
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The contributions of molecular vibrations and higher triplet levels to the intersystem crossing mechanism in metal-free organic emitters†
Rongjuan
Huang
a,
João
Avó
b,
Thomas
Northey
c,
E.
Chaning-Pearce
a,
Paloma L.
dos Santos
a,
Jonathan S.
Ward
 d,
Przemyslaw
Data
ae,
Marc K.
Etherington
a,
Mark A.
Fox
d,
Thomas J.
Penfold
c,
Mário N.
Berberan-Santos
f,
João C.
Lima
b,
Martin R.
Bryce
d and
Fernando B.
Dias
*a
d,
Przemyslaw
Data
ae,
Marc K.
Etherington
a,
Mark A.
Fox
d,
Thomas J.
Penfold
c,
Mário N.
Berberan-Santos
f,
João C.
Lima
b,
Martin R.
Bryce
d and
Fernando B.
Dias
*a
aPhysics Department, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: f.m.b.dias@durham.ac.uk
bREQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal
cSchool of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
dChemistry Department, Durham University, South Road, Durham, DH1 3LE, UK
eSilesian University of Technology, Faculty of Chemistry, Strzody 9, 44-100 Gliwice, Poland
fCentro de Química-Física Molecular, Instituto Superior Técnico, 1049-001 Lisboa, Portugal
First published on 23rd May 2017
Abstract
Dual luminescence, i.e. intense, simultaneous, room temperature phosphorescence (RTP) and thermally activated delayed fluorescence (TADF) is observed in a series of donor–acceptor–donor (D–A–D) molecules. This dual luminescence is stronger in the “angular” isomers, compared to their “linear” regioisomers, which is consistent with an enhanced intersystem crossing (ISC) in the former. Herein, we demonstrate that the small energy gap between the triplet levels, T1–Tn, below the lowest singlet state, S1, in the “angular” regioisomers, enhances the coupling between S1 and T1 states and favors ISC and reverse ISC (rISC). This is consistent with a spin–vibronic mechanism. In the absence of this “triplet ladder”, due to the larger energy difference between T1 and Tn in the “linear” regioisomers, the ISC and rISC are not efficient. Remarkably, the enhancement of the ISC rate in the “angular” regioisomers is accompanied by an increase of the rate of internal conversion (IC). These results highlight the contributions of higher triplet excited states and molecular vibronic coupling to the harvest of triplet states in organic compounds, and cast the TADF and RTP mechanisms into a common conceptual framework.
Introduction
Metal-free organic compounds with dual emission from singlet and triplet excited states, e.g. fluorescence and phosphorescence at room temperature (RTP) or prompt and delayed fluorescence due to thermally activated delayed fluorescence (TADF),1 have attracted great attention in recent years due to their potential for application in photonic and optoelectronic devices, including sensing of organic solvents2 and oxygen,3,4 optical thermometry,5 optical storage,6 mechanoluminochromic materials,7 counterfeit labeling,8 luminescent probes for biological imaging,9 and organic light emitting diodes (OLEDs).10,11Detailed investigations aimed at revealing the fundamental mechanisms of TADF and RTP are also topics of strong research focus, because they offer excellent opportunities to investigate the dynamics of excited triplet states in organic materials.12 These studies can, therefore, provide valuable information about the role of molecular conformation and connectivity, and coupling between electronic states for promoting triplet harvesting. These aspects are still not completely understood, and are extremely relevant when the properties of the triplet excited state are of importance, such as in photon up-conversion due to triplet–triplet annihilation,13 triplet harvesting in OLEDs,14–16 and organic spintronics.17
One of the major problems affecting the current state-of-the-art OLEDs is the large roll-off observed in most devices. This is primarily caused by the long lifetime of the emitting excitons used in TADF molecules and heavy metal complexes, and also makes the current metal-free RTP emitters unsuitable for application in OLEDs. Consequently, developing strategies to accelerate the triplet harvesting rate in both metal-free TADF and RTP emitters is a major objective, and requires detailed investigations of the intersystem crossing (ISC) and reverse intersystem crossing (rISC) mechanisms.
The observation of dual fluorescence–phosphorescence at room temperature in pure organic materials is a rare phenomenon.18 This is because good fluorescent emitters generally show very small yields of triplet formation. At the other extreme, strongly phosphorescent molecules, mostly containing heavy metals, have triplet yields close to 100%, and thus fluorescence is not observed.19 A delicate balance between the intersystem crossing rate, radiative decay rate and the non-radiative internal conversion (IC) pathways is therefore required to generate simultaneous emission from the singlet and triplet states,18,20 as shown schematically in Fig. 1a. However, in most purely organic compounds the rate of intersystem crossing, from S1 to T1, kISC, and the rate for radiative decay from T1, kph, are very slow, and cannot compete with fluorescence. Therefore, to achieve RTP, the triplet formation yield has to be enhanced, and the non-radiative deactivation pathways from T1 to S0 need to be suppressed.
 | ||
| Fig. 1 (a) Energy level diagram describing the photophysical mechanisms that give rise to RTP and TADF emissions. (b) Molecular structures of the compounds used in this work, and numbering code for molecular isomers used in this work (top). (c) Frontier molecular orbitals, HOMOs and LUMOs, for compounds 1 and 2. | ||
Concurrently, similar conditions are required for efficient TADF to be observed. However, the two mechanisms differ in that efficient reverse intersystem crossing, krISC, is required for TADF but not for RTP, see Fig. 1a. TADF and RTP are therefore often seen as two contrasting mechanisms. We show here that this is not the case. Instead, the TADF and RTP mechanisms share the same fundamental requirements at the molecular level, and TADF or RTP are different responses obtained in a common framework, which can be tuned by selecting the host properties. This reduces the emphasis upon the energy gap between the emitting singlet and triplet states, and crucially introduces the complementarity between the two mechanisms that otherwise would not exist. This complementarity means that it is feasible that both the TADF and RTP mechanisms can be operative in the same molecule, as demonstrated in Cu(I) complexes,21 and recently in a pure organic emitter.22 However, the emergence of this dual luminescence is strongly dependent on the ability to fine tune the kinetic parameters, which can be achieved by the molecular structure of the emitter and the host in which it is dispersed. Interestingly, in dual emitters showing simultaneously both long-lived forms of luminescence, the ratiometric nature of the dual-emission would be preserved during the entire luminescence lifetime, which makes these materials very convenient in gated-acquisition luminescence methods for sensing/imaging. Here, the ratio between the RTP and TADF intensities is preserved even in the microsecond time range, and can be used as an internal calibration. This is in clear contrast with conventional RTP and TADF emitters where the prompt fluorescence decays in just a few nanoseconds, and the phosphorescence or delayed fluorescence lives for microseconds or milliseconds.
Results and discussion
The development of pure organic RTP compounds has progressed significantly in recent years. These have adopted a number of different strategies, but have mainly targeted restricting the intramolecular vibrations of the emitters using intermolecular interactions, either in rigid hosts,10,23,24 crystalline form,8,25,26 or induced by aggregation.8 All of these methods restrict thermal vibrations and increase the phosphorescence lifetime and quantum yield. This suppresses the rate of T1 non-radiative decay, which in organic materials is usually faster than the radiative rate. However, it is not clear whether or not the intersystem crossing rate is also affected. Interestingly, RTP has been reported recently in a brominated fluorene derivative,27 and deuterium substitution has been also used to diminish internal conversion due to molecular vibrations, and thus enhance RTP emission.20,24Despite this strong interest in RTP materials, dual fluorescence–phosphorescence at RT has only been reported in general when the emitter is dispersed in a few selected hosts, which are able to suppress vibrations, such as poly(vinyl alcohol), poly(methyl methacrylate), β-estradiol, micelles, cyclodextrin or inorganic crystals.18,20 Remarkably, reports of RTP in conjugated hosts are extremely rare, which has limited their application in OLEDs. To the best of our knowledge, only two publications report RTP in conjugated hosts.10,14
A comprehensive investigation of molecular structure–property relationships is, therefore, still needed to guide synthetic strategies for the design of more efficient RTP emitters.28,29 There is no clear understanding how the host–guest interactions influence the intersystem crossing rate and the pathways for non-radiative decay of the excited states. For example, it is not clear why some molecules exhibit intense RTP when dispersed in particular hosts but not in others, or why some molecular structures exhibit intense RTP and structurally similar analogues exhibit only fluorescence.20,29 This clearly shows that besides suppressing internal vibrations, the rational molecular design of emitters and hosts can be used to tune the electronic energy levels and activate RTP. However, while manipulation of electronic properties to achieve RTP is very attractive, it is also extremely challenging.29
In contrast, guidelines for designing efficient TADF molecules are beginning to emerge,1,30 and delayed fluorescence is now routinely observed in organic compounds that comprise covalently linked electron donor (D) and electron acceptor (A) units, in solution and also in several solid hosts, including those with extended conjugation. In addition, the fine details determining the mechanism for efficient thermally activated reverse intersystem crossing in TADF emitters are beginning to be understood.16,31–34 This includes work illustrating that the energy alignment between the singlet state with charge transfer character and a local triplet state was proposed as a key requirement for efficient TADF.16,31 However, not all TADF materials may have a local triplet state in near-resonance with the CT states, which may be particularly valid in blue TADF emitters, where the singlet emissive state has to be kept at high energies. Therefore, more general requirements are required to explain the observation of TADF. Moreover, in the design of most TADF emitters, the attempts to minimize the singlet–triplet energy gap often have a negative impact on the fluorescence yield. Strategies to design TADF molecules with “strong” fluorescence yields and shorter luminescence lifetimes are, therefore, required.33 Moreover, as in OLEDs the triplet yield is fixed at 75%, there is still potential to improve the performance of OLED emitters by achieving a proper balance between the yield of reverse intersystem crossing and the fluorescence yield.35 These are issues that need to be resolved and require a detailed understanding of the mechanisms of triplet harvesting at the molecular level.
Herein, we explore factors influencing the rate of intersystem crossing and reverse intersystem crossing in pure organic emitters. We show that simple manipulations of the way electron donor (D) and electron acceptor (A) units are linked in D–A–D compounds have a profound influence on the activity of both RTP and TADF. When the emitters are dispersed in suitable hosts and depending on the polarity of the local surroundings, simultaneous RTP and TADF are observed. This illustrates the similarity of the two mechanisms and properties required to facilitate high intersystem crossing and reverse intersystem crossing rates in organic molecules and brings TADF and RTP into a common conceptual framework. Crucially, these synergies can be used to create and guide the design of novel emitters.
Fig. 1b shows the molecular structures of the compounds studied herein. Previously we have reported strong TADF emission in ethanol solution for compounds 2 and 4 at room temperature, and intense dual fluorescence–phosphorescence in compounds 2, 4 and 6, in ethanol solution at 100 K.36 The dual fluorescence–phosphorescence in these compounds is so intense that it is observed in a normal fluorimeter without using gated luminescence methods. In contrast, their regioisomers, 1, 3 and 5, exhibit no phosphorescence, even at low temperature and their delayed fluorescence in ethanol solution at 300 K is due to triplet–triplet annihilation,36 (see Fig. S1 and S2, ESI†). This markedly different isomer effect was not understood at the time, and an intermediate triplet level, between the singlet and lower triplet states, was introduced ad hoc to justify the enhanced TADF in 2 and 4. However, this reveals little information about the mechanism sustaining reverse intersystem crossing. In the present work, we unravel fundamental concepts on the way triplets are harvested in purely organic compounds, providing a new understanding of the mechanisms of RTP and TADF in general.
The only difference in the molecular structure of these regioisomers is the linking position of the carbazole, diphenylamine and fluorene electron donor (D) units and the dibenzothiophene-S,S-dioxide electron acceptor unit (A). In 1, 3 and 5, the acceptor is substituted at positions C-3 and C-7, whereas in 2, 4 and 6, the substituents are at positions C-2 and C-8 of the A unit. For easy identification, these different isomers are hereafter referred to as “linear” and “angular”, respectively. Despite this seemingly small change, the effect on the dynamics of the excited states is very significant in the context of RTP and TADF luminescence. Compounds 7, 8 and 9, also with “angular” substitution, show intense dual fluorescence–phosphorescence at 100 K in ethanol solution, but in the “angular” compound 10 no phosphorescence is observed.36 This illustrates that the presence of heteroatoms is crucial for the observation of dual fluorescence–phosphorescence, as previously identified by others.28 However, this is not the only required structural feature for the observation of phosphorescence, as confirmed by the absence of strong phosphorescence in the “linear” compounds 1, 3 and 5. Intrinsic molecular-structure relations must therefore be tailored to create intense RTP and TADF emitters.29
Absorption and emission in solution
The absorption and emission spectra of compounds 1 and 2, 3 and 4, 5 and 6, and of 7, 8, 9 and 10 in hexane solution are shown in Fig. 2. The absorption of the “linear” compounds 1, 3 and 5 is clearly red-shifted compared to their “angular” counterparts 2, 4 and 6, respectively (see Fig. 2a–c). This is expected from the shorter conjugation length of the “angular” emitters, which pushes the singlet and triplet states to higher energies, also see Table 1. Compounds 7, 8, 9 and 10 all have “angular” geometry, and are based on a carbazole donor unit, (except for 10), but differ in their acceptor units. All of these show blue-shifted absorption, similar to 2, 4 and 6. They also have less excited state charge transfer character, as evidenced by their blue-shifted and well-resolved emission, see Fig. 2f. This is due to a lower electron affinity of fluorene, dibenzothiophene, and carbazole units compared to the dibenzothiophene-S,S-dioxide unit. | ||
| Fig. 2 Absorption spectra of (a) 1 and 2, (b) 3 and 4, (c) 5 and 6, and (d) 7, 8, 9 and 10. Emission spectra of compounds (e) 1–6, and (f) 7–10. All spectra were collected in dilute hexane solution at room temperature. | ||
| S1 ± 0.02 (eV) | T1 ± 0.02 (eV) | ΔEST ± 0.03 (eV) | Φ f ± 0.05 | τ f ± 0.05 (ns) | Φ T ± 0.1 | k f × 108 (s−1) | k ISC × 108 (s−1) | k IC × 108 (s−1) | |
|---|---|---|---|---|---|---|---|---|---|
| a Obtained from ref. 36. | |||||||||
| 1 | 3.07 | 2.54 | 0.53 | 0.94 | 3.08a | — | 3.1 | — | 0.2 |
| 2 | 3.24 | 2.70 | 0.54 | 0.10 | 1.60a | 0.32 | 0.6 | 1.9 | 3.6 |
| 3 | 2.84 | 2.20 | 0.64 | 0.72 | 3.66a | — | 1.9 | — | 0.8 |
| 4 | 3.05 | 2.48 | 0.57 | 0.10 | 1.13a | 0.19 | 0.9 | 1.7 | 6.3 |
| 5 | 3.21 | 2.36 | 0.85 | 0.80 | 1.05a | — | 7.6 | — | 1.9 |
| 6 | 3.54 | 2.48 | 1.06 | 0.39 | 1.02a | 0.41 | 3.8 | 4.0 | 1.9 |
| 7 | 3.42 | 3.02 | 0.40 | 0.05 | 0.88 | 0.42 | 0.6 | 4.8 | 6.0 |
| 8 | 3.62 | 2.82 | 0.80 | 0.22 | 3.81 | 0.26 | 0.6 | 0.7 | 1.4 |
| 9 | 3.33 | 2.97 | 0.36 | 0.14 | 3.99 | 0.33 | 0.4 | 0.8 | 1.3 |
Table 1 shows the photophysical data in solution for compounds 1–9. The triplet energies of compounds 7–9 are high, above 2.8 eV, and in the case of 7 as high as 3.02 eV. For compounds 1–6, the triplet state appears at lower energies, ranging from 2.20 eV in 3 to 2.70 eV in 2. As a general trend, the “angular” substitution shifts the singlet and triplet states to higher energies relative to the “linear” counterparts, i.e.2, 4 and 6 have higher singlet and triplet energies than 1, 3 and 5, respectively. However, as the change in the position of substitution affects the singlet and triplet energies almost equally, the singlet–triplet energy gaps of the corresponding isomers are very similar in non-polar hexane. Because the singlet and triplet states are affected in a similar manner, this indicates that both emitting states (S1 and T1) have a similar electronic character. In the present series, compounds 7 and 9 have the smallest ΔES1–T1 gap, around 0.4 eV, and compounds 5, 6 and 8 have the largest ΔES1–T1 gap, above 0.8 eV.
The fluorescence yields follow a clear pattern. The “angular” compounds that show intense phosphorescence at low temperatures also show weak fluorescence. The fluorescence lifetimes are also significantly shorter in the “angular” substituted compounds, when compared with their “linear” counterparts, see compounds 1 to 6 in Table 1. The yield of triplet formation is also significantly higher in the “angular” oligomers. While isomers 1, 3 and 5 show no triplet population at room temperature in hexane solution, their “angular” analogues show large triplet yields. This occurs despite the singlet–triplet energy gap being very similar. Marked differences are also observed in the rate constants for radiative decay, kf, intersystem crossing, kISC, and non-radiative internal conversion, kIC. Remarkably, the radiative decay rate decreases significantly when going from “linear” to “angular” form. This drop in the radiative rates is accompanied by an increase in the intersystem crossing and internal conversion rates.
In summary, despite not significantly affecting the S1–T1 energy gap, the “angular” substitution is very effective in decreasing the radiative rate and accelerating the rates of intersystem crossing and internal conversion. This explains the observation of strong phosphorescence in the “angular” isomers, as at low temperatures internal conversion is greatly suppressed.
The simultaneous increase in internal conversion and intersystem crossing is a general rule that points to a common intermediary state for both processes, i.e., a low-lying CT state responsible for both increased internal conversion and intersystem crossing. However, a closer inspection shows another important aspect to take into account when designing novel TADF/RTP emitters. Comparison of 2, 4 and 6, in which the acceptor is conserved, shows that there is a significant increase in the rate of intersystem crossing for 6 with respect to 2 and 4. This is accompanied by a decrease in the internal conversion rate to the ground state. While in 6 the donor and acceptor moieties are linked through a C(sp2)–C(sp2) bond, in 2 and 4, a N(sp3)–C(sp2) bond performs the same function. This difference impacts both the vibrational deactivation of the singlet state and the donor–acceptor coupling needed to obtain the CT state. While the main degree of freedom in 6 is rotation around the C–C bond coupling the donor and acceptor groups, additional vibrational modes related to inversion around N (sp3–sp2 transition) exist in 2 and 4. The contribution of these additional vibrational modes can be further observed when comparing 2 and 4. In 2, the donor group is a carbazole unit, which is more rigid than the diphenylamine unit in 4. Increasing the flexibility of the donor group (and on the donor–acceptor linking motif) yields higher kIC and lower kISC.
As previously stated, the change in the substitution from “linear” to “angular” has the strongest influence on the yield of triplet formation, giving stronger triplet yields in 2, 4 and 6 and activating intense phosphorescence that is not observed in linear isomers 1, 3 and 5. Strong dual fluorescence–phosphorescence is also observed in ethanol solution at low temperatures in compounds 7, 8 and 9, see Fig. S1 (ESI†), showing that the “angular” substitution is equally effective in providing dual fluorescence–phosphorescence, even if the sulfone functionality is not present. However, it is noted that when the carbazole donors in 8 are replaced by fluorene units, i.e. no heteroatoms exist in the trifluorene structure (10), the phosphorescence is switched off, and only fluorescence is observed. The presence of heteroatoms in the “angular” molecular structures is, therefore, of fundamental importance for the observation of strong phosphorescence in these materials, but not a sole requirement.
Compounds 2 and 4 in ethanol solution have a singlet state of charge transfer character (CT) at much lower energies than the emissive S1 state in hexane and other non-polar environments. In this case, the ΔES1–T1 energy gap becomes very small in polar environments, around 0.1 eV and 0.21 eV in ethanol, for 2 and 4, respectively. As the reverse intersystem crossing rate, krISC, and the intersystem crossing rate, kISC, are proportional to each other,35 the “angular” isomers also have stronger reverse intersystem crossing rates than their “linear” counterparts. This isomer effect and the smaller ΔES1–T1 energy gap created under the influence of the stronger polarity in ethanol, explain why these compounds show strong TADF in ethanol solutions, while compounds 1 and 3, are not TADF active.36 Compound 2 was used previously in OLEDs giving EQEs of 14%.37
The correlation between the ISC and IC rates that is observed is also important when comparing compounds 2, 7, 8 and 9. These compounds all have the same carbazole donor, but different acceptors, where the presence of C, N, SO2 and S promotes increasing ISC. Fig. 3 shows that the rates of ISC and IC in these compounds show a clear positive correlation, i.e. increasing ISC rate is linked with increasing IC rate. This points to a similar mechanism and would suggest that the vibrational modes responsible for IC also work to enhance ISC, by spin–vibronic coupling and mixing low–lying triplet states and S1. This is consistent with recent findings where considering Tn, as well as S1, T1 and S0 is essential to understand the exciton decay dynamics in TADF emitters.12,16
 | ||
| Fig. 3 Dependence of the IC rate on the ISC rate in the series of compounds 2, 7, 8 and 9, all with the same donor (carbazole) but different acceptors. The size of the dot represents the CT character of the singlet state as evaluated by solvatochromism in the fluorescence spectra. | ||
It is also important to highlight that this effect is not correlated with the charge transfer character of the singlet and triplet states. Fig. 3, shows a graph of the IC rate against the ISC rate for compounds 2, 7, 8 and 9. The charge transfer character of the singlet excited state, as evaluated by fluorescence solvatochromism, is represented by the size of each dot. Compound 2 with the strongest CT character in the series shows intermediate values for both ISC and IC rates. Therefore, the variation in the ISC and IC rates is not due to increasing CT character of the excited state.
The data in Table 1 give significant information regarding the way the change in molecular connectivity influences the intersystem crossing and internal conversion rates. However, we have still not explained what causes the intersystem crossing rate to increase in the “angular” compounds. This crucial point is addressed in the next sections.
Room-temperature phosphorescence in Zeonex films
The steady-state emission spectra of compound 2 dispersed in Zeonex films are shown at 300 K in Fig. 4a and as a function of temperature in Fig. 4b. Under degassed conditions, compound 2 dispersed in Zeonex exhibits dual emission, i.e. strong fluorescence and phosphorescence at room temperature. The room-temperature phosphorescence (RTP) emission is so strong that it is easily measured in a normal fluorimeter, without the need of using time-gated acquisition methods and is higher in intensity than the fluorescence. Strong RTP is also observed for compounds 4, 6, 7 and 8, see Fig. 5, but no RTP is observed in compounds 1, 3, 5, and 9. However, the fluorescence in 9 is strongly quenched by oxygen, see Fig. S3 (ESI†). This indicates that there is a significant delayed fluorescence (DF) contribution to the fluorescence in 9, which involves triplet states. Compound 9 has the smallest ΔES1–T1 energy gap in the series, and triplet harvesting through reverse intersystem crossing may be active. Note that the excited states of 9 have no CT character.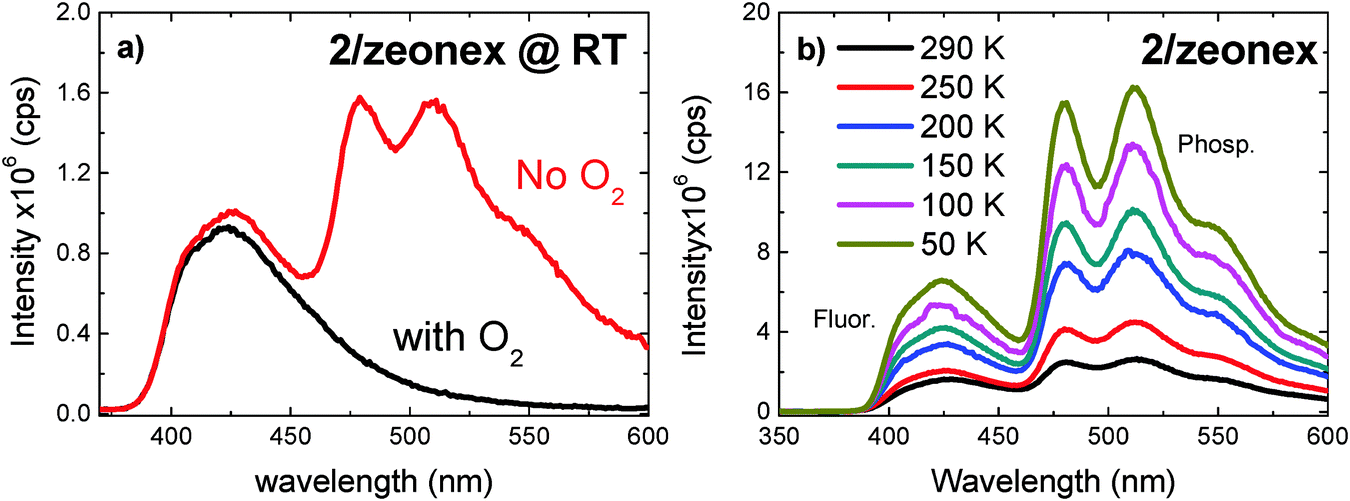 | ||
| Fig. 4 Steady-state emission spectra of 2 in Zeonex at (a) room temperature, and (b) as a function of temperature. | ||
 | ||
| Fig. 5 Steady-state emission spectra for compounds 1–9 dispersed in Zeonex at RT, in vacuum. | ||
A clear trend emerges from Fig. 5. Intense RTP is observed only for the “angular” compounds and is much stronger in 2, 4 and 7 than in 6 and 8. In the latter compound, the low RTP emission is explained by a weaker ISC rate, which could be potentially due to the lack of heteroatoms in the fluorene acceptor. However, this is not the case in 6. For this compound, the ISC rate is strong, and the IC rate is weaker than in 2 and 4. Therefore, compound 6 should be a strong RTP emitter, but it is not. When compared with the other “angular” compounds, 6 is different due to a higher radiative rate in the S1, kf, and a significantly larger ΔES1–T1 gap. Moreover, compound 6 shows a very long phosphorescence lifetime in ethanol at 100 K.36 This indicates that the radiative rate of 6 in the T1 state is significantly slower than in compounds 2 and 4, which contributes to the decrease in the phosphorescence yield.
Remarkably, compounds that are substituted at the C-3 and C-7 positions of the dibenzothiophene-S,S-dioxide unit, i.e. “linear”, show no RTP at all. This is evidenced in Fig. S8 (ESI†), in which compounds 1 and 3 show no RTP emission even when dispersed in poly(4-bromostyrene), a host that is known to enhance triplet formation due to the external heavy-atom effect.38 Therefore, the “angular” substitution and the presence of the low atomic weight heteroatoms, sulfur or nitrogen, are clearly fundamental for strong RTP observation in pure organic materials.
The strong quenching effect of oxygen on the red-shifted emission band in compounds 2, 4, 6, 7 and 8, shown in Fig. S3 (ESI†), confirms that the extra emission band is due to phosphorescence at room temperature. Also in Zeonex, compounds 2 and 4 show significantly shorter fluorescence decays than 1 and 3, see Fig. S4 in the ESI.† This is entirely consistent with the stronger triplet formation yield, see Table 1.
We turn now to discuss compounds 1–4, in which the regioisomerization effect is strongest, to understand the reasons why these compounds have such marked differences in regard to their RTP emission. A summary of the photophysical data in Zeonex films, concerning the observation of RTP, is given in Table 2. With decreasing temperature, the intensities of both the fluorescence and phosphorescence bands increase, (see Fig. 4b). This clearly indicates that the quenching effect due to vibrations is being suppressed. Moreover, while the phosphorescence in the presence of oxygen is, as expected, completely quenched, see Fig. S3 (ESI†), there is practically no difference between the intensities of the fluorescence band of the spectra collected in the presence of oxygen and under degassed conditions (with the exception of compound 9, as discussed). This shows that triplet harvesting due to TADF is not operative in these compounds when dispersed in Zeonex.
 . All were determined in Zeonex films at room temperature
. All were determined in Zeonex films at room temperature
| Φ f ± 0.05 | Φ ph ± 0.05 | Φ ph/Φf ± 0.07 | τ f ± 0.03 (ns) | τ p ± 0.5 (ms) | k f × 108 (s−1) |
k
ISC × 108![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (s−1) b (s−1) |
k
IC × 108b (s−1) |
k Ph (s−1) |
|
|
|---|---|---|---|---|---|---|---|---|---|---|
| a The phosphorescence decay in 2 is bi-exponential, the value given is the average lifetime. b Determined from the triplet yield in solution. | ||||||||||
| 1 | 0.93 | — | — | 4.75 | — | 1.9 | — | 0.1 | ||
| 2 | 0.09 | 0.26 | 2.9 | 1.68 | 34.3a | 0.5 | 1.9 | 3.5 | 23.7 | 5.5 |
| 3 | 0.56 | — | — | 4.71 | — | 1.2 | — | 0.9 | ||
| 4 | 0.06 | 0.14 | 2.3 | 0.98 | 29.8 | 0.6 | 1.9 | 7.6 | 24.7 | 8.9 |

Table 2 shows a consistent trend with the data discussed in Table 1. The intersystem crossing and internal conversion rates are higher in the “angular” isomers when compared with their “linear” counterparts, even in solid Zeonex films.
Calculations
The Kohn–Sham HOMOs and LUMOs for compounds 1–4 are shown in Fig. 1c and Fig. S6 (ESI†). These dominate the lowest transition responsible for the S1 state. All four compounds show charge transfer (CT) character, which can be quantified using the absolute orbital overlap O = 〈|ψ_LUMO|||ψ_HOMO|〉, for which smaller overlaps implies a stronger CT character.39 Therefore, the CT character at the ground state geometry is larger for compound 2 (O = 0.41) than 1 (O = 0.49), and for compound 4 (O = 0.58) than 3 (O = 0.67). To determine the excited state and emission properties, the molecules were optimized in the lowest triplet state using unrestricted Kohn–Sham (UKS) DFT. Importantly, the oscillator strength for the S1 state is over an order of magnitude larger for compound 1 (ES1 = 2.64 eV, f = 0.9053) than 2 (ES1 = 2.75 eV, f = 0.0276). This corresponds to a radiative lifetime for 1 of ∼3 ns, in good agreement with the experimentally determined fluorescence lifetime, shown in Table 1. In contrast, the smaller oscillator strength of 2 corresponds to an excited state lifetime of ∼50 ns. As the fluorescence lifetime reported for compound 2 is <2 ns (Table 1), this illustrates that the S1 state in 2 is susceptible to other processes, namely ISC, consistent with the observation of phosphorescence in 2. In addition, the energetics agrees well with the maxima of the emission spectra shown in Fig. 5. The same trend is observed for structures 3 (ES1 = 2.61 eV, f = 0.9845) and 4 (ES1 = 2.82 eV, f = 0.020). Again, the oscillator strength of the S1 states is much smaller in 4 than in 3, allowing ISC to out-compete radiative decay.To further interrogate the mechanism that is causing enhanced ISC in 2 and 4, we turn to the energy diagram in Fig. 6. Within each pair, the “angular” substituted compounds, 2 and 4, show in the calculations consistently smaller ΔES1–T1 gaps compared to their “linear” counterparts. However, this does not “per se” explain the observation of strong RTP emission (or TADF emission). When comparing across the four structures, compound 4, a good RTP emitter in Zeonex (and a good TADF emitter in ethanol solution at room temperature) has a ΔES1–T1 of 0.65 eV, whereas compound 1, which shows neither RTP nor TADF in any medium, has a similar ΔES1–T1 of 0.64 eV. Therefore, clearly the ΔES1–T1 gap is not the dominant effect, and probably more pertinent is the gap T1–Tn, between the manifold of triplet states closest to the S1 state, from where a clear pattern emerges between the “linear” and “angular” compounds.
 | ||
| Fig. 6 Energy level diagram of the singlet and triplet states in compounds 1–4, obtained from TDDFT calculations. | ||
For both 1 and 3, which do not exhibit RTP or TADF, the gap between the T1 and T2 states is large (>0.7 eV), whereas for 2 and 4, both good RTP/TADF emitters, the gap between T1 and T2 is small (∼0.3 eV). This provides the potential for efficient internal conversion between the triplet states and a decay route of the initially populated triplet to the lowest, T1, triplet state to emit, according to Kasha's rule. In this case, recent work has also demonstrated that the spin–vibronic mechanism, involving both spin–orbit coupling and non-adiabatic coupling31,32 significantly increases the rate of ISC. The close proximity of all of the triplet states makes this mechanism much more likely in 2 and 4, and in combination with the slower radiative rates of the S1 state in 2 and 4, these data sets provide strong support for the observation and mechanism of RTP. This mechanism also explains why compounds 2 and 4 are good TADF emitters in ethanol. In this case, the strong solvent polarity shifts the S1 to lower energies, without affecting significantly the energy of the T1 state, which does not have CT character. This makes the ΔES1–T1 gap smaller and facilitates rISC. To test this hypothesis, compounds 2 and 4 were dispersed in hosts of stronger polarity than Zeonex aiming to shift the S1 to lower energies. With the correct tuning of the energy levels dual luminescence, RTP and TADF, is observed, as shown below. This again casts the TADF and RTP mechanisms into a common conceptual framework, which had not been described previously.
Dual luminescence, RTP and TADF, in organic hosts
Here, compounds 7 and 9, and two other large band gap hosts diphenylphosphine oxide-4-(triphenylsilyl)phenyl (TSPO1),40 and poly(4-bromostyrene),38 are used as hosts in blends with compounds 2 and 4; see Fig. S7 (ESI†) for the molecular structures of TSPO1,40 and poly(4-bromostyrene). All of these host materials have a triplet energy higher than 2 and 4, and are able to act as donors to promote energy transfer to compounds 2 and 4. TSPO1 in particular has been used as a host in blue OLEDs.40In Fig. 7, the steady-state emission spectra of compound 4 in blends with the hosts mentioned above are compared in vacuum and air. The emission spectra are obtained at 300 K in a normal fluorimeter. The presence of dual luminescence is clearly observed. The phosphorescence band in 4 peaks at around 515 nm in the four hosts, 7 nm below the phosphorescence maximum observed in Zeonex. The fluorescence band peaks at 466 nm for blends of 4 with 7 and 9 and at 480 nm in TSPO1 and PBrS. This represents a shift of 16 and 30 nm to the red in comparison with the fluorescence maximum in Zeonex at 450 nm.
 | ||
| Fig. 7 Steady-state emission spectra of compound 4 dispersed in different hosts (a) 7, (b) 9, (c) TSPO1 and (d) poly(4-bromostyrene), identified as PBrS, in vacuum (red) and under air equilibrated (black) conditions. | ||
Interestingly, when compared with the emission in Zeonex, where no variation in the intensity of the fluorescence band is observed when oxygen is removed, in blends of 4 with 7, 9, TSPO1 and in PBrS, a marked increase in the fluorescence intensity occurs upon removing oxygen. This is observed together with the phosphorescence emission, and indicates that triplet states are being harvested in two different ways: one due to reverse intersystem crossing, giving delayed fluorescence (DF), and the other by directly decaying radiatively to the ground state, giving rise to room-temperature phosphorescence (RTP). For 4 dispersed in TSPO1, the overall luminescence decreases more than four-fold upon introducing oxygen.
The S1 and T1 energies of 4 in Zeonex, determined from the onset of the fluorescence and phosphorescence bands, are 3.05 eV and 2.63 eV, respectively. For blends of 4 in 7 and 9, the S1 shifts to 2.92 eV and the triplet state shows no shift. In blends of 4 with TSPO1 and PBrS, both the S1 and T1 energies shift to 2.82 eV and 2.53 eV, respectively. Therefore, compared with the situation in Zeonex, the S1 energy of 4 has shifted by 0.13 eV in 7 and 9, and by 0.23 eV in TSPO1 and PBrS. However, the triplet energy exhibits no shift for blends of 4 with 7 and 9, and shifts by only 0.1 eV in TSPO1 and PBrS. Therefore, the ΔES1–T1 gap decreases from 0.42 eV in Zeonex to approx. 0.29 eV in all the other hosts. This explains the turn-on of the delayed fluorescence, and the observation of dual luminescence, TADF and RTP, as shown in Fig. 8. However, it is stressed that given the manifold of triplet states below S1 in the case of 4, we cannot determine at present from which triplet the S1 is being thermally activated, only that the significant quenching of fluorescence on addition of oxygen reveals an emission mechanism which passed through the triplet states.
 | ||
| Fig. 8 (a) Time-resolved luminescence decay of 4 in TSPO1, in vacuum (red) and in the presence of oxygen (black), showing the decay of the prompt fluorescence and long-lived TADF and RTP. (b) Time-resolved spectra of 4 in TSPO1, showing the spectra of the prompt fluorescence (blue), and long-lived TADF and RTP (red). Both spectra are compared with the steady-state emission of 4 in Zeonex. (c and d) Steady-state emission spectra of 4 in TSPO1 (c) and 4 in PBrS as a function of temperature (d). The contributions of TADF and RTP are clearly observed in both situations, confirming the presence of dual long-lived luminescence. | ||
The time-resolved luminescence of compound 4 in TSPO1 is shown in Fig. 8a. The luminescence decays, obtained in vacuum and in the presence of oxygen, show a fast decay component due to prompt fluorescence. This component shows no variation in the presence of oxygen. A long-lived component due to TADF and RTP is also observed. This is completely quenched by oxygen, confirming that it originated from triplet states. In Fig. 8b, the time-resolved emission spectra obtained in the prompt fluorescence and TADF/RTP regions are compared with the steady-state emission obtained in Zeonex. The spectrum at early times is dominated by the fluorescence band, as expected, whereas the delayed component is dominated by TADF and RTP, appearing with a emission spectrum similar to the prompt fluorescence and phosphorescence.
Fig. 8c and d show the steady-state emission spectra of 4 dispersed in TSPO1 and PBrS, respectively. In both cases, dual emission is observed, with a fluorescence band peaking at 480 nm, which is due to TADF, as confirmed by its temperature dependence, decreasing in intensity at low temperatures, and well-resolved phosphorescence peaking at 523 nm in TSPO1, and at 528 nm in PBrS, which, in contrast, increases in intensity with decreasing temperature. This convincingly confirms the dual nature of the luminescence of compound 4 in these hosts. Similar observations were made for blends of compound 2 and are shown in Fig. S9–S11 (ESI†).
Conclusions
In summary, we have shown that the ISC and rISC mechanisms are enhanced in pure organic molecules by using a simple strategy based on changing the molecular connectivity of carbazole, diphenylamine and fluorene donors to a dibenzothiophene-S,S-dioxide acceptor, from “linear” to “angular”. The latter substitution of the A unit, breaks effective molecular conjugation and shifts the singlet and triplet states to higher energy. However, more importantly, the “angular” substitution gives rise to a small energy gap between triplet levels below S1 and to a slower radiative rate, making the S1 state susceptible to other decay processes such as ISC and IC. In contrast, in the “linear” compounds the radiative decay rate of the S1 state is an order of magnitude faster and in addition, the energy separation between the consecutive triplet states is much larger. The small energy difference between triplet states below S1, present in the “angular” isomers has profound implications for the ISC and rISC mechanisms. Non-adiabatic vibronic coupling between the T1 and the upper triplet levels lying below S1, creates a fast T1–Tn equilibrium, and strong mixing between these states. This enhances coupling between S1 and T1, as described recently.32 In the absence of the strong mixing between the triplet states lying below S1, due to the larger energy difference between T1 and T2, for compounds 1 and 3, the ISC is not efficient, and in combination with the increased S1 radiative decay rate produces a lower triplet yield, and consequently RTP and TADF are not observed, even when 1 and 3 are dispersed in poly(4-bromostyrene), a host in which the external heavy-atom effect is used to promote intersystem crossing. Finally, using the host to fine-tune the energy difference between S1 and T1, dual long-lived luminescence, TADF and RTP in 2 and 4 is activated simultaneously in different hosts. This work, therefore, provides guidelines that can be used to design novel molecules showing alternative routes to harvest triplet states and enhance their potential for application in photonic and optoelectronic devices.Experimental methods
Optical spectroscopy
Absorption and emission spectra were collected using a UV-3600 double beam spectrophotometer (Shimadzu), and a Fluorolog fluorescence spectrometer (Jobin Yvon), respectively. Time-resolved fluorescence decays were collected using the picosecond time-correlated single-photon counting technique (impulse response function, IRF: 21 ps). The vertical polarization excitation source was the second or third harmonic from a picosecond Ti:sapphire laser (Coherent). Emission was collected using a polarizer at magic angle and detected using a double subtractive monochromator, (Acton Research Corporation), coupled to a microchannel plate photo multiplier tube (Hamamatsu R3809U-50). Signal acquisition was performed using a TCSPC module (Becker & Hickl SPC-630). Temperature-dependent measurements were acquired using a model liquid nitrogen cryostat (Janis Research). Phosphorescence, time-resolved spectra and decays were recorded using nanosecond gated luminescence and lifetime measurements (from 400 ps to 1 s) using a high energy pulsed Nd:YAG laser emitting at 355 nm (EKSPLA), or a pulsed N2 laser emitting at 337 nm. Emission was focused onto a spectrograph and detected on a sensitive gated iCCD camera (Stanford Computer Optics) having sub-nanosecond resolution. Solution measurements used concentrations in the 10−5–10−4 M range, and samples were degassed using 5 freeze/thaw cycles, when required. Films for optical characterization were prepared in Zeonex and other hosts by drop-casting or spin-coating in a quartz substrate with an emitter/host ratio of 1:20 (w/w).
Flash photolysis
Triplet-formation quantum yields and lifetimes were measured using a flash photolysis setup composed of a LKS. 60 ns laser photolysis spectrometer from Applied Photophysics, with a Brilliant Q-Switch Nd:YAG laser from Quantel, using the third harmonics (λex = 355 nm, laser pulse half-width equal to 6 ns). First-order kinetics was observed for the decay of the lowest triplet state (T–T annihilation was prevented by the low excitation energy and/or low analyte concentration), see Fig. S12 in the ESI.† The triplet lifetimes were measured at an absorbance ca. 0.1 in degassed n-hexane. The transient spectra were obtained by monitoring the optical density change at intervals of 10 nm over the 280–600 nm range and averaging at least 5 decays at each wavelength. The ΦT values were measured by comparing the change in the absorbance of the β-carotene triplet (λ = 510 nm) sensitized by energy transfer from a substrate, ΔOD, and from 2-naphthone, ΔOD(N), used as a standard (ΦT(N) = 0.46)41 under the same experimental conditions (absorbance at 355 nm and concentration of β-carotene).42 The efficiency of energy transfer from the substrate, PET, and from 2-naphthone, PET(N), was also taken into account by measuring the triplet lifetime of the donor in the absence (τ0T) and in the presence (τT′) of β-carotene: | (1) |
 | (2) |
Computational details
The geometries of compounds 1–4 were optimized using (U)DFT(PBE0)43–45 and a 6-31g* basis set as implemented within the Q-Chem code.46 The lowest six singlet and six triplet states were calculated in every TDDFT calculation. The emission properties, calculated using TDDFT, were mimicked by optimizing the gas-phase molecules 1–4 in their lowest T1 states using UKS DFT. These T1 optimized geometries were also confirmed as true minima by frequency calculations.Author contributions
R. H. performed the majority of the optical measurements with P. L. S.; M. K. E. and E. C. performed photoinduced absorption studies and luminescence spectroscopy in Zeonex. J. A. and J. C. L. determined the fluorescence yields in Zeonex and the triplet formation yields. P. D. performed cyclic voltammetry experiments. M. N. B. S. investigated the temperature dependence of the luminescence of compound 2 in Zeonex. J. W. synthesized molecules 1–4 under the guidance of M. R. B., M. A. F. performed preliminary calculations and revised the manuscript. The full physical model was devised by J. C. L., T. J. P. and F. B. D., and it was theoretically tested within a quantum chemistry framework by T. N. and T. J. P., M. R. B. initially designed the regioisomeric emitter structures; F. B. D. conceived the original idea and the optical experiments for investigation, and analysed experimental data. F. B. D. also wrote the manuscript, which was revised by T. J. P., J. W., M. R. B., and J. C. L.Data availability
Data supporting this publication are openly available under the ‘Open Data Commons Open Database License’. Additional metadata are available at: http://dx.doi.org/10.17634/153015-3. Please contact Newcastle Research Data Service at rdm@ncl.ac.uk for access instructions.Acknowledgements
R. H. acknowledges a PhD grant funded by Durham University. P. L. S. thanks the CAPES Foundation, Ministry of Education of Brazil, for a PhD studentship, Proc. 12027/13-8, Science without Borders Program and thanks to networking action funded from the European Union's Horizon 2020 research and innovation programme under grant agreement No 691684. TJP acknowledges the EPSRC, Project EP/N028511/1 for funding. P. D. thanks the EU for a Marie Curie Fellowship H2020 research and innovation programme under grant agreement No. 659288. M. K. E. acknowledges EU's Horizon 2020 for funding the PHEBE project under grant no. 641725. F. B. D. acknowledges Samsung-SAIT for funding this work using their Global Research Outreach (GRO) Program, and also the EPSRC for funding under grant number EP/L02621X/1. We thank Dr Kathryn C. Moss and Professor Igor F. Perepichka for the initial synthesis of some of the emitters.References
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 12001 CrossRef PubMed.
- M. S. Kwon, J. H. Jordahl, A. W. Phillips, K. Chung, S. Lee, J. Gierschner, J. Lahann and J. Kim, Chem. Sci., 2016, 7, 2359–2363 RSC.
- Y. Feng, J. Cheng, L. Zhou, X. Zhou and H. Xiang, Analyst, 2012, 137, 4885–4901 RSC.
- O. S. Wolfbeis, Adv. Mater., 2008, 20, 3759–3763 CrossRef CAS.
- L. Jethi, M. M. Krause and P. Kambhampati, J. Phys. Chem. Lett., 2015, 6, 718–721 CrossRef CAS PubMed.
- A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS PubMed.
- Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 6270–6273 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y. C. Wu, P. Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- D. Chaudhuri, E. Sigmund, A. Meyer, L. Rock, P. Klemm, S. Lautenschlager, A. Schmid, S. R. Yost, T. Vanvoorhis, S. Bange, S. Hoger and J. M. Lupton, Angew. Chem., Int. Ed., 2013, 52, 13449–13452 CrossRef CAS PubMed.
- T. Kobayashi, A. Niwa, K. Takaki, S. Haseyama, T. Nagase, K. Goushi, C. Adachi and H. Naito, Phys. Rev. Appl., 2017, 7, 34002 CrossRef.
- Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817–20830 RSC.
- W. Ratzke, L. Schmitt, H. Matsuoka, C. Bannwarth, M. Retegan, S. Bange, P. Klemm, F. Neese, S. Grimme, O. Schiemann, J. M. Lupton and S. Höger, J. Phys. Chem. Lett., 2016, 7, 4802–4808 CrossRef CAS PubMed.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080, DOI:10.1002/advs.201600080.
- V. A. Dediu, L. E. Hueso, I. Bergenti and C. Taliani, Nat. Mater., 2009, 8, 707–716 CrossRef CAS PubMed.
- S. Reineke, N. Seidler, S. R. Yost, F. Prins, W. A. Tisdale and M. A. Baldo, Appl. Phys. Lett., 2013, 103, 093302 CrossRef.
- S. R. Forrest, M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley and M. E. Thompson, Nature, 1998, 395, 151–154 CrossRef.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef PubMed.
- T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS PubMed.
- S. Hirata and M. Vacha, Adv. Opt. Mater., 2017, 5, 1600996 CrossRef.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- S. Hirata, K. Totani, H. Kaji, M. Vacha, T. Watanabe and C. Adachi, Adv. Opt. Mater., 2013, 1, 438–442 CrossRef.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- J. Xu, A. Takai, Y. Kobayashi and M. Takeuchi, Chem. Commun., 2013, 49, 8447–8449 RSC.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem., 2016, 1, 592–602 CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- J. Gibson and T. J. Penfold, Phys. Chem. Chem. Phys., 2017, 19, 8428–8434 RSC.
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef CAS PubMed.
- T. Palmeira and M. N. Berberan-Santos, J. Phys. Chem. C, 2017, 121, 701–708 CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- V. Jankus, P. Data, D. Graves, C. McGuinness, J. Santos, M. R. Bryce, F. B. Dias and A. P. Monkman, Adv. Funct. Mater., 2014, 24, 6178–6186 CrossRef CAS.
- S. Reineke and M. A. Baldo, Sci. Rep., 2014, 4, 3797 CrossRef PubMed.
- T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535–13544 CAS.
- I. Lee and J. Y. Lee, Org. Electron., 2016, 29, 160–164 CrossRef CAS.
- A. A. Lamola and G. S. Hammond, J. Chem. Phys., 1965, 43, 2129–2135 CrossRef CAS.
- C. V. Kumar, L. Qin and P. K. Das, J. Chem. Soc., Faraday Trans. 2, 1984, 80, 783–793 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V Lawler, S. V Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tc01958k |
| This journal is © The Royal Society of Chemistry 2017 |