
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic structure design for nanoporous, electrically conductive zeolitic imidazolate frameworks†
Keith T.
Butler
 *a,
Stephen D.
Worrall
b,
Christopher D.
Molloy
a,
Christopher H.
Hendon
c,
Martin P.
Attfield
b,
Robert A. W.
Dryfe
d and
Aron
Walsh
ef
*a,
Stephen D.
Worrall
b,
Christopher D.
Molloy
a,
Christopher H.
Hendon
c,
Martin P.
Attfield
b,
Robert A. W.
Dryfe
d and
Aron
Walsh
ef
aDepartment of Chemistry, University of Bath, BA2 7AY, UK. E-mail: k.t.butler@bath.ac.uk
bCentre for Nanoporous Materials, School of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK
cDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
dSchool of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK
eDepartment of Materials, Imperial College London, Exhibition Road, London, SW7 2AZ, UK
fDepartment of Materials Science and Engineering, Yonsei University, Seoul, South Korea
First published on 1st August 2017
Abstract
Electronic structure calculations are used to develop design rules for enhanced electrical conductivity in zeolitic imidazolate frameworks. The electrical resistivity of Co2+ based zeolitic imidazolate frameworks has previously been found to be ∼1000 times lower than that of Zn2+ based materials. The electrical conductivity of the frameworks can also be tuned by ligand molecule selection. Using density functional theory calculations, this controllable electrical conductivity is explained in terms of tuneable conduction band edge character, with calculations revealing the improved hybridisation and extended band character of the Co2+ frameworks. The improvements in the methylimidazolate frameworks are understood in terms of improved frontier orbital matching between metal and ligand. The modular tuneability and previously demonstrated facile synthesis provides a route to rational design of stable framework materials for electronic applications. By outlining these design principles we provide a route to the future development of stable, electrically conductive zeolitic imidazolate frameworks.
Metal–organic frameworks (MOFs) are nanoporous materials with intrinsically ordered framework structures based on modular metal cluster and organic ligand compositions. The combination of topological control and chemical diversity make MOFs attractive candidates for a variety of applications. To date, much of the research in the area of MOFs has concentrated on exploiting their large surface area and great success has been achieved in fields such as gas-storage, separation and chemical catalysis1–3 and as super-capacitors for energy storage.4 Recent reports of electrical conductivity in MOF structures could open the door to many new applications, such as battery cathodes,5,6 electrocatalysts,7,8 transistors9–12 and photovoltaics.13,14 MOFs have already made an impact as components in thermoelectrics15 and memory storage.16–18 Despite the promise held by MOFs for the design of modular electronic components with molecular tunability, they are often mechanically and thermally unstable, hampering widespread application.
The exciting advances in conductive MOFs have resulted in the emergence of design principles for enhancing electrical properties by modular tuning.19,20 The conduction mechanisms of MOFs have been divided into three broad classes: (i) through space transport, via π-stacking; (ii) through bond transport, relying on covalent bond networks and (iii) hopping transport, where charge carriers make discrete jumps between structural units.21 The first two mechanisms (band transport) rely on delocalised wavefunctions across the system and are associated with higher carrier mobilities than hopping transport.22 Typically in MOFs, however, band transport is difficult to achieve, due to poor hybridisation between metal linker and organic ligand units. Possible design strategies for achieving conductive MOFs include the addition of electro-activating molecules,23 or designing structures with conductive (MX)∞ chains21 – in both cases tunable conductivity is achieved by establishing conductive pathways of continuous wavefunction overlap throughout the structure.
Zeolitic imidazolate frameworks (ZIFs) maintain the chemical tunability of MOFs with the virtue of possessing the thermal and chemical stability of zeolite-like materials.24–28 ZIFs are a sub-class of MOFs, built around the M–Im–M motif, where M is a divalent tetrahedral cation (typically, but not exclusively, Zn2+ or Co2+) and Im is an imidazolate anion.29,30 There have been few reports of electronic device applications for ZIFs; they are typically viewed as poor electrical conductors. Nonetheless, their use as high-κ dielectrics,31 in photoelectrochemical core–shell heterostructures32 and super-capacitors33 has been recently been reported, which challenges the perception of ZIFs as electronically inert materials.
Here, we set out to elucidate molecular design principles for achieving conductive ZIF materials using electronic structure calculations. We concentrate on four ZIFs, comprised of two metal cations, Zn2+(d10) and Co2+(d7) and two substituted imidazolate ligands, methylimidazolate (mIM) and benzimidazolate (bIM). Some of us have previously demonstrated how the Co2+-based ZIF-67 has more than 1000 times greater electrical conductivity than Zn2+-based ZIF-8 or ZIF-7. In this work we additionally characterise ZIF-9, which is Co2+-based, but has the same ligand as ZIF-7, see Fig. 1. The comparison of four materials based on permutations of two metals and two ligands ensures frameworks with similar geometric structures, consisting of metal ions in a tetrahedral environment, bridged by organic linkers, which bond to the metals through N lone pairs, see Fig. 1. The homogeneity of the geometries allows us to isolate the electronic structure influence on electrical conductivity, highlighting the role of orbital symmetry, electron configuration, ionisation potential and electron affinity. We demonstrate how choice of metal (based on ionisation potential) and design of ligands (for improved hybridisation) can be used to lead to the development of conductive, stable ZIF materials.
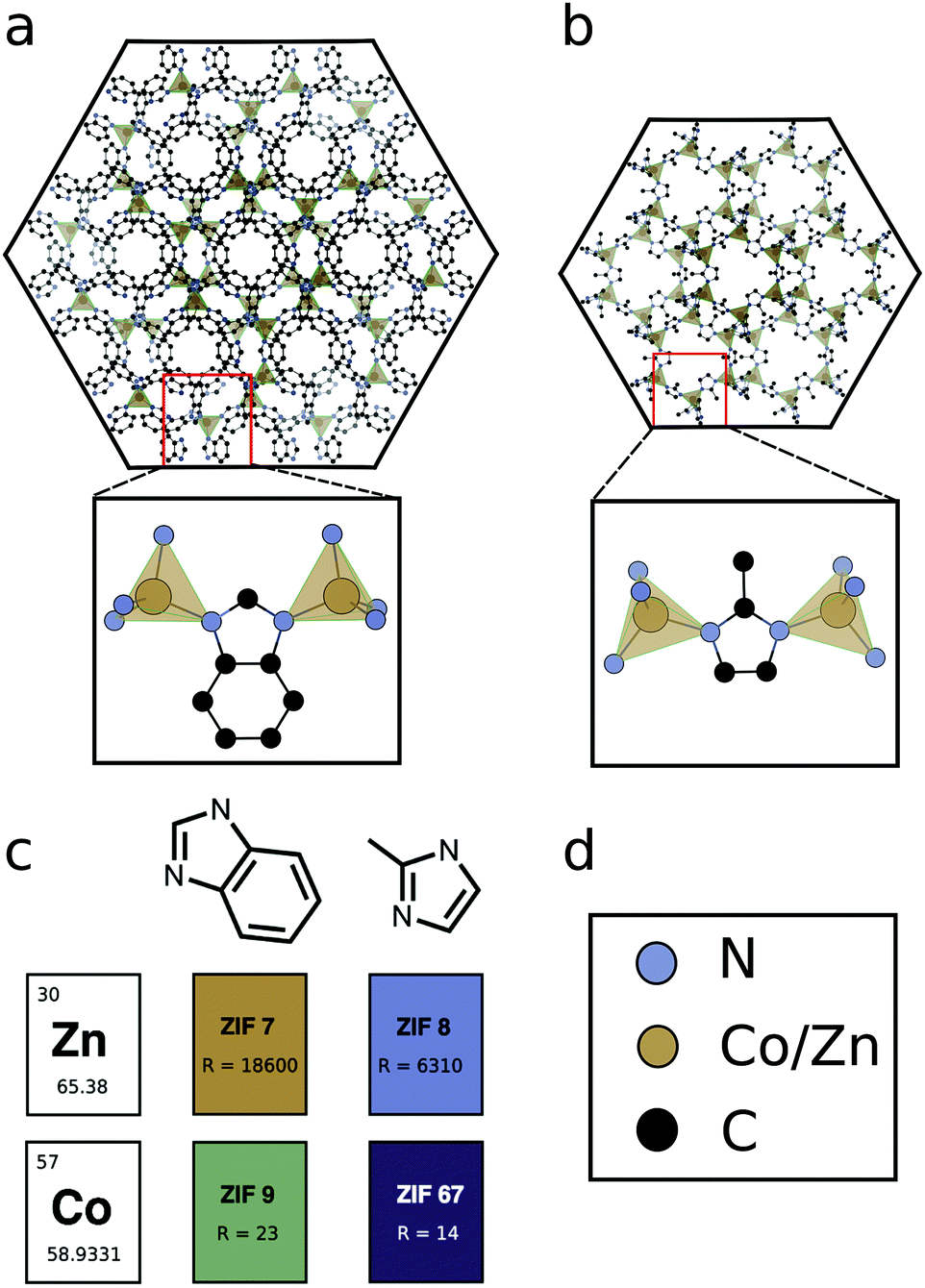 | ||
| Fig. 1 (a) The structure of ZIF-7(9), with a close up of the Zn(Co)–ligand–Zn(Co) conductive bridge. (b) The structure of ZIF-8(67), with a close up of the Zn(Co)–ligand–Zn(Co) conductive bridge. (c) The modular make-up of ligand and metal for construction of the four frameworks studies, each framework is reported along with its room temperature electrical resistance in Ω. (d) Colour key for (a) and (b) carbon: black; nitrogen: petrol blue; M(II): gold. | ||
ZIF coated electrodes were prepared by a process of electrochemical deposition, which has been previously applied to the growth of ZIF thin films.34,35 This approach to thin film growth has the great advantage of not requiring the high temperatures or long synthesis times of many other MOF thin film preparation routes and has been extensively reported for porous material synthesis.36,37 The use of mild conditions and lack of requirement of any specialised equipment also makes electrochemically deposited ZIFs promising from a scale-up and applications perspective. Frameworks can easily be coated onto electrodes where epitaxial relations between the substrate and framework exist.38 ZIF coated electrodes were synthesised by holding two metal foil electrodes in a heated, de-aerated electrolyte solution containing linker under a fixed potential difference of 2.5 V for a set time. The synthesis and characterisation of ZIF-7, ZIF-8 and ZIF-67 have been reported previously by some of us,33 the details for ZIF-9 are presented in the ESI.†
We have previously reported the electrochemical impedance spectroscopy (EIS) results for ZIF-7, ZIF-8 and ZIF-67,33 here we have repeated the experiments, including ZIF-9 in the set for consistency. EIS in a symmetrical two electrode configuration, of ZIF-7, ZIF-8, ZIF-9 and ZIF-67, was performed to obtain electrical resistance characteristics, full details are available in the ESI.† Nyquist plots of the two Zn2+ ZIFs (ZIF-7 and ZIF-8; Fig. 2a) display a clear semi-circular shape, those of the Co2+ ZIFs (ZIF-9 and ZIF-67) are instead composed almost entirely of a ∼45°, linear, diffusion dominated region with almost no discernible semi-circular character. The data were fitted with a Randles circuit to obtain values for the solution resistance (Rs, determined by the electrolyte) and the charge-transfer resistance (RCT, determined by the framework).
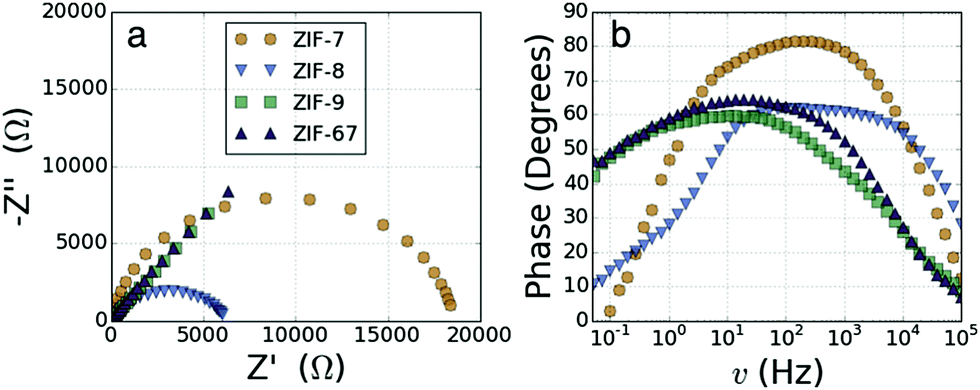 | ||
| Fig. 2 (a) Nyquist plots of ZIF-7, ZIF-8, ZIF-9 and ZIF-67. (b) Bode phase plots of ZIF-7, ZIF-8, ZIF-9 and ZIF-67. | ||
The resistance of the Co2+ based materials is around 1000 times smaller than that of the Zn2+ based materials, as previously reported,33Table 1. A new finding is that the resistance of the mIM frameworks is significantly smaller for both metals than the resistance in the bIM frameworks. The Bode phase plots (Fig. 2b) similarly show a significant difference in the behaviour between the Zn2+ and Co2+ ZIFs. The shape of the Co2+ ZIF plots are characteristic of pseudocapacitive behaviour,39 with the initial plateau in the phase at ∼45° followed by a steady decrease in phase towards 0° with increasing frequency. The shape of the Zn2+ ZIF plots show a different behaviour with the phase starting close to 0° at low frequency, before rising to plateau between 60° and 80° at medium frequencies and then finally decreasing again at higher frequencies.
| Material | R CT (Ω) | σ (S cm−1) | IP (eV) | E g (eV) |
|---|---|---|---|---|
| ZIF-7 | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
4.5–9.0 × 10−10 | 5.61 | 4.82 |
| ZIF-8 | 6310 | 1.3–2.6 × 10−9 | 5.73 | 5.29 |
| ZIF-9 | 23 | 3.6–7.3 × 10−7 | 5.21 | 3.01 |
| ZIF-67 | 14 | 0.6–1.2 × 10−6 | 5.62 | 4.25 |
Following the analysis of Jamnik40 and developed by Wang and Hong,41 the electronic and ionic contributions to mixed conductivity can be separated by analysis of the impedance frequency response. The conductivities, reported in Table 1, of the samples are obtained by normalising for the MOF electrode area (1.2 cm2) and film thickness (10–20 μm). We note that even the highest conductivity is significantly lower than some of the champion MOF conductors reported to date;19 however, RCT represents an upper limit to the resistance of the ZIF and includes contributions from interface contact resistance and other sources extrinsic to the ZIF. Therefore, the conductivities reported here are a conservative estimate of the framework conductivity and are nonetheless of the order of conductivities found in other promising MOFs and organic conductors.42
To understand the differences in conductivity of the frameworks, we have performed density functional theory (DFT) calculations. We first consider the full framework, employing periodic boundary conditions and projector augmented waves43 within the vasp44 code, we use a hybrid functional (HSE0645) to accurately represent the electronic structure, full details are available in the ESI.† The electronic density of states (DOS) of the four materials are presented in Fig. 3, the DOS and electronic energy levels are calculated based on evacuated pore structures, available as .cif files (see data access statement). We begin by examining the effect of metal substitution, ZIF-7 and ZIF-9 are structurally analogous, as are ZIF-8 and ZIF-67. In both cases there is a marked increase in the metal character at the conduction band edge of the electronic DOS comparing the Co2+ frameworks to the Zn2+ frameworks. In both Co2+ based materials there is close to 50% metal contribution to the conduction band edge, this results from the d7 electron configuration of Co2+, compared to the d10 configuration of Zn2+. In the Co2+ frameworks the empty d-states of the metal are available to hybridise with the ligand LUMO levels. The tetragonal environment results in a crystal field splitting on the Co2+ with partially occupied dxy, dxz and dyz states, which can interact with the sp2 hybridised N. This hybridisation results in extended conduction states, bridging the imidazolate and the metal (Fig. 4b) and results in the greatly improved conductivity in the Co2+ frameworks. This finding highlights the importance of choosing metal ions with relatively shallow ionisation potentials, which allow them to participate in the formation of band edges in the framework, we note that a similar improvement in conductivity was reported when replacing Mn2+ by Fe2+, when the loosely bound Fe2+ electrons form the valence band edge.42
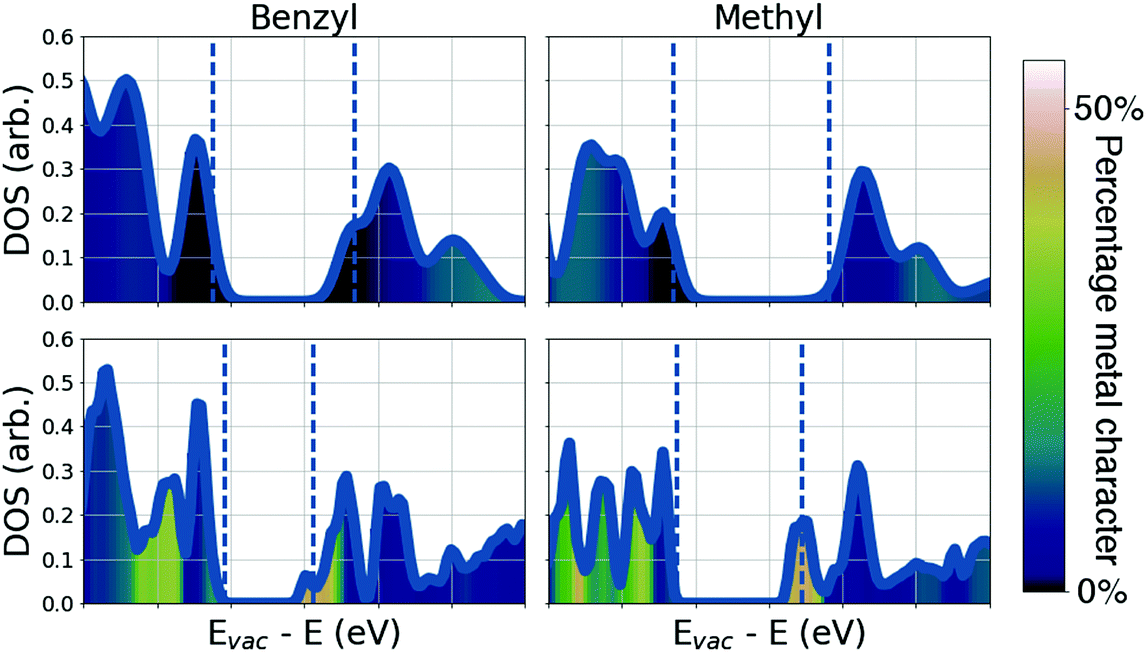 | ||
| Fig. 3 Electronic density of states plots of the four materials studied, showing the degree of metal contribution to the electronic states. The valence and conduction bad edges are highlighted by dashed blue lines. Upper row, Zn2+ ZIFs; lower Co2+ ZIFs. | ||
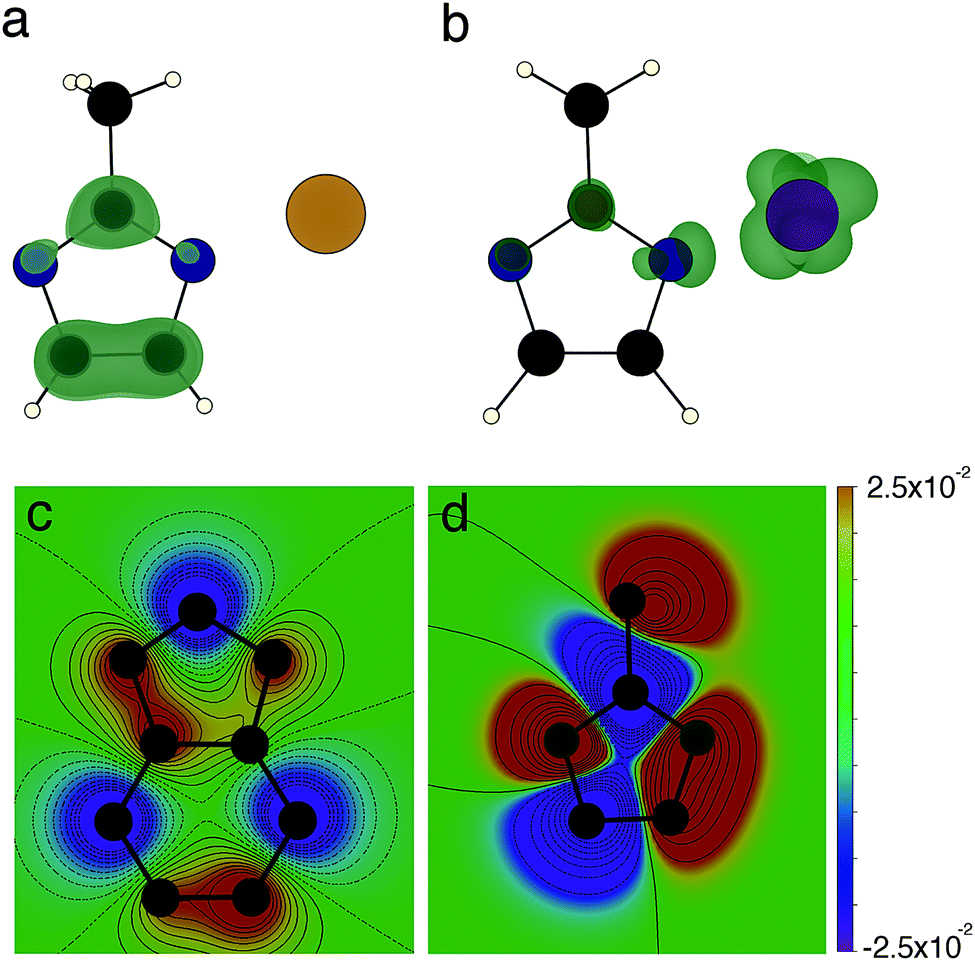 | ||
| Fig. 4 The effects of changing linker and ligand. (a) Conduction band minimum isosurface in ZIF-8; (b) conduction band minimum isosurface in ZIF-67. Both isosurfaces are at 0.00025 e Å−3 and concentrate on the structural unit displayed in Fig. 1, N atoms are in blue, C atoms are black, H atoms are white, Zn2+ is beige and Co2+ is indigo. (c) Contour plot of the of the LUMO orbital coefficients of bIm, C is black and N is grey (H atoms are suppressed) (d) contour plot of the of the LUMO orbital coefficients of mIm, C is black and N is grey (H atoms are suppressed). Wavefunction coefficients are in atomic units. | ||
Another important difference between Co2+ and Zn2+ is the availability of alternative oxidation states for Co, i.e. Co3+. The availability of multiple oxidation states promotes hopping transport in a framework,46,47 with charges being transferred between metal centres via the organic ligand. This type of conduction is dependent on the overlap in energy of the ligand and linker frontier orbitals.46 In ZIFs we know the configuration of the metal atomic orbitals, in order to assess the possibility of hopping of oxidation states we require knowledge of the ligand frontier orbitals.
We now turn our attention to the role of the ligand in determining the framework conductivity. In the case of both metals the framework with the mIm shows superior conductivity. This cannot be ascribed to a separation effect, as the M⋯M distance in the presence of both ligands is very similar; we therefore suggest that the origin of this effect is related to the electronic structure of the ligand. The conduction pathway in the ZIF frameworks depends on the ligand N to metal interaction. The strength of the overlap between the ligand and the metal depends on the extent to which their wavefunctions overlap; in this case ligand N wavefunction with metal dxy, dxz or dyz orbitals. Given the orientation of the ligands tetrahedrally about the cation, the overlap of the orbitals with the d-orbitals of the metal is highly dependent on the spatial extent of the ligand LUMO. The spin resolved density of states (ESI,† Fig. S4) shows that in each of the Co materials the conduction band minimum is formed from a single spin-down channel on the Co, hybridised with a spin-down channel on the N. This spin-dependent band edge structure can have important consequences for the carrier transport and suggests the intriguing possibility of Co-based ZIF spin filters.
We have performed quantum chemical calculations of the charged ligand molecules to provide an understanding of the spatial resolution of the ligand wavefunctions. These calculations of the molecules in isolation are performed using Gaussian09,48 with the B3LYP functional49,50 and 6-31G* basis sets.51 Using the macrodensity package,52 we analyse wavefunctions of the LUMO levels of the mIm and bIm studied here, calculating the spherical average of the wavefunction coefficients about the N atoms, with a radius of 2 Å. The extended π system of the bIm withdraws density away from the N sites, resulting in an average of coefficients centred at the N site of 2.5 × 10−4 a.u., compared to 2.2 × 10−3 a.u. at the mIm N sites. The contours of the wavefunction coefficients are plotted in Fig. 3(c and d); the greater density in the mIm is clear from this plot.
Of the three conduction mechanisms outlined earlier the conduction in ZIFs cannot be via π-stacking and is unlikely to occur through extended covalent bonds; therefore, it is likely to be mediated by a hopping mechanism. As outlined in a recent review of these mechanisms the overlap of ligand/linker wavefunctions and the availability of variable linker oxidation states are crucial for the efficacy of hopping transport in frameworks. We propose that conductive ZIFs can be rationally designed by choosing: (i) a metal cation with a shallow second ionisation potential and partially occupied/unoccupied dxy, dxz, dyz orbitals and (ii) a ligand with high LUMO density at the N sites the degree of hybridisation can be maximised, promoting the formation of an extended conduction band. A systematic search of metal species screening for (II) oxidation states in tetrahedral environments, with a suitable second ionisation potential was performed using our recently published smact code,53 suggesting Fe2+ as a promising cation replacement for Co2+. Screening of candidate ligands can be achieved through relatively low-cost calculations of molecular imidazolate species.
We have studied the electrical conductivity of four ZIF materials, using electronic structure calculations to explain observed trends. The four frameworks are made from a combination of two metals (Zn2+ and Co2+) and two ligands (bIm and mIm). The Co2+ frameworks display a significantly greater electrical conductivity than the Zn2+ based frameworks, by 3 to 4 orders of magnitude, as reported previously.33 Here, we explain how this increased conductivity in Co2+ based frameworks is linked to the availability of empty d-states for forming the conduction band edge. We find that mIm frameworks are more conductive than the bIm frameworks by 1.5 to 2 times; this is explained by the wavefunction shape of LUMO level of mIm, which offers improved hybridisation with the metal linkers to form conduction pathways. This general principle of ensuring unoccupied d-states with the correct orbital symmetry to overlap with ligands, along with ligand frontier orbital levels optimised through the addition of electron donating or withdrawing groups, offers a route to rational design of electrically conductive zeolitic imidazolate frameworks. Such electrically conductive ZIFs would open the way for enhanced functionality of frameworks, where they are used in fields such as gas separation,54 CO2 catalysis,55 energy storage,56 mixed matrix membranes57 and drug delivery.58
We acknowledge membership of the UK′s HPC Materials Chemistry Consortium (EPSRC EP/L000202) and access to computational resources through PRACE. A. W. acknowledges support from the Royal Society for a University Research Fellowship and K. T. B. is funded by EPSRC (EP/M009580/1 and EP/J017361/1). SDW acknowledges the NoWNANO DTC for funding. RAWD would like to thank the EPSRC (UK, grant references EP/K039547/1 and EP/K016954/1) for support.
Data access statement: analysis scripts used examine the density of states, and the optimised crystal structures are available on-line, free of charge, from http://https://github.com/keeeto/conductive-zifs-data.
References
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. Gascon, A. Corma, F. Kapteijn and F. X. L. i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS PubMed.
- G. Férey, F. Millange, M. Morcrette, C. Serre, M.-L. Doublet, J.-M. Grenèche and J.-M. Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259–3263 CrossRef PubMed.
- B. Nohra, H. E. Moll, L. M. R. Albelo, P. Mialane, J. Marrot, C. Mellot-Draznieks, M. O'Keeffe, R. N. Biboum, J. Lemaire, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2011, 133, 13363–13374 CrossRef CAS PubMed.
- S. R. Ahrenholtz, C. C. Epley and A. J. Morris, J. Am. Chem. Soc., 2014, 136, 2464–2472 CrossRef CAS PubMed.
- M. Usman, S. Mendiratta and K.-L. Lu, Adv. Mater., 2017, 29, 1605071 CrossRef PubMed.
- K. T. Butler, C. H. Hendon and A. Walsh, ACS Appl. Mater. Interfaces, 2014, 6, 22044–22050 CAS.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC.
- K. T. Butler, C. H. Hendon and A. Walsh, J. Am. Chem. Soc., 2014, 136, 2703–2706 CrossRef CAS PubMed.
- K. J. Erickson, F. Léonard, V. Stavila, M. E. Foster, C. D. Spataru, R. E. Jones, B. M. Foley, P. E. Hopkins, M. D. Allendorf and A. A. Talin, Adv. Mater., 2015, 27, 3453–3459 CrossRef CAS PubMed.
- L. Pan, Z. Ji, X. Yi, X. Zhu, X. Chen, J. Shang, G. Liu and R.-W. Li, Adv. Funct. Mater., 2015, 25, 2677–2685 CrossRef CAS.
- S. M. Yoon, S. C. Warren and B. A. Grzybowski, Angew. Chem., Int. Ed., 2014, 53, 4437–4441 CrossRef CAS PubMed.
- S. D. Worrall, M. A. Bissett, W. Hirunpinyopas, M. P. Attfield and R. A. W. Dryfe, J. Mater. Chem. C, 2016, 4, 8687–8695 RSC.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- R. Grau-Crespo, A. Aziz, A. W. Collins, R. Crespo-Otero, N. C. Hernández, L. M. Rodriguez-Albelo, A. R. Ruiz-Salvador, S. Calero and S. Hamad, Angew. Chem., Int. Ed., 2016, 55, 16012–16016 CrossRef CAS PubMed.
- S. S. Park, E. R. Hontz, L. Sun, C. H. Hendon, A. Walsh, T. Van Voorhis and M. Dincă, J. Am. Chem. Soc., 2015, 137, 1774–1777 CrossRef CAS PubMed.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS PubMed.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- B. Chen, Z. Yang, Y. Zhu and Y. Xia, J. Mater. Chem. A, 2014, 2, 16811–16831 CAS.
- L. B. du Bourg, A. U. Ortiz, A. Boutin and F.-X. Coudert, APL Mater., 2014, 2, 124110 CrossRef.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS PubMed.
- S. Moggach, T. Bennett and A. Cheetham, Angew. Chem., 2009, 121, 7221–7223 CrossRef.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- D. W. Lewis, A. R. Ruiz-Salvador, A. Gomez, L. M. Rodriguez-Albelo, F.-X. Coudert, B. Slater, A. K. Cheetham and C. Mellot-Draznieks, CrystEngComm, 2009, 11, 2272–2276 RSC.
- S. Eslava, L. Zhang, S. Esconjauregui, J. Yang, K. Vanstreels, M. R. Baklanov and E. Saiz, Chem. Mater., 2013, 25, 27–33 CrossRef CAS.
- W. Wen Zhan, Q. Kuang, J. Zhang Zhou, X. Jian Kong, Z. Xiong Xie and L. Sun Zheng, J. Am. Chem. Soc., 2013, 135, 1926–1933 CrossRef PubMed.
- S. D. Worrall, H. Mann, A. Rogers, M. A. Bissett, M. P. Attfield and R. A. Dryfe, Electrochim. Acta, 2016, 197, 228–240 CrossRef CAS.
- W.-W. Zhan, Q. Kuang, J.-Z. Zhou, X.-J. Kong, Z.-X. Xie and L.-S. Zheng, J. Am. Chem. Soc., 2013, 135, 1926–1933 CrossRef CAS PubMed.
- H. Al-Kutubi, A. Dikhtiarenko, H. R. Zafarani, E. J. R. Sudholter, J. Gascon and L. Rassaei, CrystEngComm, 2015, 17, 5360–5364 RSC.
- M. Li and M. Dincă, J. Am. Chem. Soc., 2011, 133, 12926–12929 CrossRef CAS PubMed.
- M. Li and M. Dincă, Chem. Mater., 2015, 27, 3203–3206 CrossRef CAS.
- K. T. Butler, Y. Kumagai, F. Oba and A. Walsh, J. Mater. Chem. C, 2016, 4, 1149–1158 RSC.
- P. L. Taberna, P. Simon and J. F. Fauvarque, J. Electrochem. Soc., 2003, 150, A292 CrossRef CAS.
- J. Jamnik, Solid State Ionics, 2003, 157, 19–28 CrossRef CAS.
- C. Wang and J. Hong, Electrochem. Solid-State Lett., 2007, 10, A65–A69 CrossRef CAS.
- L. Sun, C. H. Hendon, M. a. Minier, A. Walsh and M. Dincă, J. Am. Chem. Soc., 2015, 137, 6164–6167 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Matter. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse, Phys. Rev. B: Condens. Matter Matter. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- M. Dincă and F. Léonard, MRS Bull., 2016, 41, 854–857 CrossRef.
- A. Walsh, K. T. Butler and C. H. Hendon, MRS Bull., 2016, 41, 870–876 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09 Revision A.02, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Matter. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- K. T. Butler, J. Buckeridge, C. R. A. Catlow and A. Walsh, Phys. Rev. B: Condens. Matter Matter. Phys., 2014, 115320, 1–6 Search PubMed.
- D. W. Davies, K. T. Butler, A. J. Jackson, A. Morris, J. M. Frost, J. M. Skelton and A. Walsh, Chemistry, 2016, 1, 617–627 CrossRef CAS PubMed.
- C. Gücüyener, J. van den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef PubMed.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed.
- D. Yu, B. Wu, L. Ge, L. Wu, H. Wang and T. Xu, J. Mater. Chem. A, 2016, 4, 10878–10884 CAS.
- L. Hao, P. Li, T. Yang and T.-S. Chung, J. Membr. Sci., 2013, 436, 221–231 CrossRef CAS.
- C.-Y. Sun, C. Qin, X.-L. Wang, G.-S. Yang, K.-Z. Shao, Y.-Q. Lan, Z.-M. Su, P. Huang, C.-G. Wang and E.-B. Wang, Dalton Trans., 2012, 6906–6909 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tc03150e |
| This journal is © The Royal Society of Chemistry 2017 |