
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spin-dimer networks: engineering tools to adjust the magnetic interactions in biradicals†
Yulia B.
Borozdina
ab,
Evgeny A.
Mostovich
ac,
Pham Thanh
Cong
d,
Lars
Postulka
d,
Bernd
Wolf
d,
Michael
Lang
d and
Martin
Baumgarten
 *a
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: martin.baumgarten@mpip-mainz.mpg.de
bInstitute of Biochemistry, Ernst-Moritz-Arndt University Greifswald, Felix-Hausdorff-Straße 4, 17487 Greifswald, Germany
cNovosibirsk Institute of Organic Chemistry Siberian Branch of the Russian Academy of Science, Lavrentiev avenue 9, 630090 Novosibirsk, Russia
dPhysikalisches Institut, Johann Wolfgang Goethe-University Frankfurt, 60438 Frankfurt am Main, Germany
First published on 15th August 2017
Abstract
Magneto-structural correlations in stable organic biradicals have been studied on the example of weakly exchange coupled models with nitronyl nitroxide and imino nitroxide spin-carrying entities. Here, heteroatom substituted 2,2′-diaza- and 3,3′-diaza-tolane bridged biradicals were compared with the hydrocarbon analogue, while a biphenyl model with its 2,2′-bipyridine counterpart. For a 3,3′-diazatolane bridge the torsional angle between the nitronyl nitroxides and the pyridyl rings increased heavily (∼52–54°) leading to a smaller theoretical intra-dimer exchange coupling value. However, a very large antiferromagnetic coupling was obtained experimentally. This could be appropriately explained by the presence of dominating inter-dimer exchange between the molecules. For the bis(imino nitroxide) with tolane bridge a field induced ordered state between 1.8 to 4.3 T in AC-susceptibility measurements was observed. In terms of a Bose Einstein condensate (BEC) of triplons this phenomenon could be described as a magnetic field induced ordered phase with 3D character.
Introduction
Magnetic dimers formed by weakly interacting antiferromagnetically coupled spin S = 1/2 centres are recognized as suitable candidates for exploring critical phenomena under well-controlled conditions.1 When these systems are placed in a magnetic field strong enough to close the dimer gap, a gas of triplet excitations (triplons) is formed.1 Depending on the topology of the dimer–dimer couplings, various scenarios can be observed. Prominent examples include the Bose-Einstein condensation (BEC) of triplons in three-dimensionally-coupled dimer systems, as described by Tchernyshyov et al.,2 and the Luttinger-liquid behavior revealed in a one-dimensional spin-ladder system by Krämer and co-workers.3 More recently, a Berezinskii–Kosterlitz–Thouless scenario was observed by Tutsch et al. in a two-dimensional coordinated copper polymer with large interlayer spacing.4In general, organic magnetic materials possess properties not shown by traditional inorganic compounds.5–8 Plasticity, flexibility and solubility in common organic solvents define the ease of various device fabrication. Advanced chemistry techniques permit small structural modifications in already achieved model systems in order to fine-tune their physical properties. In addition, organic-based magnetic materials offer a number of convenient tools extremely helpful in controlling the intra- and inter-dimer exchange coupling, such as π–π interactions, hydrogen bonding, etc. Nitronyl nitroxide (NN) radicals are well-known for their stability and bidentate character.9,10 NN radicals are among the most popular spin carriers used to construct molecule-based magnets.10
Importantly, in these radicals the spin density of the unpaired electron is delocalized over two semi-equivalent sites of coordination. This in turn allows the arrangement of the NN-molecules into a supramolecular network of interacting spins.11 Theoretical studies emphasized the importance of the mutual orientation and the relative distances in the crystal packing for promoting an efficient magnetic coupling.11,12 As was mentioned by Lahti,13 minor changes in the crystal packing of biradicals could result in significant alterations of the magnetic behavior in the bulk material. In this regard the influence of the π-bridges on the intra- and intermolecular exchange interactions in conjugated biradical networks was studied on the example of the diazotolane dinitroxide models (Fig. 1). It was anticipated that heteroatom substitution could offer a particularly effective pathway for transmitting the magnetic interactions.14
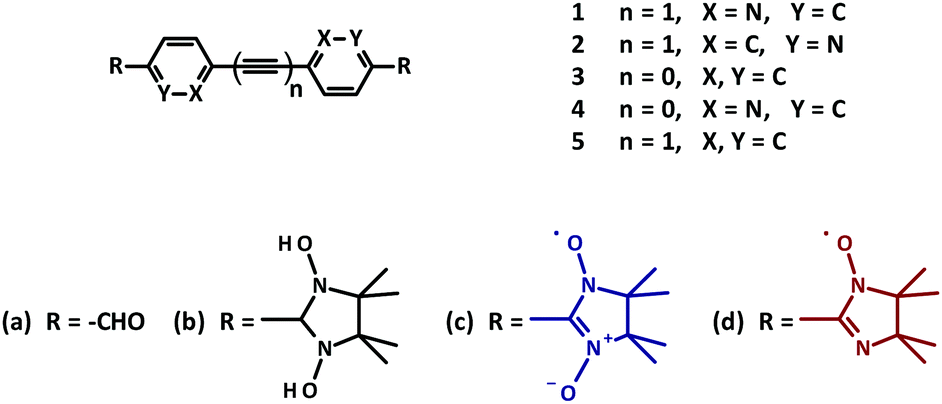 | ||
| Fig. 1 Structure of biradical model systems 1–5. | ||
Typically, imino nitroxides (INs) reveal a reduced Jdimer value when compared to the corresponding NN biradicals. Taking this fact into consideration, already known bridges could be used, where otherwise a strong intramolecular exchange coupling was observed.11 Following this strategy, three bisimino nitroxides 3d, 4d, 5d were prepared (Fig. 1).15
In the current work we report the synthesis of a series of new nitroxide biradicals (Fig. 1), their characterization, including X-ray structural analysis and their magnetic properties, which are discussed with respect to the quantum chemical calculations based on the DFT approach.
Results and discussion
Synthesis of nitroxide biradicals
Access to the family of diazatolane biradicals 1c and 2c required the synthesis of key precursors 1a and 2a, which were prepared following Sonogashira–Hagihara methodology.16 Interestingly, both structural isomers 1a and 2a could be achieved starting from the same precursor, i.e. commercially available 2,5-dibromopyridine 6 (Scheme 1). Depending on the reaction conditions (such as solvent, base) compound 6 could be selectively monolithiated.17–21 More precisely, reaction times and solvents played a crucial role in the course of electrophilic substitution, leading to predominant formation of the organolithium intermediate at the 2 or 5 position.22,23 In order to drive the reaction in the course of kinetically more favorable 2-bromo-5-carbaldehyde-pyridine 7, it was carried out in dry diethyl ether at −78 °C, and DMF was added to the mixture 20 min after the addition of n-BuLi was complete (Scheme 1).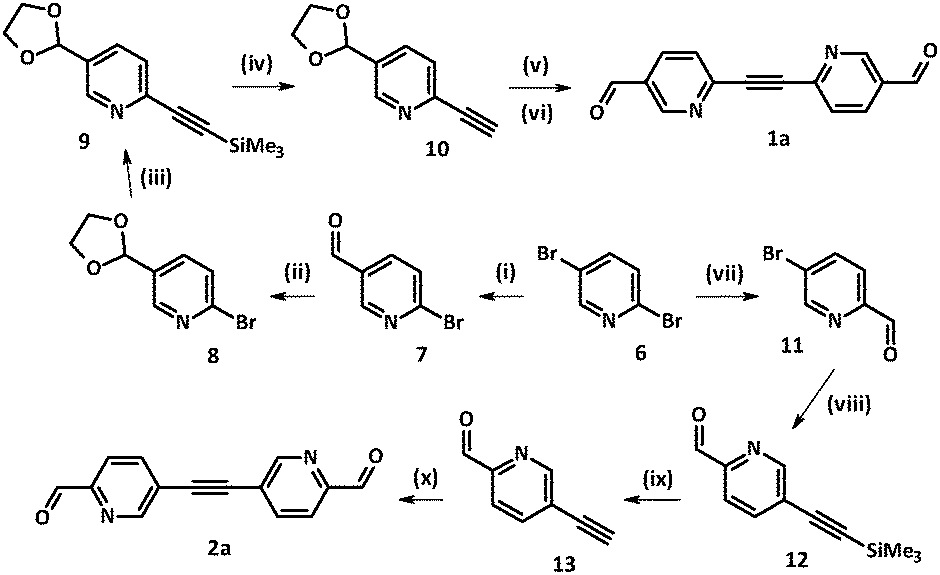 | ||
| Scheme 1 Synthesis of dialdehydes 1a and 2a. Reagents and conditions: (i) (a) n-BuLi, Et2O, −78 °C, under Ar, 45 min, (b) DMF, −78 °C, under Ar, 105 min, (c) HCl (3%), 20 min; (ii) ethyleneglycol, p-TsOH, benzene, reflux, 20 h; (iii) TMSA, Pd(PPh3)2Cl2, CuI, PPh3, DMF, Et3N, heating at 80 °C under Ar, 14 h; (iv) 1 M NaOH, H2O-THF, 5 °C, 35 min; (v) 8, Pd(PPh3)2Cl2, CuI, PPh3, CH3CN, Et3N, at rt under Ar, 18 h; (vi) p-TsOH, H2O–acetone, 84 h; (vii) (a) n-BuLi, toluene, −78 °C, under Ar 2 h, (b) DMF, −78 °C, under Ar, 2 h, (c) NH4Cl, 30 min; (viii) TMSA, Pd(PPh3)2Cl2, CuI, PPh3, DMF, Et3N, heating at 80 °C under Ar, 16 h; (ix) K2CO3, MeOH, under Ar 90 min; (x) 11, Pd(PPh3)2Cl2, CuI, PPh3, DMF, Et3N, heating at 80 °C under Ar, 12 h. | ||
According to the preliminary results obtained by our group, 2-ethynyl-5-formyl-pyridine was unstable. Therefore, at first aldehyde 7 was transformed into 2-bromo-5-[1,3]dioxolan-2-yl-pyridine 8 following the standard protocol.24 Compound 8 was involved in Pd-catalyzed Sonogashira–Hagihara coupling with trimethylsilyl acetylene (TMSA) giving rise to derivative 9. Hydrolysis of the trimethylsilyl group using 1 N NaOH solution in a deaerated THF–water mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) granted the NMR-pure ethynyl derivative 10, which was used further without additional purification.
:1) granted the NMR-pure ethynyl derivative 10, which was used further without additional purification.
Sonogashira–Hagihara coupling reactions can be successfully carried out under various conditions.16 For our systems it was found that by decreasing the reaction temperature from 80 °C (DMF/Et3N, 1:1) to ∼22 °C (CH3CN/Et3N, 1:1) the formation of several side-products was prevented. Therefore, the attachment of bromo derivative 8 to the pre-organized ethynyl-pyridine 10 was carried out at room temperature in the CH3CN/Et3N solvent mixture, which led to the corresponding product in a reasonable yield (72%). Notably, our attempts to remove dioxolane protective groups in the presence of dilute HCl (3%) resulted in precipitation of the acid by the triple bond. Thus, the final step towards 1a was performed under milder conditions. To a solution of the dioxolane precursor in an acetone–water mixture (7:1) a catalytic amount of p-TsOH acid (2 mol%) was added.24 Upon stirring the reaction mixture for 3 days at room temperature the target 2,2′-diazatolane 4,4′-dialdehyde 1a was obtained in nearly quantitative yield (81%).
The isomeric 3,3′-diazatolane dialdehyde 2a was obtained in a similar way (Scheme 1). Here, 2,5-dibromo-pyridine 6 was selectively monolithiated in position 2 in toluene media. Time of the exchange reaction with n-BuLi was increased to 90 min to ensure the desired organolithium intermediate formation, and precursor 11 was obtained in 49% yield.21 Next, standard Sonogashira–Hagihara methodology16 was employed. Final separation on a silica gel column with a hexane/EtOAc eluent mixture (3:2) yielded 5,5′-ethyne-1,2-diylbis(pyridine-2-carbaldehyde) 2a in decent yield (69%).
Several synthetic procedures towards compound 3a were reported in the literature.15 Inspired by the efficiency and simplicity of the approach sketched in Scheme 2, commercially available 4,4′-dibromobiphenyl 14 was treated with n-BuLi in the presence of a catalytic amount of TMEDA. After addition of dry DMF, typical work-up and purification, biphenyl dialdehyde 3a was isolated in 58% yield.
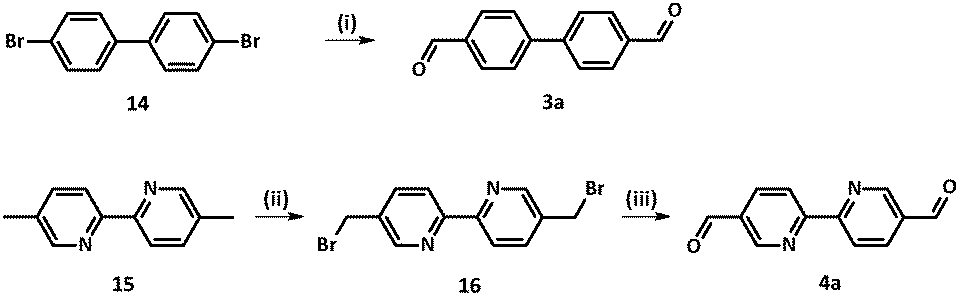 | ||
| Scheme 2 Synthesis of dialdehydes 3a and 4a. Reagents and conditions: (i) (a) n-BuLi, THF, TMEDA, under Ar, −78 °C 30 min, −50 °C 30 min; (b) DMF, −78 °C, under Ar, 1 h, (c) NH4Cl, 20 min; (ii) NBS, CCl4, benzoyl peroxide, reflux under Ar, 14 h; (iii) hexamethylenetetramine, EtOH–H2O, reflux, 60 h. | ||
Bipyridine-4,4′-dicarbaldehyde 4a was achieved upon formation of 5,5′-bis-(bromo-methyl)-2,2′-bipyridine 16 from the corresponding dimethyl-bipyridine precursor 15 following the procedure described by Vögtle.25 Then nucleophilic substitution under Sommelet conditions led to 4a.26 Synthesis of tolane dialdehyde 5a was described elsewhere.11
Final steps in the synthesis of nitroxides included condensation between 2,3-bis(hydroxyamino)-2,3-dimethylbutane (BSA) with an aldehyde, followed by the oxidation of the condensation products (i.e. imidazolidine derivatives 1–5b).11,15 Typically, the first reaction was performed under strictly anaerobic conditions in refluxing toluene (Scheme 3). Here, the corresponding N,N′-dihydroxyimidazolidines 1b, 3b, 4b, and 5b were obtained in quantitative yields. As an exception synthesis of diazotolane imidazolidine 2b was realized in absolute degassed methanol at room temperature.
 | ||
| Scheme 3 Synthesis of nitroxide biradicals 1c and 1d. Reagents and conditions: (i) BSA, toluene, reflux under Ar, 4–6 h; (ii) NaIO4, CH2Cl2–H2O, 0–5 °C, 30–40 min; (iii) MnO2, CH3NO2, 35 min. | ||
In order to avoid further oxidation, and, therefore, to diminish the loss of the radical units, oxidation of N,N′-dihydroxyimidazolidine intermediate 1b with sodium periodate was carried out at ∼0–5 °C using an ice bath.27 The progress of the reaction could be conveniently monitored by TLC analysis of the reaction mixture aliquots. Synthesis of derivative 2c was achieved using an excess of MnO2 in methanol. Imino biradicals 1d, 3d, and 4d were obtained following the procedure described by Tretyakov et al.28,29 This method helped to avoid the harmful usage of acids and to synthesize the target molecules in better yields. As an illustration, transformation of imidazolidine 1b into the corresponding imino biradical 1d with an excess of MnO2 in CH3NO2 media is depicted in Scheme 3. To aid the interpretation of magnetic properties of the isomeric NN 1c and 2c related IN biradical 1d was also prepared.
The UV-Vis absorption spectra of 1c and 2c with maxima around 600 nm were typical for the nitronyl nitroxides, while the red bisiminonitroxides absorbed around 460 nm (Table S2, ESI†). The EPR spectra of bisnitronylnitroxides exhibited nine lines with An/2 spacing for four equivalent nitrogens with two strongly coupled radicals. The bisiminonitroxides (1d, 3b, 4b, 5d) featured 13 lines, corresponding to the inequivalent nitrogens of the imidazoline ring (as presented in the Graphical abstract and Table S2, ESI†).
Crystal structure analysis
Crystals were obtained by slow diffusion of hexane in dichloromethane solutions of the nitroxide biradicals at room temperature. Deep-blue needle crystals of nitronyl nitroxides 1c and 2c and red blocks of imino nitroxides 1d, 3d, 4d, and 5d were then characterized using X-ray diffraction analysis. Selected torsion angles are reported in Table 1 (with some additional structural data given in the ESI†).| Biradical | d, Å | θ, deg | |
|---|---|---|---|

|
1c |
1.4513(15) on C18
1.4564(15) on C5 |
21.62(9)
24.84(10) |
| 2c | 1.472(18) | 53.72(11) | |
| 1d |
1.4743(12) on C25
1.4717(12) on C5 |
23.16(12)
24.85(9) |
|
| 3d | 1.471(2) |
20.2(2) O1
15.35(18) at O2 |
|
| 4d | 1.473(2) | 13.57(19) | |
| 5d | 1.4754(17) | 5.97(12) |
All nitroxide biradicals 1c, 2c, 1d, 3d, 4d, and 5d under study possess a planar backbone – the necessary prerequisite for the propagation of weak intramolecular magnetic interactions. Importantly, in such biradical models the torsion angles θ (see Table 1) play a central role in the modulation of magnetic exchange interactions.
Not surprisingly, similar torsion angles θ of about ∼24° result in a nearly identical degree of conjugation.11,15 Consequently, a comparable spin polarization and close values of the exchange integrals were expected for most of the obtained biradical models (see for comparison Table 1). In contrast to the described situation, the radical units in nitronyl nitroxide 2c were far more twisted (>50°). From this crystal structure analysis it was assumed that nitronyl biradical 2c would exhibit a unique behavior among the compounds investigated here.
Biradical 1c adopts a triclinic space group with an inversion centre residing in the middle of the acetylene bridge (Fig. 2). Remarkably, in the asymmetric unit there are two crystallographically independent molecules with a nearly equal geometry. The dihedral angles of the mean plane of the imidazolidine ring and the two coplanar pyridine rings were found to be ≈25° for O1–N2–C7 (O2N3–C7) towards C5 and 22° for O3N5–C20 (O4–N6–C20) towards C18 (see Table 1). Here, the N(2)–O(1) 1.280(1), N(3)–O(2) 1.278(2), N(5)–O(3) 1.279(1), and N(6)–O(4) 1.272(2) Å bond lengths occur in the typical range for the nitroxides.11,14,15 The N(2)–C(7) 1.355(2), N(2)–C(11) 1.503(1), N(3)–C(7) 1.351(1), N(3)–C(8) 1.503(2), N(5)–C(20) 1.358(2), N(5)–C(24) 1.502(1), N(6)–C(20) 1.354(2), and N(6)–C(21) 1.497(1) Å bond distances are elongated due to the short intermolecular contacts that stabilize the crystal packing (for instance, the ones formed by N(6)O(4)).
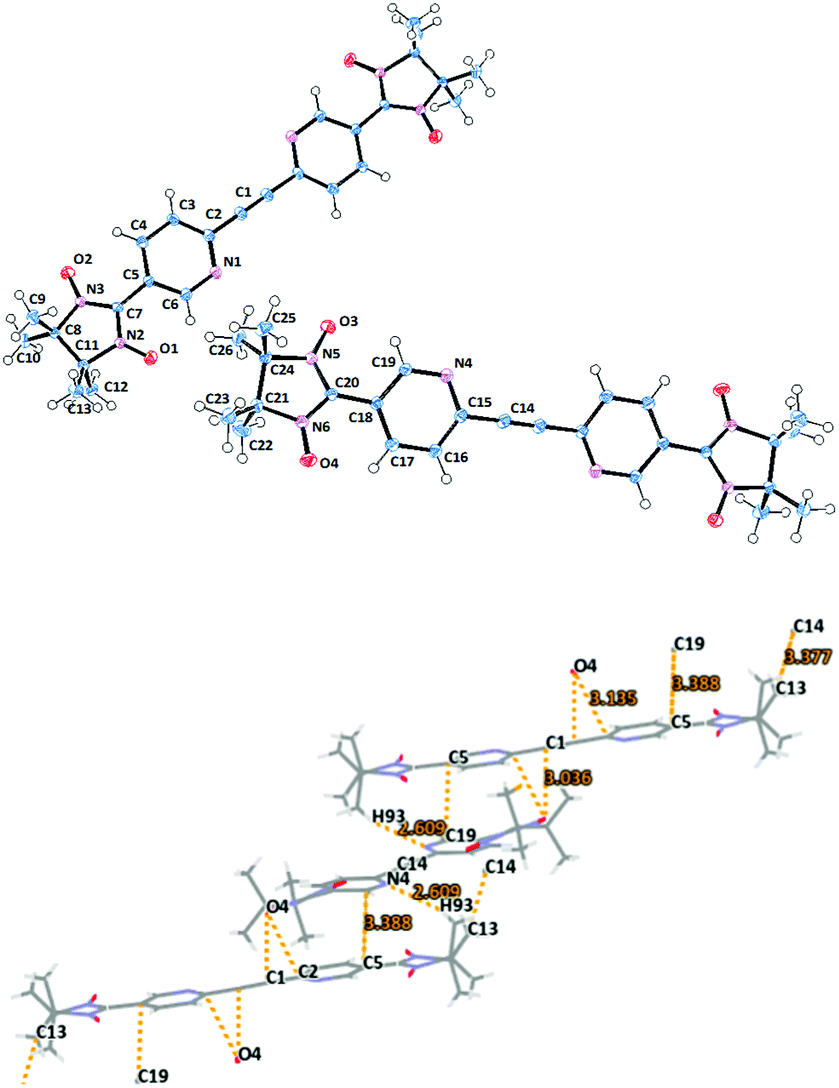 | ||
| Fig. 2 Molecular structure (top) of biradical 1c with ORTEP drawn at the 50% probability level and crystal packing (bottom) with emphasized short contacts. | ||
The intermolecular contacts between NO groups of the first molecule with the pyridine rings (O4⋯C1′ 3.036, O4⋯C2′ 3.135 Å) of the neighboring biradical lead to V-shaped alignment of dimers (Fig. 2 and Fig. S1, ESI†).
Biradical 2c crystallizes in the P21/c space group and its molecular structure is shown in Fig. 3. Compound 2c features a transoid arrangement with an inversion center of symmetry located in the C1–C1′ bond, which is typical for the derivatives containing acetylene bridges (Fig. 3). In general, the structural data of 2c are rather similar to those described for the other nitronyl nitroxide biradicals in the literature (Table 1 and Table S1, ESI†).11,14 A remarkable difference is found in the largely increased torsion between the pyridyl ring and the imidazolidine fragment O1–N2–C7 (O2–N3–C7) with a dihedral angle of 53.72(11) (Fig. 3).
 | ||
| Fig. 3 Molecular structure (top) and crystal packing (bottom) of biradical 2c (view along the a axes). | ||
The crystal packing of 2c presents a beautiful example where the bidentate character of the nitronyl nitroxide fragment plays a major role in the spatial arrangement of the biradical chains. Here, the molecules of 2c form infinite zig-zag ribbons along the b axes (Fig. 3 and Fig. S3, ESI†). Notably, the neighboring molecules are organized in an alternate fashion, placing NN groups next to the pyridine plane and, thereby, stabilizing the crystal packing (Fig. S3, ESI†).
Imino biradical 1d crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . There are two independent centrosymmetric half molecules in the asymmetric unit cell. (Fig. 4). The main structural characteristics are similar to those described for the corresponding nitronyl nitroxide derivative 1c. Thus, the pyridine rings form angles of ≈23° and ≈25° with the imidazolidine plane (Table 1). The N(2)–O(1) 1.269(1), N(221)–O(211) 1.277(2), and N(222)–O(212) 1.257(5) Å bond distances featured minor differences.
. There are two independent centrosymmetric half molecules in the asymmetric unit cell. (Fig. 4). The main structural characteristics are similar to those described for the corresponding nitronyl nitroxide derivative 1c. Thus, the pyridine rings form angles of ≈23° and ≈25° with the imidazolidine plane (Table 1). The N(2)–O(1) 1.269(1), N(221)–O(211) 1.277(2), and N(222)–O(212) 1.257(5) Å bond distances featured minor differences.
 | ||
| Fig. 4 Molecular structure (top) with two biradical molecules per asymmetric unit of biradical 1d and short contacts and π-stacking specified in fragments of molecules from crystal packing. | ||
The packing arrangement of radical dimers 1d is shown in Fig. 4, and can be described as a ladder-like structure with biradicals of the first kind forming a stair, and nitroxides of the second type extending from both sides of each step (Fig. S4, ESI†). The structure is supported by numerous hydrogen bonds arising from the pyridine π-bridges and the imino nitroxide chelating units. The short contacts of the NO groups and the hydrogen atoms of the pyridine core O211⋯H261′ = 2.456, O212⋯H261′ = 2.538 Å (Fig. 4) seems to be the most important. The shortest interchain distances of 3.352 Å (C6⋯C25′) are found between the two neighboring diazatolane bridges (Fig. 4). The closest intermolecular contacts between NO groups and a pyridine ring are slightly larger, and are within the range of ∼2.4–2.7 (O1⋯ H41′, O1⋯H31′, respectively) and ∼3.1 Å (O1⋯C4′).
Imino biradicals 3d and 4d feature isomorphic crystal structures. Thus, they possess the P21/n space group with a center of symmetry located in the middle of the aromatic C–C′ bond (Fig. 5 and Fig. S4, ESI†). The biphenyl bridge in 3d is surprisingly planar, and the dihedral angle between the mean plane of the benzene ring and the imidazolidine moiety is only 20.2°. 3d is slightly disordered, since the five membered ring can flip around 180° such that the two NO groups could be oriented ‘E’ or ‘Z’ with 50% probability. The N(1)–O(1) 1.226(3) and N(2)–O(2) 1.213(3) Å bond lengths are in the standard range.15
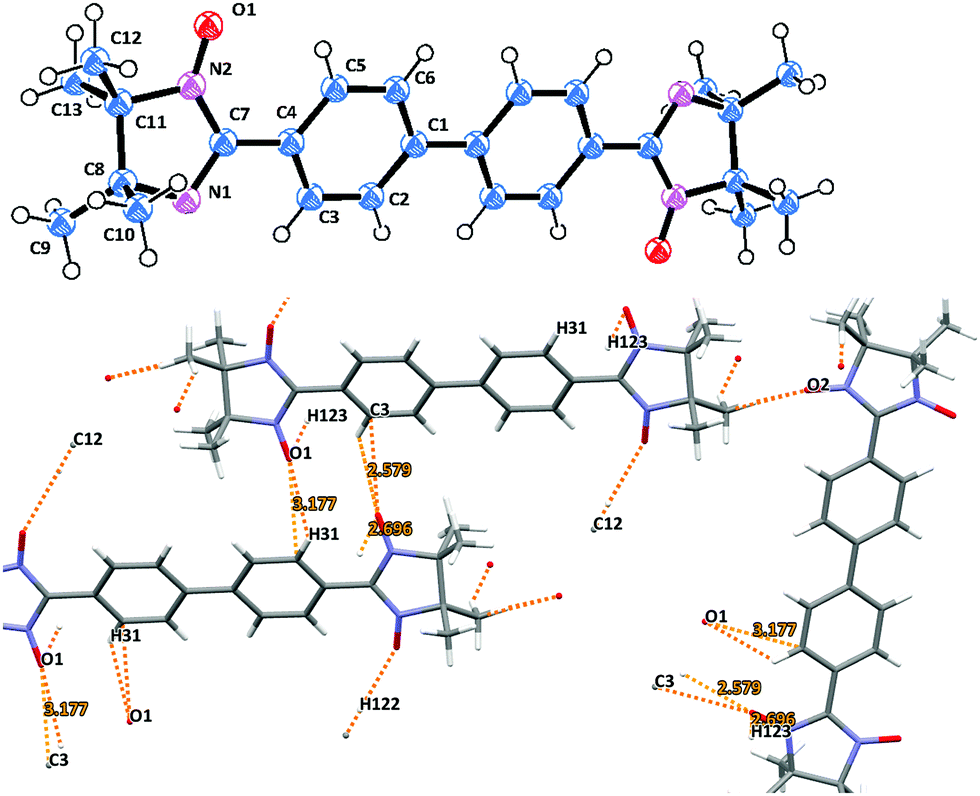 | ||
| Fig. 5 Molecular structure (top) and crystal packing (bottom) of biradical 3d. | ||
Naturally, for the isomorphic imino nitroxide derivatives 3d and 4d the main structural characteristics are very similar.14,27 Thus, in 4d the O–N–C–N–O moiety is planar, with the N(3)–O(2) bond being slightly shorter than N(2)–O(1) 1.167(4) and 1.215(3) Å, respectively (Fig. S4, ESI†). Furthermore, in the asymmetric unit of 4d there are two crystallographically independent forms with the ratio of ∼2:3 belonging to different biradical chains (Fig. S4, ESI†).
An important feature already mentioned for the other biradicals of the current series is the abundance of close intermolecular contacts between the N–O entities and the aromatic moieties in the asymmetric unit. In particular, the hydrogen bonding O1⋯H31′ 2.580, O1⋯H123′ 2.696, and O1⋯C3′ 3.177 Å between the neighboring oxygen atoms and phenyl rings in 3d defines the formation of ribbon-like structures which are further organized in a zig-zag pattern. Likewise, multiple oxygen contacts (i.e. O1⋯H91′ 2.302, O2⋯H51′ 2.473, O2⋯H93′ 2.600, O1⋯C9′ 3.199 and O2⋯C5′ 3.085 Å) accompany the molecular ordering in the case of imino derivative 4d (Fig. S4, ESI†). Notably, the neighboring chains in 4d are connected with close O2⋯O2′ 2.801 Å contacts.
Imino nitroxide biradical 5d crystallizes in a monoclinic space group with P21/n symmetry. The structure of compound 5d is shown in Fig. 6. In contrast to the previously described biradicals compound 5d has surprisingly small torsion angles between the radical unit and the adjacent phenyl ring of only about 6°.30 The overall tolane backbone is fairly planar. The N(2)–O(1) 1.271(1) Å bond distances are similar for this type of compound.
 | ||
| Fig. 6 Molecular structure (top) and crystal packing (bottom) of biradical 5d. | ||
The oxo atoms in 5d act as bridging ligands (O1⋯H141′ 2.622, O1⋯H131′ 2.654 Å) connecting the neighboring biradical molecules into 1D zig-zag shaped chain structures in the crystal cell. These radical ribbons are then piled up along the b crystallographic axis (Fig. S5, ESI†), using C7⋯C10′ 3.381 Å close contacts to stabilize the packing (Fig. 6 and Fig. S7, ESI†).
Magnetic characterization
To gain a more complete picture of the influence of a given π-spacer on the electronic properties, and especially on the intra- and inter-dimer exchange couplings, DFT calculations were performed. To get a first idea on the sizeable changes of the exchange interaction in the studied biradical models, geometry optimization with the B3LYP hybrid function and the 6-31G* basis set was carried out.31 Then the broken symmetry approach (BS) for the singlet and triplet states with the BLYP functional (to avoid Hartree–Fock contamination) and the same basis set were applied.12 Therefore, the direct exchange interaction for a weakly coupled spin dimer can be described as J/kB = E(BS) − E(T), since the spin expectation values 〈S2(BS)〉 and 〈S2(T)〉 for the broken symmetry configuration are close to 1 and for the triplet configuration they are close to 2 such that their difference is close to unity.32 Notably, in the case of antiferromagnetic interactions J takes negative values. The calculations were then redone in accordance with the X-ray geometries, as listed in Table 2. describes the inter-dimer coupling, where z is the number of the nearest neighbors. Tmax corresponds to the maximum in the χmol(T) plot
describes the inter-dimer coupling, where z is the number of the nearest neighbors. Tmax corresponds to the maximum in the χmol(T) plot
| Biradical | T max [K] |
J
calcintraa [K] |
J
calcintrab [K] |
J expintra [K] |
|
|---|---|---|---|---|---|
| a Optimized geometries were obtained from DFT calculations (B3LYP, 6-31G*), then single point calculations for the energy of the broken symmetry and the triplet state were applied (UBLYP, 6-31G* for the evaluation of J).31 b The exchange interactions were evaluated for X-ray crystal structures applying the broken symmetry approach with the BLYP functional and the 6-31G* basis set. c Note that dominant magnetic exchange in 2c is between two adjacent NN units belonging to different biradicals (for details see the text and Fig. 3). | |||||
| 1c | 6.8 | −19.6 | −9.8 | −5.4 ± 0.2 | −1.4 ± 0.4 |
| 2c | 53 | −2.4 | −3.1 | −12.0 ± 3.0c | −42.4 ± 1.7c |
| 1d | — | −2.4 | −1.1 | <−1.5 | — |
| 3d | 4.2 | −4.7 | −5.0 | −3.3 ± 0.1 | −2.0 ± 0.5 |
| 4d | 3.2 | −3.2 | −3.6 | −2.5 ± 0.1 | −5.8 ± 1.4 |
| 5d | 2.8 | −2.6 | −1.6 | −2.2 ± 0.1 | −0.5 ± 0.1 |

Applying a Quantum Design SQUID magnetometer the temperature dependence of the molar magnetic susceptibilities χmol(T) of microcrystalline samples 1c, 2c, 1d, 3d, 4d, and 5d was determined in the range of 2 K ≤ T ≤ 270 K in a magnetic field B = 1 T. The obtained data were corrected for the temperature-independent diamagnetic core contribution of the constituents.33 The magnetic contribution of the sample holder was determined in an independent experiment without a sample. The results are graphically displayed in the form of χmolT vs. T in the main panel, and χmolvs. T in the insets of Fig. 7 and 8 and in the ESI† (Fig. S8). AC-susceptibility measurements (χac) were performed only on 5d as a function of the magnetic field at T = 0.028 K using an ultra-high resolution AC-susceptometer adapted to a 3He–4He top-loading dilution refrigerator. The compensated-coil susceptometer was optimized for measuring small single crystals in the mg range.
 | ||
| Fig. 7 χ mol T as a function of temperature (solid orange circles) of 1c. The black broken line indicates the theoretical value of 0.75 cm3 K mol−1, expected for the two uncoupled spin S = 1/2 entities. Inset: Molar susceptibility χmol as a function of temperature (solid green circles) in a semilog representation, and a theoretical fit with a mean-field correction (red solid line).33 | ||
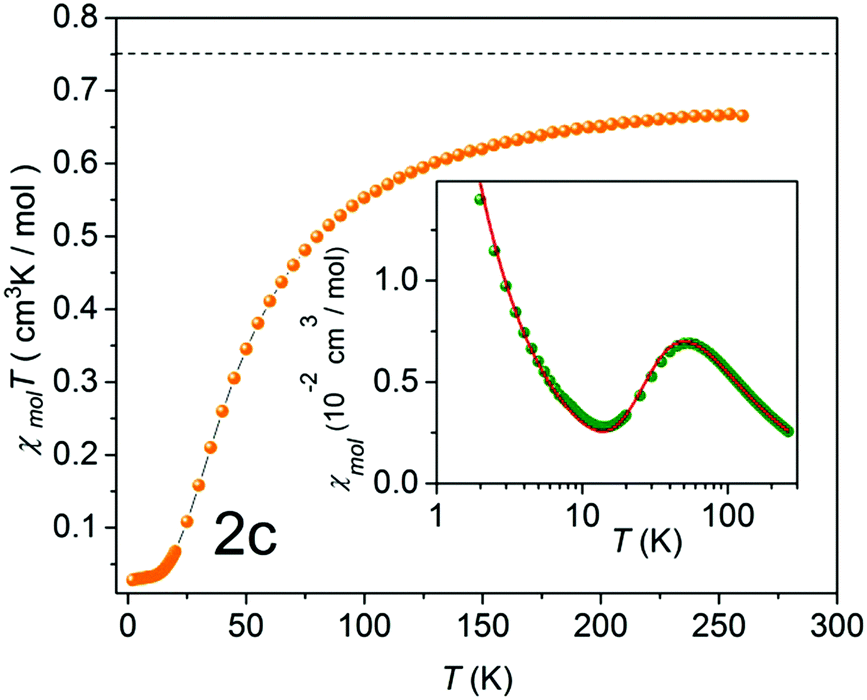 | ||
| Fig. 8 χ mol T as a function of temperature (solid orange circles) of 2c. The black broken line indicates the theoretical value of 0.75 cm3 K mol−1, expected for the two uncoupled spin S = 1/2 entities. Inset: Molar susceptibility χmol as a function of temperature (solid green circles), and a theoretical fit with a mean-field correction (red solid line),33 including a Curie term corresponding to S = 1/2 entities. | ||
Generally, all the biradicals under investigation (1c, 2c, 1d, 3d, 4d, 5d) featured a similar magnetic behavior in the studied temperature range. The observed χmolT values at 300 K of the nitroxides are around 0.7 cm3 K mol−1. Importantly, experimental data are rather close to the theoretical value of 0.75 cm3 K mol−1, which is expected for the two uncoupled spin S = 1/2 units (indicated by the broken line in Fig. 7). This indicates the high quality of the studied single crystals.
As a typical example, χmolT (T) of NN-biradical 1c is shown in Fig. 7. Upon decreasing the temperature χmolT features an approximately linear moderate decrease down to ∼120 K followed by a more pronounced drop below ∼50 K. This overall behavior reflects the dominant antiferromagnetic intra-dimer coupling Jintra. The inset of Fig. 7 shows the molar magnetic susceptibility as a function of temperature. The Bleany–Bowers equation34 for a model of an isolated dimer χiso with a mean-field correction was applied to fit the experimental data (solid line in the inset of Fig. 7). Using this fitting the sizes of the intra-dimer Jintra and inter-dimer 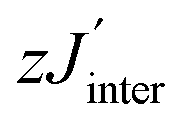 (where z is the number of the nearest neighbors) coupling constants were extracted:
(where z is the number of the nearest neighbors) coupling constants were extracted:
| χmol = χiso /[1 − (zJ′·χiso/Ng2μB2)] | (1) |
Thus, for 1c an experimental intra-dimer coupling constant Jintra/kB = −5.4 ± 0.2 K was obtained. This value is in tangible agreement with the coupling constant acquired from the broken-symmetry approach (Table 2). It should be mentioned that the experimental and theoretical Jintra values of 1c are very similar to the ones reported for tolane NN (Jintra/kB = −4.8 K).11 In light of this it is reasonable to assume that a mere substitution of N for C in position X (as indicated in Fig. 1), if it does not severely affect the geometry of a given crystal structure, has no practical influence on the magnetic interactions.
An unexpected result was acquired for NN derivative 2c where N was substituted with C in the Y position (Fig. 1). For this system the DFT calculations predict Jintra values of a few Kelvin, i.e. similar to those found for the other materials under investigation. The magnetic properties of 2c determined experimentally are shown in Fig. 8. The χmolT data decreased gradually and leveled off around 20 K at a small value of approximately 0.02 cm3 K mol−1. In the inset of Fig. 8 a broad maximum in χmol at 53 K is clearly visible. From a theoretical fit33 corrected with a Curie term for uncoupled S = 1/2 entities (solid red line in the inset of Fig. 8), an inter-dimer coupling constant of Jinter/kB = −42.4 K was obtained. This is significantly larger than Jintra of 1c and tolane NN in ref. 11.
This observation is conceivable in terms of the crystal structure peculiarities. As described above, the radical units in nitronyl nitroxide 2c are far more twisted (∼53°) in comparison to other biradical models described here. The further analysis of the X-ray data revealed exceptionally short contacts of ∼3.51 Å between two neighboring NO fragments in 3,3′-diazatolane nitronyl nitroxide 2c (Fig. S7, ESI†). Apparently, these short contacts are responsible for the unpredicted strong antiferromagnetic exchange interactions found in the solid. Taking only these intermolecular interactions into account and replacing the second radical unit by hydrogen, the value Jinter/kB = −45.6 K was calculated, which was very close to the experimentally obtained value of −42.4 ± 1.7 K (Table 2). Such short inter-dimer distances between spin centers could not be found in the other biradicals.
In accordance with the theoretical predictions and crystal structure analysis IN biradicals 1d, 3d, 4d and 5d featured weak antiferromagnetic intramolecular coupling. Their intra-dimer exchange constants are listed in Table 2 (see also Fig. S8, ESI†). Interestingly, in the case of IN-biradicals 3d and 4d the N-heteroatomic substitution has barely affected their magnetic properties in accordance to the previous statement. From the experimental data recorded for imino biradical 1d no clear maximum could be resolved, indicating an extremely weak intra-dimer coupling constant <−1.5 K.
Remarkably, a closer examination of the low-temperature (T < Jintra/kB) susceptibility data for IN-biradical 5d revealed a significant deviation from the isolated-dimer model. Typically, magnetization of the isolated-dimer systems changes in a step-wise manner at the saturation field Bs and temperatures T < Jintra/kB. Such field-temperature induced transitions correspond to the alteration in the spin-state population of the dimers, namely, the occupation of the low-lying triplet state. This phenomenon is reflected in the form of a single peak in susceptibility χ, the field derivative of magnetization. Fig. 9 shows the AC-susceptibility of IN biradical 5d as a function of the magnetic field at 0.028 K (right scale) together with the magnetization of the material at the same temperature (left scale).
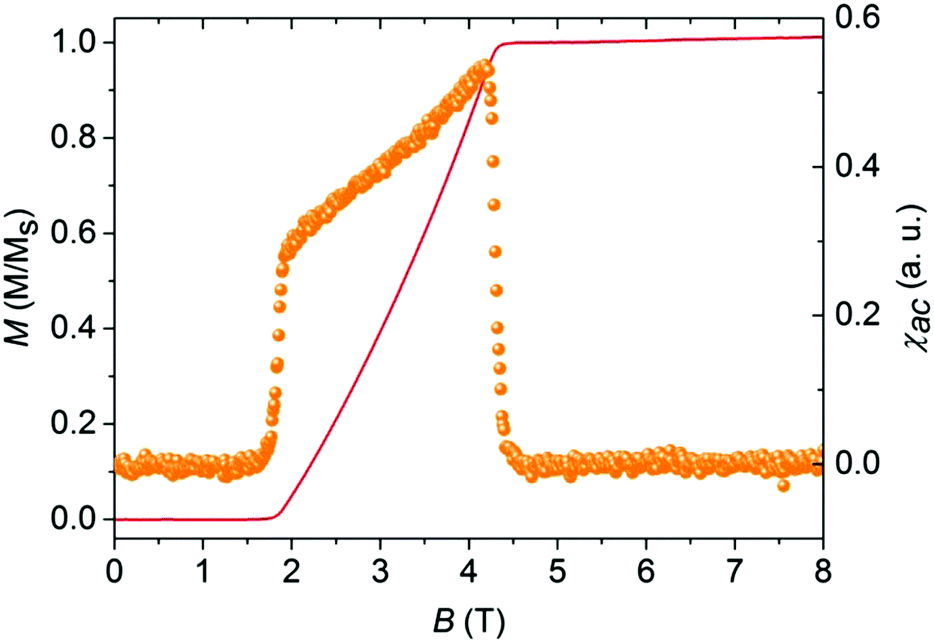 | ||
| Fig. 9 Magnetization M(B) of 5d (solid red line, left scale) at 28 mK normalized to the saturation magnetization together with the magnetic AC-susceptibility χac(B) (orange full circles, right scale). | ||
The AC susceptibility was measured only for IN biradical 5d since only for this sample a large single crystal was available. Polycrystalline samples have been shown to be critical for their intercrystalline contacts and depend on their powder to crystallinity content and several mg are still needed. The data highlight a well-pronounced double-peak feature in χac, an evidence of a field-induced ordered state between Bc1 = 1.8 T and Bc2 = 4.3 T at 0.028 K. Such field-induced phases are expected for a coupled-dimer system where the lower edge of a band of magnetic excitations crosses the ground state at Bc1. Here, Bc2 corresponds to the saturation field Bs where the full magnetization of the system is obtained. The magnetization exhibits no hysteretic behavior. This feature is attributed to small magnetic intermolecular interactions between the neighboring biradical molecules mediated via hydrogen bonds. The strength of these interactions depends on the distance and the relative orientation of the radicals.35,36 The anomalies at the critical fields Bc1 and Bc2 are much sharper than the one found in a quasi-2D system.11 More likely, the inter-dimer coupling in 5d has a 3D character, and therefore, this field-induced ordered phase can be described in terms of a BEC of triplons.
Experimental section
Materials and methods
All chemicals and reagents were used as received from commercial sources (Acros Organics, Aldrich, Fluka, Lancaster, Merck and Strem) without additional purification. Solvents for synthesis were used as received, unless otherwise mentioned. ESR spectra were recorded in dilute, oxygen-free solutions in toluene, concentrations ∼10−4 mol L−1, using a Bruker ESP300 E X-band spectrometer equipped with an NMR gaussmeter (Bruker ER035), a frequency counter (Bruker ER041XK) and a variable temperature control continuous flow N2 cryostat (Bruker B-VT 2000). The g-factor corrections were obtained using DPPH (g = 2.0037) as the standard. UV-Vis spectra were recorded in toluene solutions with a PerkinElmer spectrometer (UV/Vis/NIR Lambda 900) using a 1 cm optical path quartz cell at room temperature, unless otherwise specified. 1H and 13C NMR spectra were recorded on a Bruker DPX 250, Bruker DMX 300 spectrometer. Solid powders were pressed and IR spectra of the samples were recorded as they were (Nicolet 730 FT-IR spectrometer). Mass spectra (FDMS) were obtained on a VG Instruments ZAB-2 mass spectrometer. Elemental analyses were performed at the University of Mainz, Faculty of Chemistry and Pharmacy on a Foss Heraeus Varieo EL. The melting points were measured on a Büchi B-545 apparatus (uncorrected) by using open-ended capillaries. Crystallographic data for the reported structures of the biradicals have been deposited at the Cambridge Crystallographic Data Centre.‡ 2,3-Bis(hydroxyamino)-2,3-dimethylbutane (BHA) synthesis was described elsewhere.11,15:1) solvents was added through the rubber septum. The solution was carefully deaerated by purging with argon for 20–25 min, and a catalytic mixture of Pd(PPh3)2Cl2 (1.1 mmol, 0.05 eq.), PPh3 (2.2 mmol, 0.1 eq.), and CuI (1.1 mmol, 0.05 eq.) was added at once. The resulting mixture was slightly heated (to 45 °C) and ethynyltrimethylsilane (32.6 mmol, 1.5 eq.) was added through the septum. After that the heating was increased to 80 °C. A white precipitate began to form after ∼15 min of heating. After the reported time (4 to 16 h), the mixture was cooled to ambient temperature, and the crystalline white solid of triethylamine hydrobromide was isolated by filtration. The orange-brown filtrate was concentrated, mixed with NH4Cl saturated aqueous solution (50 mL), and extracted with dichloromethane or diethyl ether (3 × 40 mL). The organic fractions were combined, dried over magnesium sulfate, and concentrated with silica gel in vacuo. The residue was purified using column chromatography.
:1) as the solvent media was preferred. Here, after 5 minutes of stirring at room temperature formation of triethylamine hydrobromide salt was observed. The light yellow reaction mixture was stirred at room temperature for 17–19 h. The work-up was done in accordance with protocol A1.
:2) afforded precursor 9 in 89% yield. 1H-NMR (CDCl3, 250 MHz, 298 K, 16 scan), δ ppm: 0.28 (s, 9H, –CH3), 4.09 (m, 4H, –CH2), 5.85 (s, 1H, H-7), 7.47 (d, 1H, 3J = 8 Hz, H-3), 7.74 (dd, 1H, 3J = 8 Hz, H-4), 8.65 (s, 1H, H-6). 13C-NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 0 (–CH3), 67.1 (–CH2), 94.8 (C-9), 102.2 (C-7), 104.1 (C-8), 126.7 (C-3), 132.6 (C-5), 134.9 (C-4), 143.4 (C-2), 148.6 (C-6). MS-FD (70 eV, CH2Cl2) m/z: 247.1 (M+), MW calculated C13H17NO2Si (MW+) 247.37.
Next, to the solution of the corresponding dioxolane precursor (0.38 g, 1.35 mmol) in an acetone–water mixture (7:1, 40 mL) para-toluene-sulfonic acid (52 mg, 0.027 mmol, 2 mol%) was added. The progress of the reaction was monitored by TLC (SiO2, hexane/ethyl acetate, 3:1). After 84 h of stirring at room temperature the reaction was complete. Acetone was evaporated, and the residue was diluted with dichloromethane (30 mL). The organic layer was separated, and the water phase was extracted with dichloromethane (3 × 30 mL). The combined organic extracts were dried over MgSO4. The solvent was removed in vacuo giving 0.26 g (81%) of dialdehyde 1a as a yellow solid. 1H-NMR ((CD3)2SO, 250 MHz, 298 K, 16 scan), δ ppm: 8.01 (d, 2H, 3J = 8 Hz, H-2), 8.37 (dd, 2H, 3J = 8 Hz, H-3), 9.19 (s, 2H, H-6), 10.20 (s, 2H, –CHO). 13C-NMR ((CD3)2SO, 63 MHz, 298 K, 256 scan), δ ppm: 90.1 (C-8), 128.8 (C-3), 131.1 (C-5), 137.3 (C-4), 145.7 (C-2), 152.2 (C-6), 192.2 (–CHO). MS-FD (70 eV, CH2Cl2) m/z: 236.3 (M+), MW calculated C14H8N2O2 (MW+) 236.23.
:3). 5-Bromo-2-pyridinecarbaldehyde 11 was synthesized in 49% yield. 1H-NMR (CDCl3, 250 MHz, 298 K, 16 scan), δ (ppm): 7.8 (d, 1H, H-6), 7.9 (dd, 1H, 3J = 8 Hz, H-4), 8.78 (d, 1H, H-3), 9.97 (s, 1H, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 13C-NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 122.7 (C-5), 126.2 (C-3), 139.9 (C-4), 151.1 (C-2), 151.5 (C-6), 192.3 (–CHO). MS-FD (70 eV, CH2Cl2) m/z: 187.3 (M+), MW calculated C6H4BrNO (MW+) 186.01.
:100). 1H-NMR (CDCl3, 250 MHz, 298 K, 16 scan), δ ppm: 0.26 (s, 9H, –CH3), 7.86 (d, 2H, 3J = 2 Hz, H-3, H-4), 8.77 (s, 1H, H-4), 8.65 (s, 1H, H-6), 10.02 (s, 1H, –CO). 13C-NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 0 (–CH3), 98.8 (C-8), 101.9 (C-7), 121.1 (C-3), 122.4 (C-5), 140.4 (C-4), 150.8 (C-2), 151.7 (C-6), 192.7 (CO). MS-FD (70 eV, CH2Cl2) m/z: 202.7 (M+), MW calculated C11H13NOSi (MW+) 203.31.
:1 v/v) was stirred at rt for 1.5 h. Brine was added to the mixture, the organic layer was separated, and the water phase was extracted with Et2O (4 × 30 mL). Combined organic extracts were dried over Na2SO4, and the solvents were evaporated under reduced pressure. The light-brown solid was washed with a hot hexane/acetone mixture (∼20:1) to give a pale yellow precipitate in 51% yield (0.47 g). 1H-NMR (CDCl3, 250 MHz, 298 K, 16 scan), δ ppm: 3.36 (s, 1H, H-8), 4 7.87 (d, 2H, 3J = 8 Hz, H-3, H-4), 8.79 (t, 1H, 3J = 8 Hz, H-6), 10.0 (s, 1H, –CO). 13C-NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 78.7 (C-7), 83.6 (C-8), 119.9 (C-3), 122.7 (C-5), 139.2 (C-4), 150.4 (C-2), 152.0 (C-6), 191.4 (–CHO). MS-FD (70 eV, CH2Cl2) m/z: 130.3 (M+), MW calculated C8H5NO (MW+) 131.13.
:4:1), and recrystallized from the hexane/acetone/THF mixture (2:1:1) to give 1,2-(bis(2-formyl-5-yl)pyridinyl)ethyne 2a as a yellow solid in 62% yield. 1H-NMR (CDCl3, 250 MHz, 298 K, 64 scan), δ ppm: 8.04 (d, 2H, 3J = 8 Hz, H-4), 8.33 (d, 2H, 3J = 8 Hz, H-3), 9.09 (s, 2H, H-6), 10.03 (s, 2H, –CHO). 13C-NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 59.3 (C-7), 121.6 (C-4), 125.1 (C-5), 138.8 (C-3), 150.2 (C-2), 150.5 (C-6), 191.2 (–CHO). MS-FD (70 eV, CH2Cl2) m/z: 236.0 (M+), MW calculated C14H8N2O2 (MW+) 236.23.
:2), yielding 0.78 g (58%) of light-yellow dialdehyde 3a. 1H NMR (CDCl3, 250.13 MHz), δ ppm: 7.71–7.75 (d, 4H, 3J = 8.0 Hz, H-2), 7.92–7.95 (d, 4H, 3J = 8.0 Hz, H-3), 10.02 (s, 2H, –CHO). 13C-NMR (CDCl3, 63 MHz, 298 K, 206 scan), δ ppm: 128.1 (C-1), 130.4 (C-2), 136 (C-3), 145.6 (C-4), 191.7 (−CHO). MS-FD (70 eV, CH2Cl2) m/z: 210.1 (M+), MW calculated C14H10O2 (MW+) 210.1.
:1) for 60 h. The mixture was extracted with ethyl acetate/toluene (1:1) (4 × 50 mL) to afford effective phase separation. The organic extracts were collected and dried over MgSO4. The filtrate was concentrated in vacuo to a volume of ca. 2 mL, placed on a column with silica, and eluted with an ethyl acetate/hexane gradient solvent mixture (1:4, 1:3). The colorless fractions were collected and evaporated leading to product 4a in 57% overall yield. 1H NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 7.60–7.64 (d, 2H, 3J = 8.02 Hz, H-4), 7.94–7.98 (dd, 2H, 3J = 8.02 Hz, H-6), 8.76 (d, 2H, 3J = 1.6 Hz, H-3), 10.02 (s, 2H, –CHO). 13C-NMR (THF-d8, 63 MHz, 298 K, 338 scan), δ ppm: 121.3 (C-3), 127.6 (C-5), 129.4 (C-4), 131.9 (C-6), 136.4 (C-2), 189.7 (–CHO). MS-FD (70 eV, CH2Cl2) m/z: 211.6 (M+), MW calculated C14H10O2 (MW+) 212.1.
O). 13C-NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 122.7 (C-5), 126.2 (C-3), 139.9 (C-4), 151.1 (C-2), 151.5 (C-6), 192.3 (–CHO). MS-FD (70 eV, CH2Cl2) m/z: 187.3 (M+), MW calculated C6H4BrNO (MW+) 186.01.
:100). 1H-NMR (CDCl3, 250 MHz, 298 K, 16 scan), δ ppm: 0.26 (s, 9H, –CH3), 7.86 (d, 2H, 3J = 2 Hz, H-3, H-4), 8.77 (s, 1H, H-4), 8.65 (s, 1H, H-6), 10.02 (s, 1H, –CO). 13C-NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 0 (–CH3), 98.8 (C-8), 101.9 (C-7), 121.1 (C-3), 122.4 (C-5), 140.4 (C-4), 150.8 (C-2), 151.7 (C-6), 192.7 (CO). MS-FD (70 eV, CH2Cl2) m/z: 202.7 (M+), MW calculated C11H13NOSi (MW+) 203.31.
:1 v/v) was stirred at rt for 1.5 h. Brine was added to the mixture, the organic layer was separated, and the water phase was extracted with Et2O (4 × 30 mL). Combined organic extracts were dried over Na2SO4, and the solvents were evaporated under reduced pressure. The light-brown solid was washed with a hot hexane/acetone mixture (∼20:1) to give a pale yellow precipitate in 51% yield (0.47 g). 1H-NMR (CDCl3, 250 MHz, 298 K, 16 scan), δ ppm: 3.36 (s, 1H, H-8), 4 7.87 (d, 2H, 3J = 8 Hz, H-3, H-4), 8.79 (t, 1H, 3J = 8 Hz, H-6), 10.0 (s, 1H, –CO). 13C-NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 78.7 (C-7), 83.6 (C-8), 119.9 (C-3), 122.7 (C-5), 139.2 (C-4), 150.4 (C-2), 152.0 (C-6), 191.4 (–CHO). MS-FD (70 eV, CH2Cl2) m/z: 130.3 (M+), MW calculated C8H5NO (MW+) 131.13.
:4:1), and recrystallized from the hexane/acetone/THF mixture (2:1:1) to give 1,2-(bis(2-formyl-5-yl)pyridinyl)ethyne 2a as a yellow solid in 62% yield. 1H-NMR (CDCl3, 250 MHz, 298 K, 64 scan), δ ppm: 8.04 (d, 2H, 3J = 8 Hz, H-4), 8.33 (d, 2H, 3J = 8 Hz, H-3), 9.09 (s, 2H, H-6), 10.03 (s, 2H, –CHO). 13C-NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 59.3 (C-7), 121.6 (C-4), 125.1 (C-5), 138.8 (C-3), 150.2 (C-2), 150.5 (C-6), 191.2 (–CHO). MS-FD (70 eV, CH2Cl2) m/z: 236.0 (M+), MW calculated C14H8N2O2 (MW+) 236.23.
:2), yielding 0.78 g (58%) of light-yellow dialdehyde 3a. 1H NMR (CDCl3, 250.13 MHz), δ ppm: 7.71–7.75 (d, 4H, 3J = 8.0 Hz, H-2), 7.92–7.95 (d, 4H, 3J = 8.0 Hz, H-3), 10.02 (s, 2H, –CHO). 13C-NMR (CDCl3, 63 MHz, 298 K, 206 scan), δ ppm: 128.1 (C-1), 130.4 (C-2), 136 (C-3), 145.6 (C-4), 191.7 (−CHO). MS-FD (70 eV, CH2Cl2) m/z: 210.1 (M+), MW calculated C14H10O2 (MW+) 210.1.
:1) for 60 h. The mixture was extracted with ethyl acetate/toluene (1:1) (4 × 50 mL) to afford effective phase separation. The organic extracts were collected and dried over MgSO4. The filtrate was concentrated in vacuo to a volume of ca. 2 mL, placed on a column with silica, and eluted with an ethyl acetate/hexane gradient solvent mixture (1:4, 1:3). The colorless fractions were collected and evaporated leading to product 4a in 57% overall yield. 1H NMR (CDCl3, 63 MHz, 298 K, 256 scan), δ ppm: 7.60–7.64 (d, 2H, 3J = 8.02 Hz, H-4), 7.94–7.98 (dd, 2H, 3J = 8.02 Hz, H-6), 8.76 (d, 2H, 3J = 1.6 Hz, H-3), 10.02 (s, 2H, –CHO). 13C-NMR (THF-d8, 63 MHz, 298 K, 338 scan), δ ppm: 121.3 (C-3), 127.6 (C-5), 129.4 (C-4), 131.9 (C-6), 136.4 (C-2), 189.7 (–CHO). MS-FD (70 eV, CH2Cl2) m/z: 211.6 (M+), MW calculated C14H10O2 (MW+) 212.1.
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –CH3), 88.1 (C-7), 127.2 (C-3), 136.8 (C-4), 137.9 (C-5), 140.9 (C-2), 150.7 (C-6). MS-FD (70 eV, CH2Cl2) m/z: 496.4 (M+), MW calculated C26H36N6O4 (MW+) 496.60.
–CH3), 88.1 (C-7), 127.2 (C-3), 136.8 (C-4), 137.9 (C-5), 140.9 (C-2), 150.7 (C-6). MS-FD (70 eV, CH2Cl2) m/z: 496.4 (M+), MW calculated C26H36N6O4 (MW+) 496.60.
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –N), 7.42–7.76 (m, 8H, ArH), 7.83 (s, 4H, –OH). 13C NMR ((CD3)2SO, 63 MHz, 298 K, 256 scan), δ ppm: 17.6, 24.8 (CH3), 66.5 (–CH3), 90.4 (N–C–N), 126.3, 129.5 (CHAr), 139.8, 141.4 (CAr). MS-FD (70 eV, CH2Cl2) m/z: 470.1 (M+), MW calculated C26H30N6O4 (MW+) 470.60.
–N), 7.68 (s, 4H, –OH), 7.73–7.77 (d, 2H, ArH), 8.12–8.15 (d, 2H, ArH), 8.48 (s, 2H, ArH). 13C NMR ((CD3)2SO, 63 MHz, 298 K, 256 scan), δ ppm: 17.3, 20.8 (CH3), 66.3 (–CH3), 87.9 (N–C–N), 125.8, 127.9, 131.9 (CHAr), 137.4, 146.5 (CAr). MS-FD (70 eV, CH2Cl2) m/z: 471.8 (M+), MW calculated C26H30N6O4 (MW+) 472.28.
–N), 7.42–7.76 (m, 8H, ArH), 7.83 (s, 4H, –OH). 13C NMR ((CD3)2SO, 63 MHz, 298 K, 256 scan), δ ppm: 17.6, 24.8 (CH3), 66.5 (–CH3), 90.4 (N–C–N), 126.3, 129.5 (CHAr), 139.8, 141.4 (CAr). MS-FD (70 eV, CH2Cl2) m/z: 470.1 (M+), MW calculated C26H30N6O4 (MW+) 470.60.
–N), 7.68 (s, 4H, –OH), 7.73–7.77 (d, 2H, ArH), 8.12–8.15 (d, 2H, ArH), 8.48 (s, 2H, ArH). 13C NMR ((CD3)2SO, 63 MHz, 298 K, 256 scan), δ ppm: 17.3, 20.8 (CH3), 66.3 (–CH3), 87.9 (N–C–N), 125.8, 127.9, 131.9 (CHAr), 137.4, 146.5 (CAr). MS-FD (70 eV, CH2Cl2) m/z: 471.8 (M+), MW calculated C26H30N6O4 (MW+) 472.28.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–), 90.1 (Cimid-2), 121.6 (Ar-C), 128.2, 131.0 (ArC–H), 143.5 (Ar-C). MS-FD (70 eV, (CD3)2SO) m/z: 494.293 (M+), MW calculated C28H38N4O4 (MW+) 494.291.
:2) mixture (∼50 mL) was cooled down to 0–5 °C using an ice bath. To that mixture a 5% aqueous solution of NaIO4 (0.8 eq.) was added dropwise. The progress of the reaction was monitored by TLC (SiO2, hexane/ethyl acetate or dichloromethane with 5% methanol). After the reaction was complete (0.5–3 h), a dark-blue organic layer was separated. The aqueous layer was extracted with dichloromethane (3 × 25 mL). The combined organic extracts were additionally washed with water (2 × 30 mL), brine (1 × 50 mL), and dried over MgSO4 (in some cases – Na2SO4). The residue after filtration was reduced to a volume of 2 mL, and purified on a chromatographic column (SiO2, hexane/ethyl acetate or CH2Cl2/MeOH).
:3) afforded 54 mg of the bluish crystals of 1c (13%). M.p.: 219–220 °C. UV-Vis (CHCl3) λ/nm (ε, mol−1 cm−1): 612 (808), 344 (55554); (toluene) λ/nm (ε, mol−1 cm−1) 623 (483), 345 (50402). FT-IR (solid; ν/cm−1): 3108, 3072, 2986, 2939 (s, Py–C–H stretching), 2144 (w, CC), 1734 (m, CO), 1583 (s, PyCC stretching), 1549 (s, CNimid), 1347 (s, N–O). Elemental analysis: C 62.7; H 5.96; N 17.02; calculated for C26H30N6O4: C 63.66; H 6.16; N 17.13.
:1). After evaporation of the solvent blue crystals were obtained in 34% total yield. M.p.: 236–239 °C. UV-Vis (CHCl3) λ/nm (ε, mol−1 cm−1): 591 (503), 341 (38929). FT-IR (powder, ν/cm−1): 3048, 2985, 2933, 2868 (s, Py–C–H stretching), 2141 (w, CC), 1733 (m, CO), 1588 (m, PyCC stretching), 1560 (s, CNimid), 1360 (s, N–O). Elemental analysis: C 63.1; H 6.22; N 16.62; calculated for C26H30N6O4: C 63.66; H 6.16; N 17.13.
207). FT-IR (powder, ν/cm−1): 3067, 2969, 2924 (s, Py–C–H stretching), 2136 (w, CC), 1591 (s, PyCC stretching), 1555 (s, CNimid), 1368 (m, N–O). Elemental analysis: C 67.6; H 6.43; N 18.07; calculated for C26H30N6O2: C 68.10; H 6.59; N 18.30.
:1, 3:1). The yield of orange crystals of 3d was 0.14 g (47%). M.p.: 248.5–250 °C. UV-Vis (CHCl3) λ/nm (ε, mol−1 × cm−1): 461 (1090), 486 (1016). FT-IR (powder, ν/cm−1): 3069, 2973, 2926, 2862 (s, Ph–C–H stretching), 1611 (s, Ph CC stretching), 1567 (w, CN), 1364 (s, N–O). Elemental analysis: C 71.86; H 7.32; N 12.66; calculated for C26H32N4O2: C 72.19; H 7.46; N 12.95.
C stretching), 1562 (s, CNimid), 1366 (s, N–O). Elemental analysis: C 65.36; H 7.02; N 19.11; calculated for C24H30N6O2: C 66.34; H 6.96; N 19.34.
:2) eluent mixture afforded red crystals of 5d in 58% total yield. M.p.: compound began decomposing above 195 °C. UV-Vis (toluene) λ/nm (ε, mol−1 cm−1): 467 (944), 326 (35207). FT-IR (powder, ν/cm−1): 3067, 2969, 2924 (s, Py–C–H stretching), 2136 (w, CC), 1591 (s, PyCC stretching), 1555 (s, CNimid), 1368 (m, N–O). Elemental analysis: C 73.15; H 6.76; N 12.11; calculated for C28H32N4O2: C 73.66; H 7.06; N 12.27.
C–), 90.1 (Cimid-2), 121.6 (Ar-C), 128.2, 131.0 (ArC–H), 143.5 (Ar-C). MS-FD (70 eV, (CD3)2SO) m/z: 494.293 (M+), MW calculated C28H38N4O4 (MW+) 494.291.
:2) mixture (∼50 mL) was cooled down to 0–5 °C using an ice bath. To that mixture a 5% aqueous solution of NaIO4 (0.8 eq.) was added dropwise. The progress of the reaction was monitored by TLC (SiO2, hexane/ethyl acetate or dichloromethane with 5% methanol). After the reaction was complete (0.5–3 h), a dark-blue organic layer was separated. The aqueous layer was extracted with dichloromethane (3 × 25 mL). The combined organic extracts were additionally washed with water (2 × 30 mL), brine (1 × 50 mL), and dried over MgSO4 (in some cases – Na2SO4). The residue after filtration was reduced to a volume of 2 mL, and purified on a chromatographic column (SiO2, hexane/ethyl acetate or CH2Cl2/MeOH).
:3) afforded 54 mg of the bluish crystals of 1c (13%). M.p.: 219–220 °C. UV-Vis (CHCl3) λ/nm (ε, mol−1 cm−1): 612 (808), 344 (55554); (toluene) λ/nm (ε, mol−1 cm−1) 623 (483), 345 (50402). FT-IR (solid; ν/cm−1): 3108, 3072, 2986, 2939 (s, Py–C–H stretching), 2144 (w, CC), 1734 (m, CO), 1583 (s, PyCC stretching), 1549 (s, CNimid), 1347 (s, N–O). Elemental analysis: C 62.7; H 5.96; N 17.02; calculated for C26H30N6O4: C 63.66; H 6.16; N 17.13.
:1). After evaporation of the solvent blue crystals were obtained in 34% total yield. M.p.: 236–239 °C. UV-Vis (CHCl3) λ/nm (ε, mol−1 cm−1): 591 (503), 341 (38929). FT-IR (powder, ν/cm−1): 3048, 2985, 2933, 2868 (s, Py–C–H stretching), 2141 (w, CC), 1733 (m, CO), 1588 (m, PyCC stretching), 1560 (s, CNimid), 1360 (s, N–O). Elemental analysis: C 63.1; H 6.22; N 16.62; calculated for C26H30N6O4: C 63.66; H 6.16; N 17.13.
207). FT-IR (powder, ν/cm−1): 3067, 2969, 2924 (s, Py–C–H stretching), 2136 (w, CC), 1591 (s, PyCC stretching), 1555 (s, CNimid), 1368 (m, N–O). Elemental analysis: C 67.6; H 6.43; N 18.07; calculated for C26H30N6O2: C 68.10; H 6.59; N 18.30.
:1, 3:1). The yield of orange crystals of 3d was 0.14 g (47%). M.p.: 248.5–250 °C. UV-Vis (CHCl3) λ/nm (ε, mol−1 × cm−1): 461 (1090), 486 (1016). FT-IR (powder, ν/cm−1): 3069, 2973, 2926, 2862 (s, Ph–C–H stretching), 1611 (s, Ph CC stretching), 1567 (w, CN), 1364 (s, N–O). Elemental analysis: C 71.86; H 7.32; N 12.66; calculated for C26H32N4O2: C 72.19; H 7.46; N 12.95.
C stretching), 1562 (s, CNimid), 1366 (s, N–O). Elemental analysis: C 65.36; H 7.02; N 19.11; calculated for C24H30N6O2: C 66.34; H 6.96; N 19.34.
:2) eluent mixture afforded red crystals of 5d in 58% total yield. M.p.: compound began decomposing above 195 °C. UV-Vis (toluene) λ/nm (ε, mol−1 cm−1): 467 (944), 326 (35207). FT-IR (powder, ν/cm−1): 3067, 2969, 2924 (s, Py–C–H stretching), 2136 (w, CC), 1591 (s, PyCC stretching), 1555 (s, CNimid), 1368 (m, N–O). Elemental analysis: C 73.15; H 6.76; N 12.11; calculated for C28H32N4O2: C 73.66; H 7.06; N 12.27.
Conclusion
We have demonstrated that the synthesis and study of structurally similar compounds exhibiting different magnetic behaviors provide a reasonable approach for understanding the relationship between the substituents in the aromatic ring and the intramolecular exchange constant Jintra. Unfortunately, the magnetic interactions based on structural peculiarities are difficult to predict as their strength strongly depends on the relative orientation between the interacting magnetic orbitals. Here, the torsion angles θ have crucial impacts on the overall π-conjugation in the nitroxide biradical systems. The latter in turn is responsible for the efficient communication between the nitroxide fragments. The crystal structure analysis shows that the synthesized biradicals are planar with relatively small torsions between the radical fragments and the aromatic bridges (with an exception in the case of derivative 2c). These are the essential prerequisites for obtaining a weak intramolecular coupling. A rapid increase of the torsion hinders the conjugation within the biradical system 2c, decreasing the intramolecular exchange interactions within the molecule. This structural feature causes an enhancement of the intermolecular coupling within 2c. As a result, an unprecedentedly high value of the coupling constant is observed in NN biradical 2c. This work illustrates the difficulties in the design and prediction of a target structure, and the need for an experimental proof.Magnetic measurements, carried out on single-crystalline samples, confirmed that for biradicals 1c, 1d, 3d, 4d and 5d, weak antiferromagnetic intramolecular interactions are predominant. According to the magnetic characterization studied π-bridged nitroxides possess a moderate intra-dimer coupling in the range −2 to −6 K as derived from the fits based on an isolated dimer model. Furthermore, magnetic measurements and their interpretation revealed that NN biradical 2c exhibits surprisingly strong antiferromagnetic interactions, owing to the short (∼3.5 Å) interdimer interaction. IN biradical 5d features a field-induced ordered phase assigned to 3D inter-dimer couplings, which accounts for a description in terms of a BEC of triplons.
In summary, we have found promising candidates in the quest for purely organic molecular magnets and crystalline networks with higher ordering.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank Dr Dieter Schollmeyer (Johannes Gutenberg-University Mainz) and Dr Volker Enkelmann for crystallographic data analysis. The authors are pleased to acknowledge continued support from the Max Planck Society and the DFG-SFB TR49. Open Access funding provided by the Max Planck Society.Notes and references
- S. Sachdev, Nat. Phys., 2008, 4, 173 CrossRef CAS.
- T. Giamarchi, Ch. Rüeggv and O. Tchernyshyov, Nat. Phys., 2008, 4, 198–204 CrossRef CAS.
- C. Rüegg, K. Kiefer, B. Thielemann, D. F. McMorrow, V. Zapf, B. Normand, M. B. Zvonarev, P. Bouillot, C. Kollath, T. Giamarchi, S. Capponi, C. Poilblanc, D. Biner and K. W. Krämer, Phys. Rev. Lett., 2008, 101, 247202 CrossRef PubMed.
- U. Tutsch, B. Wolf, S. Wessel, L. Postulka, Y. Tsui, H. O. Jeschke, I. Opahle, T. Saha-Dasgupta, R. Valenti, A. Brühl, K. Removic-Langer, T. Kretz, H.-W. Lerner, M. Wagner and M. Lang, Nat. Commun., 2014, 5, 5169 CrossRef CAS PubMed.
- J. Veciana, J. Cirujeda, C. Rovira, E. Molins and J. J. Novoa, J. Phys., 1996, 6, 1967 CAS.
- H. Sasaki, L.-R. Lin, T. Yokoyama, M. D. Sevilla, V. N. Reddy and F. J. Giblin, Invest. Ophthalmol. Visual Sci., 1998, 39, 544 CAS.
- W. Bi, J. Cai, Y. Zhang, S. Liu, S. A. Green and L. Bi, Bioorg. Med. Chem. Lett., 2008, 18, 1788 CrossRef CAS PubMed.
- M. C. Krishna and W. DeGraff, J. Med. Chem., 1998, 41, 3477 CrossRef CAS PubMed.
- J. H. Osiecki and E. F. Ullman, J. Am. Chem. Soc., 1968, 90, 1078 CrossRef CAS.
- A. Caneschi, D. Gatteschi, P. Rey and R. Sessoli, Inorg. Chem., 1988, 27, 1756 CrossRef CAS; A. Caneschi, D. Gatteschi, R. Sessoli and P. Rey, Acc. Chem. Res., 1989, 22, 392 CrossRef.
- Y. B. Borozdina, E. A. Mostovich, V. Enkelmann, B. Wolf, P. T. Cong, U. Tutsch, M. Lang and M. Baumgarten, J. Mater. Chem. C, 2014, 2, 6618 RSC.
- K. Yamaguchi, M. Okumura, J. Maki, T. Noro, H. Namimoto, M. Nakano, T. Fueno and K. Nakasuji, Chem. Phys. Lett., 1992, 190, 353 CrossRef CAS.
- G. Seber, R. S. Fretas, J. T. Mague, A. Paduan-Filho, X. Gratens, V. Bindilatti, N. F. Oliveira Jr., N. Yoshioka and P. M. Lahti, J. Am. Chem. Soc., 2012, 134, 3825 CrossRef CAS PubMed.
- G. Zoppellaro, A. Geies and M. Baumgarten, Eur. J. Org. Chem., 2004, 2367 CrossRef CAS.
- E. Mostovich, Y. Borozdina, V. Enkelmann, K. Remović-Langer, B. Wolf, M. Lang and M. Baumgarten, Cryst. Growth Des., 2012, 12, 54 CAS.
- S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Communications, 1980, 627 CAS.
- C. Bolm, M. Ewald, M. Felder and G. Schlinglo, Chem. Ber., 1992, 125, 1169 CrossRef CAS.
- F. C. Alderweireldt, I. Vrijens, E. L. Esmans and E. D. Clercq, Nucleosides Nucleotides, 1989, 8, 891 Search PubMed.
- J. Wicha and M. Masnyl, Heterocycles, 1981, 16, 521 CrossRef CAS.
- F. J. Romero-Salguero and J.-M. Lehn, Tetrahedron Lett., 1999, 40, 859 CrossRef CAS.
- X. Wang, P. Rabbat and P. O'Shea, Tetrahedron Lett., 2000, 41, 4335 CrossRef CAS.
- A. Landa, A. Minkkila and G. Blay, Chem. – Eur. J., 2006, 12, 3472 CrossRef CAS PubMed.
- M. A. Peterson and J. Mitchell, J. Org. Chem., 1997, 62, 8237 CrossRef CAS PubMed.
- T. W. Greene and P. G. Wuts, Protective groups in organic synthesis, 1991 Search PubMed.
- F. Ebmeyer and F. Vögtle, Chem. Ber., 1989, 122, 1725 CrossRef CAS.
- I. Karamé, M. Jahjah and M. Lemaire, Tetrahedron: Asymmetry, 2004, 15, 1569 CrossRef.
- R. Ziessel and G. Ulrich, J. Mater. Chem., 1999, 9, 1435 RSC.
- E. V. Tretyakov, S. Tolstikov and A. Mareev, Eur. J. Org. Chem., 2009, 2548 CrossRef CAS.
- E. V. Tretyakov and V. I. Ovcharenko, Russ. Chem. Rev., 2009, 78, 971 CrossRef CAS.
- E. Önal, Y. Yerli, B. Cosut, G. Pilet, V. Ahsen, D. Luneau and C. Hirel, New J. Chem., 2014, 38, 4440 RSC.
- DFT calculations were carried out using Gaussian 09, B3LYP hybrid functionals for geometry optimization and the BLYP functional for energy calculation of broken symmetry vs. triplet states together with 6-31g(d) basis sets. See M. J. Frisch et al. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013.
- M. E. Ali and S. N. Datta, J. Phys. Chem. A, 2006, 110, 2776 CrossRef CAS PubMed.
- O. Kahn, Molecular Magnetism, VCH, Weinheim-New York, 1993 Search PubMed.
- B. Bleaney and D. K. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CrossRef CAS.
- T. Mitsumori, K. Inoue, N. Koga and H. Iwamura, J. Am. Chem. Soc., 1995, 117, 2467 CrossRef CAS.
- R. Akabane, M. Tanaka, K. Matsuo, N. Koga, K. Matsuda and H. Iwamura, J. Org. Chem., 1997, 62, 8854 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Selected spectroscopic data, additional crystallographic data, structural details and magnetic susceptibility plots of 1d, 3d, 4d and 5d (pdf). CCDC 823716, 816632, 816630, 823717, 858078 and 810139. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7tc03357e |
| ‡ Crystallographic data for the reported structures of biradicals 1c, 2c, 1d, 3d, 4d and 5d have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 823716, 816632, 816630, 823717, 858078, 810139, respectively. |
| § The 13C-NMR spectrum of 2b was not obtained, as the sample appeared to be unstable in DMSO and began to oxidize in the NMR tube upon measurement. |
| This journal is © The Royal Society of Chemistry 2017 |