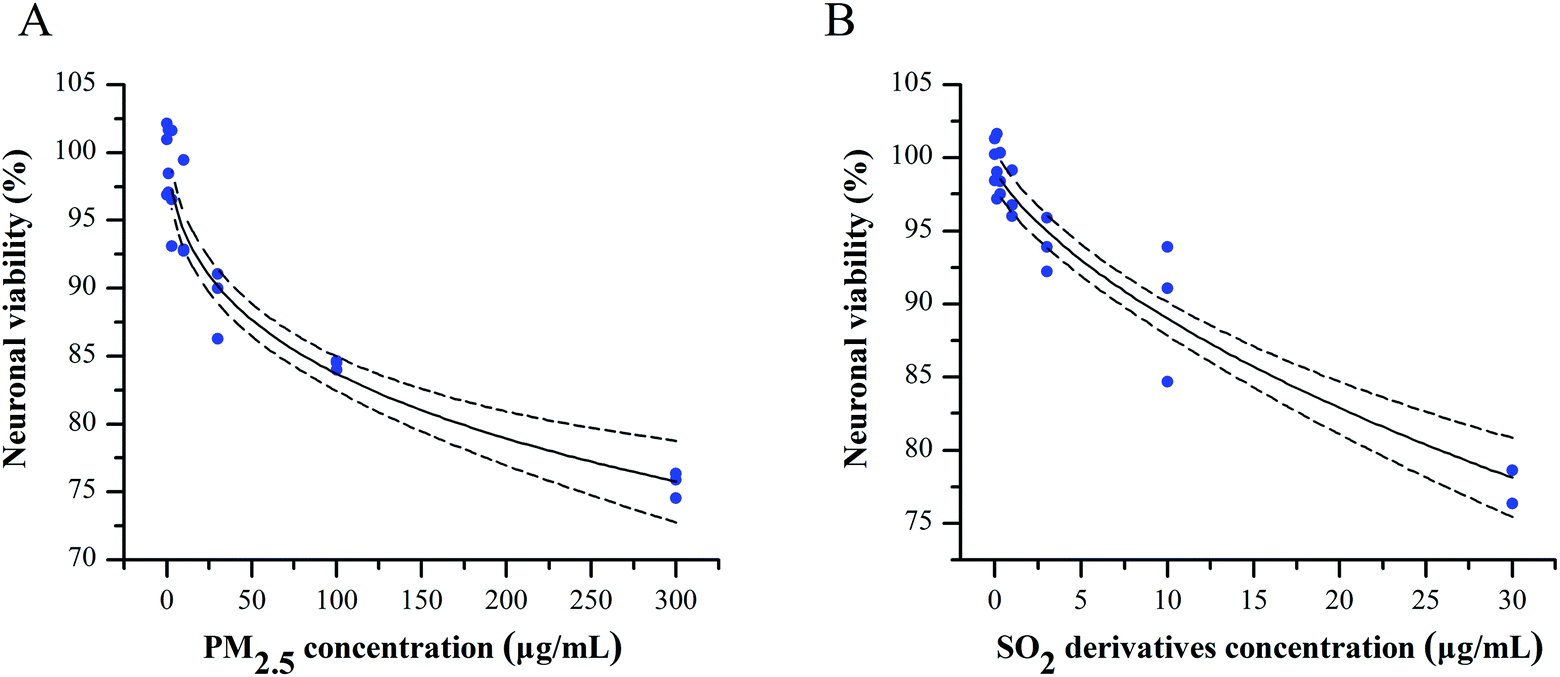 | ||
| Fig. 1 Concentration–response analyses for the PM2.5 (A) and SO2 derivatives (B) in the neuronal survival assay. The concentration–response curves for the effects of the PM2.5 and SO2 derivatives on neuronal survival were fitted with the Langmuir function. The solid curved lines represent the best fits to the Langmuir functions; the dotted lines show the upper and lower 95% confidence intervals (CIs) of the mean responses. Three independent experiments were performed in triplicate for each treatment group. The results are expressed as the absorbance percentage of the control cells. | ||
Combined effects for PM2.5 and SO2 in promoting neurodegeneration
Considering that pollutants are present in atmospheric media at levels below their NOECs as indicated by bioassays, we applied the NOEC mixture ratio (10:1) here in a traditional “fixed ratio” approach to clarify the potential combined effects of PM2.5 and SO2 by using neuronal survival as the test endpoint. First, we predicted the concentration–response curves for mixtures at the equitoxic NOEC mixture ratios described above by applying the CA model in Fig. 2A, as well as the combined curve and its 95% confidence intervals. Importantly, the CA model-predicted concentration–response curve was observed to be above the 95% confidence intervals. These findings indicate that the combination of the two components was significantly synergistic with respect to neuronal injury in comparison with the individual treatments. To confirm this prediction, we compared the effects of the PM2.5 and the SO2 derivatives singly or in binary mixtures with the effect predicted by the CA model (Fig. 2B). The PM2.5 at 10 μg mL−1 and the SO2 derivatives at 1 μg mL−1 did not alter the neuronal viability (95 and 97% of the control, respectively), whereas co-exposure to the two components caused a significant reduction in the neuronal viability (83% of control). In comparison, the neuronal viability predicted by the CA model was 94%. These results suggest that the combined exposure to the PM2.5 and SO2 derivatives caused a synergistic effect on the survival of neurons when the level of each substance was below its individual NOEC. Moreover, morphological alterations associated with cell viability as well as dendrites and spines were observed for the cells upon single and combined treatments with PM2.5 (10 μg mL−1) and SO2 derivatives (1 μg mL−1) in Fig. 2C.
 | ||
| Fig. 2 Predicted and observed co-exposure effects from a 10:1 mixture of the PM2.5 and SO2 derivatives. (A) The observed effects of the mixture are shown as blue circles and represent three independent experiments that were performed in triplicate, and the doses shown here are the doses of each component in 1/0.1, 3/0.3, 10/1, 30/3, and 100/10 μg mL−1. The predicted effects were calculated using the CA model and are shown as a solid line. The red dashed curves show the best fit of the data to the regression model, and the black dotted lines show the upper and lower 95% CIs of the mean responses. (B) Neuronal survival was assessed after individual or combined treatment with PM2.5 (10 μg mL−1) and SO2 derivatives (1 μg mL−1) as predicted by the CA model. Three independent experiments were performed in triplicate for each treatment group. ##p < 0.01 compared with SO2 alone; bp < 0.01 compared with PM2.5 alone. (C) Morphological observations of the neurons after combined exposure to the PM2.5 (10 μg mL−1) and SO2 derivatives (1 μg mL−1). Scale bar, 20 μm. | ||
Neurodegeneration typically involves a progressive loss of neuronal integrity and function that result in neuronal death.29 The potential mechanisms underlying apoptosis and neuronal death involve the release of mitochondrial pro-apoptotic proteins,30 along with key processes involving oxidative stress31 and synaptic loss.32 Postsynaptic densities (PSD-95) and the N-methyl-D-aspartic acid (NMDA) receptor are widely used as markers for synaptic loss. The structure of post-synaptic plasticity is based on the PSD ultrastructure, and PSD-95 may contribute to synapse stabilization.33 NMDA receptors also play a pivotal role in the regulation of neuronal plasticity34 that is associated with learning, memory and development.35 The subunits of this receptor, especially NR2B, can interact with the prominent PSD-95.36 Previous studies have shown that PSD-95 expression can regulate postsynaptic clustering and activate glutamate receptors, including NR2B.37 Thus, alterations in these two proteins are signs of synaptic degeneration38 that cause neuronal injury and lead to neurodegenerative diseases.39 Therefore, to address this neurodegeneration, we further evaluated the synaptic protein expression and neuronal apoptosis in primary cultured neurons. As shown in Fig. 3A–C, individual treatments with the PM2.5 or SO2 derivatives tended to decrease the expression of PSD-95 and NR2B (0.90- and 0.96-fold of the control, respectively, for PSD-95, and 0.96- and 0.95-fold of the control, respectively, for NR2B), whereas caspase-3 was activated (1.15- and 1.09-fold of the control, respectively), but the differences from the control were not statistically significant. By contrast, the combined exposure to the PM2.5 and SO2 derivatives significantly reduced PSD-95 (0.83-fold of the control) and NR2B (0.82-fold of the control) expressions and stimulated caspase-3 activation (1.43-fold of the control) relative to the control and to each individual component. Additionally, TUNEL staining (Fig. 3D) confirmed the apoptosis. The above results imply that the combined exposure to PM2.5 and SO2 caused a synergistic effect on neurodegeneration under the concentration not causing neurodegenerative damage if it was by itself.
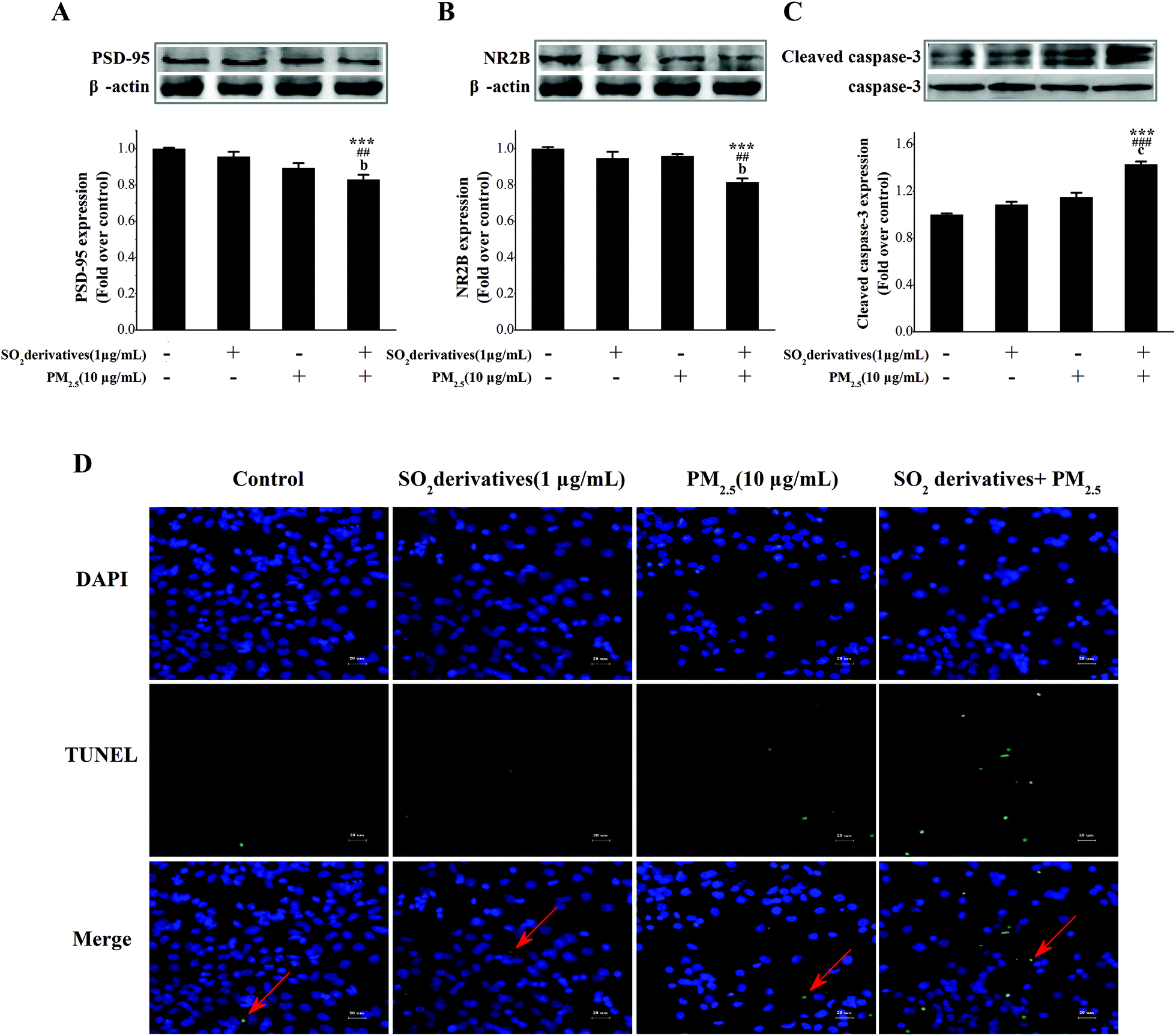 | ||
| Fig. 3 Synergistic effects of co-exposure to the PM2.5 and SO2 derivatives on neurodegeneration. The observed effects on (A) PSD-95 and (B) NR2B expression, and (C) caspase-3 activation after individual or combined treatment with the PM2.5 (10 μg mL−1) and the SO2 derivatives (1 μg mL−1); (D) TUNEL staining after individual or combined treatment with the PM2.5 (10 μg mL−1) and SO2 derivatives (1 μg mL−1). Scale bar, 20 μm. The value of each treated group was expressed as the fold increase compared with the mean value in the control group. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the negative control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with SO2 alone; and ap < 0.05, bp < 0.01, cp < 0.001 compared with PM2.5 alone. | ||
Tau phosphorylation-associated synergistic neurodegeneration induced by PM2.5 and SO2
Hyperphosphorylated tau is generally associated with synaptic loss and neuronal death.40 Tau is a major microtubule-associated protein (MAP), accounting for over 80% of the MAPs, and one of its most important functions is to regulate the stability of axonal microtubules.41 The release of tau plays a key role in synaptic plasticity.42 Therefore, defective tau proteins, which cannot properly stabilize the microtubules, will lead to synaptic loss40 or axonal transport deficits,43 thereby playing an important role in neurodegeneration.44 Therefore, we first determined the tau expression in primary cultured cortical neurons that were exposed to PM2.5 and SO2 individually and in combination (Fig. 4A and B). The PM2.5 (10 μg mL−1) or SO2 derivatives (1 μg mL−1) alone did not cause an obvious change in the total tau level, but the combined exposure to the two components statistically increased the total tau expression relative to that of the control and to each individual component. Generally, the normal tau function is primarily achieved through post-translational modifications, especially phosphorylation.40 Changes in tau phosphorylation can reportedly affect synapses, thereby causing neurodegenerative injury.45 Therefore, we further evaluated that the phosphorylation level of tau at Thr231 (Fig. 4B and C) was significantly elevated following co-combined treatment with PM2.5 (10 μg mL−1) and SO2 derivatives (1 μg mL−1) (2.05-fold of the control) in comparison with the control and with individual exposure to each component. However, individual treatments with the PM2.5 (10 μg mL−1) or SO2 derivatives (1 μg mL−1) did not affect this endpoint. | ||
| Fig. 4 The effects of single or combined exposures to PM2.5 and SO2 on the expression of total tau and tau phosphorylation. (A) Quantifications of the total tau protein expression, (B) immunoblot analysis of total tau and tau phosphorylation expression, and (C) quantifications of tau phosphorylation protein expression in the primary cultured cortical neurons before and after single or combined exposures to the PM2.5 (10 μg mL−1) and SO2 derivatives (1 μg mL−1). (D) Quantifications of the total tau protein expression, (E) immunoblot analysis of the total tau and tau phosphorylation expression, and (F) quantifications of tau phosphorylation protein expression in the cortices of mice that were exposed to PM2.5 (0.075 mg m−3), SO2 (0.5 mg m−3) or their combination. The value in each treated group was expressed as the fold increase compared with the mean value in the control group. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the negative control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with SO2 alone; and ap < 0.05, bp < 0.01, cp < 0.001 compared with PM2.5 alone. | ||
The average PM2.5 and SO2 concentration of the ambient air quality secondary standard was limited to 0.035–0.075 mg m−3 and 0.15–0.5 mg m−3 according to GB 3095-2012,46 respectively. In experimental studies, the lowest PM2.5 concentration at which a significant neuronal loss occurred was 225 μg m−3 (ref. 21) and the statistically effective concentration for SO2 neurotoxicity was 14 mg m−3 (ref. 47) for in vivo assays. To provide in vivo support for the results from the in vitro assays, we exposed animals to PM2.5 (0.075 mg m−3) and SO2 (0.5 mg m−3) individually or in combination for four weeks and evaluated tau expression and phosphorylation at Thr231 in the cortex. Therefore, lower concentrations of PM2.5 and SO2, both of which were close to environmental pollutant levels were chosen to study synergistic injury. As presented in Fig. 4D–F, neither PM2.5 nor SO2 inhalation alone caused any obvious changes in the total tau or tau phosphorylation levels, but the combined exposure to the two components induced a statistically significant enhancement of total tau (1.34-fold of the control) and tau phosphorylation (1.46-fold of the control) expression compared with the vehicle control and either individual treatment. Coupled with the in vitro data, these findings in the cortex also confirm that the combined exposure to PM2.5 and SO2 at low doses that did not induce obvious effects from individual exposures were able to produce tau phosphorylation-associated synergistic neurodegeneration.
MicroRNA modulation of tau phosphorylation in synergistic neurodegeneration induced by PM2.5 and SO2
Although they possess no protein coding sequences, the evolutionarily conserved miRNAs are involved in the developmental control of gene expression at the post-transcriptional level.48 miRNAs play a key role in biological processes49 and have the potential to act as biomarkers to diagnose different types of diseases.50 In particular, recent studies indicated that miRNAs played an important role in neurodegeneration induced by disorders of RNA processing.51 Considering the vital involvement in the neurodegenerative diseases, such as Parkinson's disease52 and multiple sclerosis,53 we focused on miR-337-5p, which was significantly decreased after co-exposure to PM2.5 and SO2 for four weeks (Fig. 5).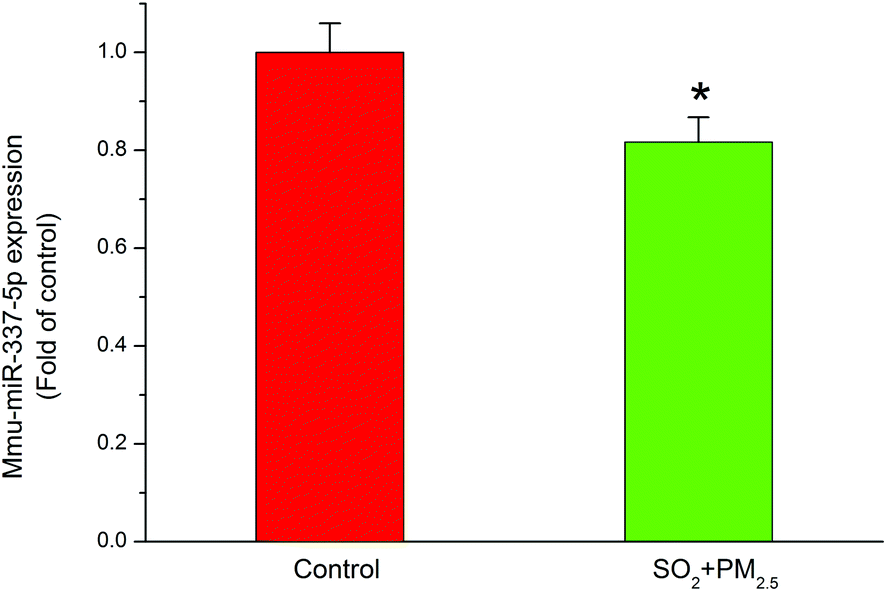 | ||
| Fig. 5 The effects of combined exposures to PM2.5 and SO2 on the expression of miR-337-5p in mice cortex. The mice in the combined group were exposed to 0.5 mg m−3 SO2 in 1 m3 exposure chambers for 28 days (6 h per day), and those in the control groups were exposed to ambient air in another 1 m3 chamber for the same period. In addition, the mice in the combined group received an intranasal instillation of PM2.5 (0.075 mg m−3) every other day. The value in each treated group was expressed as the fold increase compared with the mean value in the control group. *p < 0.05 compared with the control group. | ||
Next, to clarify the possible miRNA modulation, we predicted the potential targets for miR-337-5p by using four bioinformatics programs, namely Targetscan, RNA22, RNAhybrid and miRMap, and found that miR-337-5p was associated with Mapt (see Table S2, ESI†). miRNAs reportedly regulate downstream gene expression by binding to the 3′-untranslated regions (3′-UTR) of their targeted mRNAs. The Mapt gene encodes the tau protein, which is thought to facilitate microtubule assembly and stabilization. Therefore, we hypothesized that miR-337-5p might mediate tau-associated synergistic neurodegeneration by binding to the 3′-UTR of Mapt after SO2 and PM2.5 co-exposure. To test this possibility, we screened the 3′-UTR region of Mapt for mREs (5′-AACGGC-3′) and identified two putative binding sites for miR-337-5p using Targetscan software. Fig. 6 shows that miR-337-5p efficiently inhibited luciferase expression by binding to the Mapt 3′-UTR and significantly decreased the relative luciferase reporter activity of the wild-type Mapt 3′-UTR. By contrast, the luciferase reporter activity of the two mutants of Mapt 3′-UTR (two binding sites, position 3176–3182 and position 3697–3703) decreased and the second binding site has stronger binding force than the first with miR-337-5p. The relative luciferase reporter activity of mutant Mapt 3′-UTR did not change significantly when the binding sites in the Mapt 3′-UTR were mutated. These results suggest that miR-337-5p could directly bind to the 3′-UTR of Mapt. In addition, we pretreated HEK293T cells with a miR-337-5p inhibitor that efficiently reduced miR-337-5p binding to the Mapt 3′-UTR. Coupled with the elevation in tau phosphorylation, these data suggest that co-exposure to PM2.5 and SO2 down-regulated miR-337-5p, which then stimulated tau and its subsequent phosphorylation through the direct binding of miR-337-5p to the 3′-UTR of Mapt mRNA.
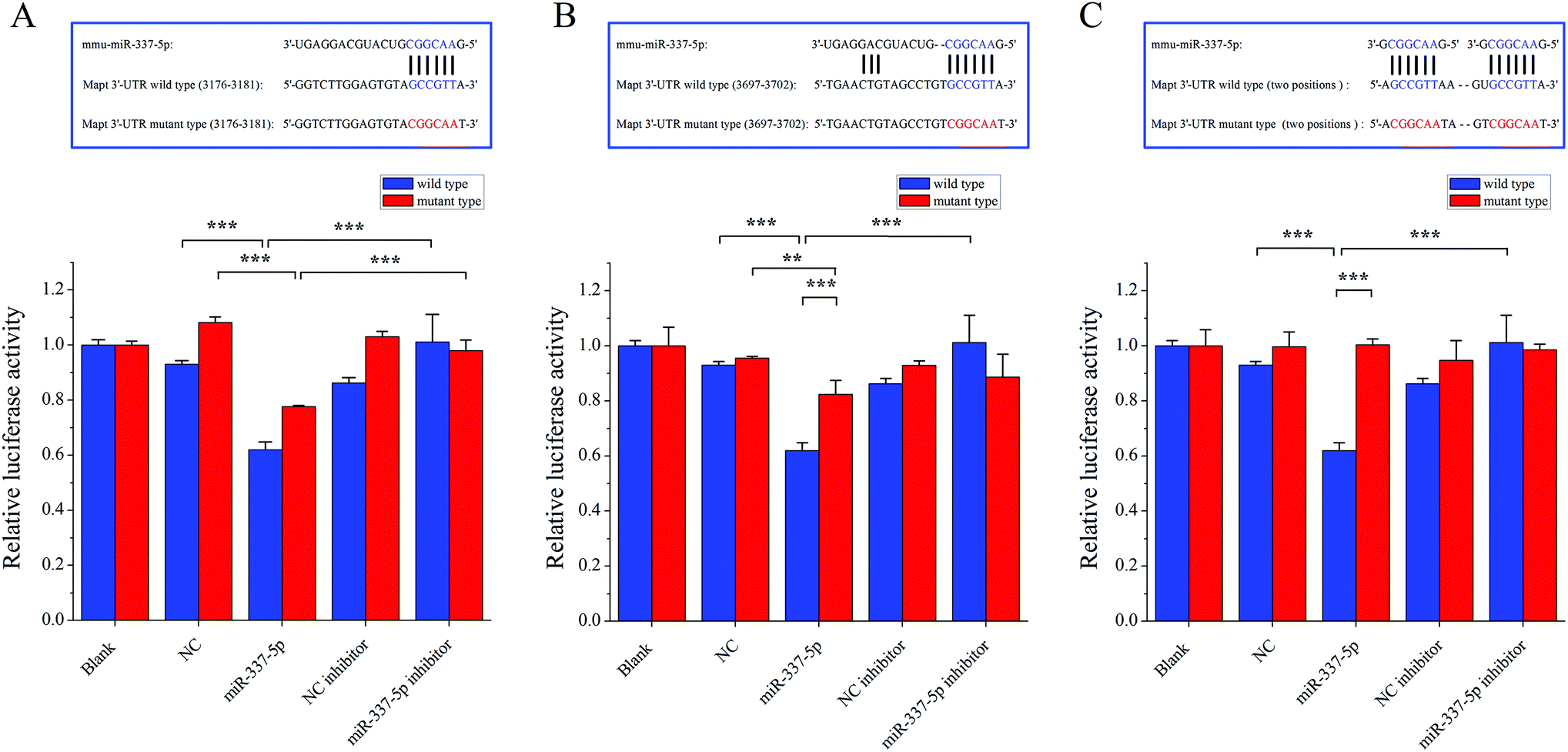 | ||
| Fig. 6 Dual-luciferase reporter gene analysis. An overview of the luciferase assay used for dual-luciferase reporter experiments. These experiments were used to measure the direct inhibition of Mapt expression by miR-337-5p. Mapt contains a conserved 3′-UTR sequence (positions 3176–3182 and 3697–3703) that perfectly complements the miR-337-5p seed sequence (both are shown in blue). The mutant Mapt 3′-UTR contains mutations in the miR-337-5p binding site that disrupt base pairing (indicated in red). The different effects of miR-337-5p on Mapt 3′-UTR and its mutant (A) position 3176–3182, (B) position 3697–3703 and (C) two binding sites are shown as the means ± SD. miR-337-5p can efficiently inhibit luciferase expression by binding to Mapt 3′-UTR, but they had less or no effect on luciferase expression when the binding sites in the Mapt 3′-UTR were mutated. *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
Conclusion
In summary, we showed that co-exposure to PM2.5 and SO2 led to neurodegeneration at low doses that did not induce obvious effects after individual exposures. We subsequently demonstrated that miR-337-5p, which is homologous to human miRNA that targets tau, was involved in the combined effect and contributed to synergistic neurodegeneration. This work implies that there is potential risk of neuronal dysfunction from the co-existence of PM2.5 and SO2 in coal-burning areas and provides new insights into the molecular markers for related diseases.Conflict of interest
The authors declare no competing financial interest.Abbreviations
| PM2.5 | Particulate matter |
| SO2 | Sulfur dioxide |
| LPS | Lipopolysaccharides |
| NF-κB | Nuclear factor-κB |
| AP-1 | Activator protein-1 |
| TNF-α | Tumor necrosis factor-α |
| IL-1α | Interleukin-1α |
| NC | Nitrocellulose |
| BSA | Bovine serum albumin |
| Mapt | Microtubule-associated protein tau |
| PSD | Postsynaptic densities |
| NMDA | N-Methyl-D-aspartic acid |
Acknowledgements
This study was supported by the National Science Foundation of China (NSFC, no. 91543203, 21477070, 21377076, 21222701), the Specialized Research Fund for the Doctoral Program of Higher Education of China (SRFDP, no. 20121401110003, 20131401110005), the Project supported by the Shanxi Young Sanjin Scholarship of China, and the Research Project supported by the Shanxi Scholarship Council of China (no. 2015-006).References
- R. A. Rohde and R. Muller, Air pollution in China: mapping of concentrations and sources, PLoS One, 2015, 10, e0135749, DOI:10.1371/journal.pone.0135749 . eCollection 2015.
- J. L. Mauderly and J. M. Samet, Is there evidence for synergy among air pollutants in causing health effects?, Environ. Health Perspect., 2009, 117, 1–6 CrossRef CAS PubMed.
- Y. T. Huang, A. G. Rappold, D. W. Graff, A. J. Ghio and R. B. Devlin, Synergistic effects of exposure to concentrated ambient fine pollution particles and nitrogen dioxide in humans, Inhalation Toxicol., 2012, 24, 790–797 CrossRef CAS PubMed.
- N. Labranche, K. C. El, L. Dewachter, C. Dreyfuss, J. van de Fontaine, P. Borne, G. Berkenboom and S. Pochet, Vascular oxidative stress induced by diesel exhaust microparticles: synergism with hypertension, J. Cardiovasc. Pharmacol., 2012, 60, 530–537 CrossRef CAS PubMed.
- J. Chen, G. Liu, Y. Kang, B. Wu, R. Sun, C. Zhou and D. Wu, Coal utilization in China: environmental impacts and human health, Environ. Geochem. Health, 2014, 36, 735–753 CrossRef CAS PubMed.
- Y. Kulahci, C. Sever, F. Zor, F. Uygur, N. Noyan, R. Evinc, S. Oksuz, C. Sahin and H. Duman, Thermal burns associated with the misuse of flammable liquids in stoves: a continuing problem, J. Burn Care Res., 2011, 32, 302–308 CrossRef PubMed.
- Y. Wang, Q. Ying, J. Hu and H. Zhang, Spatial and temporal variations of six criteria air pollutants in 31 provincial capital cities in China during 2013–2014, Environ. Int., 2014, 73, 413–422 CrossRef CAS PubMed.
- H. Zhang, J. Hu, M. Kleeman and Q. Ying, Source apportionment of sulfate and nitrate particulate matter in the Eastern United States and effectiveness of emission control programs, Sci. Total Environ., 2014, 490, 171–181 CrossRef CAS PubMed.
- L. Craig, J. R. Brook, Q. Chiotti, B. Croes, S. Gower, A. Hedley, D. Krewski, A. Krupnick, M. Krzyzanowski, M. D. Moran, W. Pennell, J. M. Samet, J. Schneider, J. Shortreed and M. Williams, Air pollution and public health: a guidance document for risk managers, J. Toxicol. Environ. Health, Part A, 2008, 71, 588–698 CrossRef CAS PubMed.
- M. Qin, X. Wang, Y. Hu, X. Huang, L. He, L. Zhong, Y. Song, M. Hu and Y. Zhang, Formation of particulate sulfate and nitrate over the pearl river delta in the fall: diagnostic analysis using the community multiscale air quality model, Atmos. Environ., 2015, 112, 81–89 CrossRef CAS.
- Y. Xie, B. Zhao, L. Zhang and R. Luo, Spatiotemporal variations of PM2.5 and PM10 concentrations between 31 Chinese cities and their relationships with SO2, NO2, CO and O3, Particuology, 2015, 20, 141–149 CrossRef CAS.
- M. Viana, W. Maenhaut, X. Chi, X. Querol and A. Alastuey, Comparative chemical mass closure of fine and coarse aerosols at two sites in south and west Europe: implications for EU air pollution policies, Atmos. Environ., 2007, 41, 315–326 CrossRef CAS.
- W. C. Lo, R. H. Shie, C. C. Chan and H. H. Lin, Burden of disease attributable to ambient fine particulate matter exposure in Taiwan, J. Formosan Med. Assoc., 2016 DOI:10.1016/j.jfma.2015.12.007.
- P. Gillespie, J. Tajuba, M. Lippmann, L. C. Chen and B. Veronesi, Particulate matter neurotoxicity in culture is size-dependent, Neurotoxicology, 2013, 36, 112–117 CrossRef CAS PubMed.
- S. Genc, Z. Zadeoglulari, S. H. Fuss and K. Genc, The adverse effects of air pollution on the nervous system, J. Toxicol., 2012, 2012, 782462 Search PubMed.
- K. R. Doty, M. Guillot-Sestier and T. Town, The role of the immune system in neurodegenerative disorders: adaptive or maladaptive?, Brain Res., 2015, 1617, 155–173 CrossRef CAS PubMed.
- L. Calderón-Garcidueñas, A. Calderón-Garcidueñas, R. Torres-Jardón, J. Avila-Ramírez, R. J. Kulesza and A. D. Angiulli, Air pollution and your brain: what do you need to know right now, Prim. Health Care Res. Dev., 2015, 16, 329–345 CrossRef PubMed.
- F. Liu, Y. Huang, F. Zhang, Q. Chen, B. Wu, W. Rui, J. C. Zheng and W. Ding, Macrophages treated with particulate matter PM2.5 induce selective neurotoxicity through glutaminase-mediated glutamate generation, J. Neurochem., 2015, 134, 315–326 CrossRef CAS PubMed.
- U. Ranft, T. Schikowski, D. Sugiri, J. Krutmann and U. Krämer, Long-term exposure to traffic-related particulate matter impairs cognitive function in the elderly, Environ. Res., 2009, 109, 1004–1011 CrossRef CAS PubMed.
- R. Field, S. Campion, C. Warren, C. Murray and C. Cunningham, Systemic challenge with the TLR3 agonist poly I: C induces amplified IFNα/β and IL-1β responses in the diseased brain and exacerbates chronic neurodegeneration, Brain, Behav., Immun., 2010, 24, 996–1007 CrossRef CAS PubMed.
- L. S. Fagundes, A. S. Fleck, A. C. Zanchi, P. H. Saldiva and C. R. Rhoden, Direct contact with particulate matter increases oxidative stress in different brain structures, Inhalation Toxicol., 2015, 27, 462–467 CrossRef CAS PubMed.
- M. Szyszkowicz, B. Rowe and G. Kaplan, Ambient sulphur dioxide exposure and emergency department visits for migraine in Vancouver, Canada, Int. J. Occup. Med. Environ. Health, 2009, 22, 7–12 Search PubMed.
- G. Yao, H. Yue, Y. Yun and N. Sang, Chronic SO2 inhalation above environmental standard impairs neuronal behavior and represses glutamate receptor gene expression and memory-related kinase activation via neuroinflammation in rats, Environ. Res., 2015, 137, 85–93 CrossRef CAS PubMed.
- B. Li, M. Chen, L. Guo, Y. Yun, G. Li and N. Sang, Endogenous 2-Arachidonoylglycerol alleviates Cyclooxygenases-2 elevation-mediated neuronal injury from SO2 inhalation via PPARγ pathway, Toxicol. Sci., 2015, 147, 535–548 CrossRef CAS PubMed.
- J. Payne, M. Scholze and A. Kortenkamp, Mixtures of four organochlorines enhance human breast cancer cell proliferation, Environ. Health Perspect., 2001, 109, 391–397 CrossRef CAS PubMed.
- N. Sang, Y. Yun, G. Yao, H. Li, L. Guo and G. Li, SO2-induced neurotoxicity is mediated by cyclooxygenases-2-derived prostaglandin E2 and its downstream signaling pathway in rat hippocampal neurons, Toxicol. Sci., 2011, 124, 400–413 CrossRef CAS PubMed.
- M. L. Block and L. Calderón-Garcidueñas, Air pollution: mechanisms of neuroinflammation and CNS disease, Trends Neurosci., 2009, 32, 506–516 CrossRef CAS PubMed.
- The Central People's Government of the People's Republic of China website; http://www.gov.cn/zwgk/2011-12/20/content_2024895.htm, 2011.
- S. Maciotta, M. Meregalli and Y. Torrente, The involvement of microRNAs in neurodegenerative diseases, Front. Cell. Neurosci., 2013, 7, 265 Search PubMed.
- S. Elmore, Apoptosis: a review of programmed cell death, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- R. Santhanasabapathy and G. Sudhandiran, Farnesol attenuates lipopolysaccharide-induced neurodegeneration in Swiss albino mice by regulating intrinsic apoptotic cascade, Brain Res., 2015, 1620, 42–56 CrossRef CAS PubMed.
- R. S. Williams and C. Bate, An in vitro model for synaptic loss in neurodegenerative diseases suggests a neuroprotective role for valproic acid via inhibition of cPLA2 dependent signaling, Neuropharmacology, 2015, 101, 66–75 Search PubMed.
- C. E. Taft and G. G. Turrigiano, PSD-95 promotes the stabilization of young synaptic contacts, Philos. Trans. R. Soc. London, Ser. B, 2013, 369, 20130134, DOI:10.1098/rstb.2013.0134.
- Y. Bu, N. Wang, S. Wang, T. Sheng, T. Tian, L. Chen, W. Pan, M. Zhu, J. Luo and W. Lu, Myosin IIb-dependent regulation of actin dynamics is required for NMDA receptor trafficking during synaptic plasticity, J. Biol. Chem., 2015, 290, 25395–25410 CrossRef CAS PubMed.
- O. A. Shipton and O. Paulsen, GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity, Philos. Trans. R. Soc. London, Ser. B, 2013, 369, 20130163, DOI:10.1098/rstb.2013.0163.
- S. L. Cousins and F. A. Stephenson, Identification of N-methyl-D-aspartic acid (NMDA) receptor subtype-specific binding sites that mediate direct interactions with scaffold protein PSD-95, J. Biol. Chem., 2012, 287, 13465–13476 CrossRef CAS PubMed.
- A. E. El-Husseini, E. Schnell, D. M. Chetkovich, R. A. Nicoll and D. S. Bredt, PSD-95 involvement in maturation of excitatory synapses, Science, 2000, 290, 1364–1368 CAS.
- Q. Jiang, Y. Wu, J. Wang, X. Wu, J. Qin and Y. Jiang, Characterization of developing rat cortical neurons after epileptiform discharges, Int. J. Dev. Neurosci., 2010, 28, 455–463 CrossRef CAS PubMed.
- M. Marttinen, K. M. Kurkinen, H. Soininen, A. Haapasalo and M. Hiltunen, Synaptic dysfunction and septin protein family members in neurodegenerative diseases, Mol. Neurodegener., 2015, 10, 16 CrossRef PubMed.
- M. Medina and J. Avila, Further understanding of tau phosphorylation: implications for therapy, Expert Rev. Neurother., 2015, 15, 115–122 CrossRef CAS PubMed.
- J. Wang and Z. Wang, Senescence may mediate conversion of tau phosphorylation-induced apoptotic escape to neurodegeneration, Exp. Gerontol., 2015, 68, 82–86 CrossRef CAS PubMed.
- S. Sokolow, K. M. Henkins, T. Bilousova, B. Gonzalez, H. V. Vinters, C. A. Miller, L. Cornwell, W. W. Poon and K. H. Gylys, Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer's disease, J. Neurochem., 2015, 133, 368–379 CrossRef CAS PubMed.
- E. Peethumnongsin, L. Yang, V. Kallhoff-Muñoz, L. Hu, A. Takashima, R. G. Pautler and H. Zheng, Convergence of presenilin- and tau-mediated pathways on axonal trafficking and neuronal function, J. Neurosci., 2010, 30, 13409–13418 CrossRef CAS PubMed.
- E. Audouard, L. Van Hees, V. Suain, Z. Yilmaz, L. Poncelet, K. Leroy and J. Brion, Motor deficit in a Tauopathy model is induced by disturbances of axonal transport leading to dying-back degeneration and denervation of neuromuscular junctions, Am. J. Pathol., 2015, 185, 2685–2697 CrossRef CAS PubMed.
- B. A. Warmus, D. R. Sekar, E. McCutchen, G. D. Schellenberg, R. C. Roberts, L. L. McMahon and E. D. Roberson, Tau-mediated NMDA receptor impairment underlies dysfunction of a selectively vulnerable network in a mouse model of frontotemporal dementia, J. Neurosci., 2014, 34, 16482–16495 CrossRef PubMed.
- Ministry of Environmental Protection of the People's Republic of China website, http://kjs.mep.gov.cn/hjbhbz/bzwb/dqhjbh/dqhjzlbz/201203/t20120302_224165.htm, (accessed 16.01.01), 2012.
- G. Li and N. Sang, Delayed rectifier potassium channels are involved in SO2 derivative-induced hippocampal neuronal injury, Ecotoxicol. Environ. Saf., 2009, 72, 236–241 CrossRef CAS PubMed.
- D. P. Bartel, MicroRNAs: target recognition and regulatory functions, Cell, 2009, 136, 215–233 CrossRef CAS PubMed.
- K. Vrijens, V. Bollati and T. S. Nawrot, MicroRNAs as potential signatures of environmental exposure or effect: A systematic review, Environ. Health Perspect., 2015, 123, 399–411 CAS.
- C. Wu, C. Wang, X. Guan, Y. Liu, D. Li, X. Zhou, Y. Zhang, X. Chen, J. Wang, K. Zen, C. Zhang and C. Zhang, Diagnostic and prognostic implications of a serum miRNA panel in oesophageal squamous cell carcinoma, PLoS One, 2014, 9, e92292 Search PubMed.
- R. Johnson, W. Noble, G. G. Tartaglia and N. J. Buckley, Neurodegeneration as an RNA disorder, Prog. Neurobiol., 2012, 99, 293–315 CrossRef CAS PubMed.
- L. F. Cardo, E. Coto, R. Ribacoba, M. Menéndez, G. Moris, E. Suárez and V. Alvarez, MiRNA profile in the substantia nigra of Parkinson's disease and healthy subjects, J. Mol. Neurosci., 2014, 54, 830–836 CrossRef CAS PubMed.
- W. J. Magner, B. Weinstock-Guttman, M. Rho, D. Hojnacki, R. Ghazi, M. Ramanathan and T. B. Tomasi, Dicer and microRNA expression in multiple sclerosis and response to interferon therapy, J. Neuroimmunol., 2016, 292, 68–78 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information on some supporting figures and tables. See DOI: 10.1039/c6tx00314a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |