
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dye functionalized-ROMP based terpolymers for the use as a light up-converting material via triplet–triplet annihilation†
M.
Hollauf
a,
P. W.
Zach
b,
S. M.
Borisov
 b,
B. J.
Müller
b,
D.
Beichel
a,
M.
Tscherner
c,
S.
Köstler
c,
P.
Hartmann
c,
A.-C.
Knall
a and
G.
Trimmel
*a
b,
B. J.
Müller
b,
D.
Beichel
a,
M.
Tscherner
c,
S.
Köstler
c,
P.
Hartmann
c,
A.-C.
Knall
a and
G.
Trimmel
*a
aInstitute for Chemistry and Technology of Materials, NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: gregor.trimmel@tugraz.at
bInstitute for Analytical Chemistry and Food Chemistry, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
cMATERIALS – Institute for Surface Technologies and Photonics, Joanneum Research, Franz-Pichler-Straße 30, 8160 Weiz, Austria
First published on 19th July 2017
Abstract
In this paper we introduce and compare different terpolymers comprising covalently attached sensitizer and emitter chromophores for the use as a light up-converting material via triplet–triplet annihilation (TTA). Using the advantages of ring opening metathesis polymerisation it was possible to prepare five different polymer architectures in order to investigate the influence of polymer architecture and chromophore arrangement on the photon up-conversion behaviour. First, two new monomers containing the chromophores have been synthesized and characterized in regard to their photophysical characteristics suitable for triplet–triplet annihilation dye pair. For this purpose, a derivative of Pt(II) meso-tetraphenyltetra(tert-butyl)benzoporphyrin as sensitizer and a perylenediester as emitter were attached to norbornene moieties via ester linkages. Polymerisations of these monomeric chromophores were performed in combination with dimethyl 5-norbornene-2,3-dicarboxylate as matrix monomer. Depending on the location of the dye molecules on the polymer chain, large differences in the TTA efficiency were observed. The best quantum yields have been achieved with a completely statistically distributed terpolymer showing an up-conversion quantum yield of up to 3% in solution.
Introduction
Anti-Stokes photoluminescence, the emission of photons at higher energy than the absorbed ones, also known as photon up-conversion (UC), is a very interesting effect due to the wide range of possible applications such as bioimaging, optical data storage, display devices, high-resolution optical microscopy, drug delivery, up-conversion layers for photovoltaics and many others.1–3 The field of anti-Stokes fluorescence imaging was traditionally dominated by inorganic crystals doped with luminescent lanthanide ions and by organolanthanide complexes, where the UC scheme requires the sequential absorption of two or more photons exciting the metastable states of the emitting ions.4,5 Today the most frequent ways to achieve a photon up-conversion are through second harmonic generation or two photon absorption (TPA).6 A big disadvantage of the TPA mechanism is that it usually requires excitation irradiances in the order of MW cm−2.7 In contrast to that, the UC based on triplet–triplet annihilation (TTA) is a promising low-power up-conversion process which already shows delayed fluorescence using excitation irradiances of less than 100 mW cm−2 (solar energy is enough).8,9 This effect requires two dyes which are called sensitizer and emitter or are also known as annihilator and acceptor. First the sensitizer absorbs incident photons that allow it to occupy its excited singlet state (1S*) that quickly relaxes into a metastable excited triplet state (3S*) caused by a spin-forbidden intersystem crossing (ISC). In presence of suitable emitter molecules, that have excited triplet levels of similar energy, a triplet–triplet energy transfer (TTET) from the sensitizer to the emitter takes place. Annihilation of the two excited emitter species (3E*) generates one emitter molecule in the singlet excited state (1E*) which eventually results in TTA up-conversion.Here one molecule relaxes to its ground state in a radiationless process while the other one shows delayed fluorescence. Hence, the TTA-UC represents another possibility to obtain anti-Stokes fluorescence. The corresponding Jablonski diagram is shown in Fig. 1.
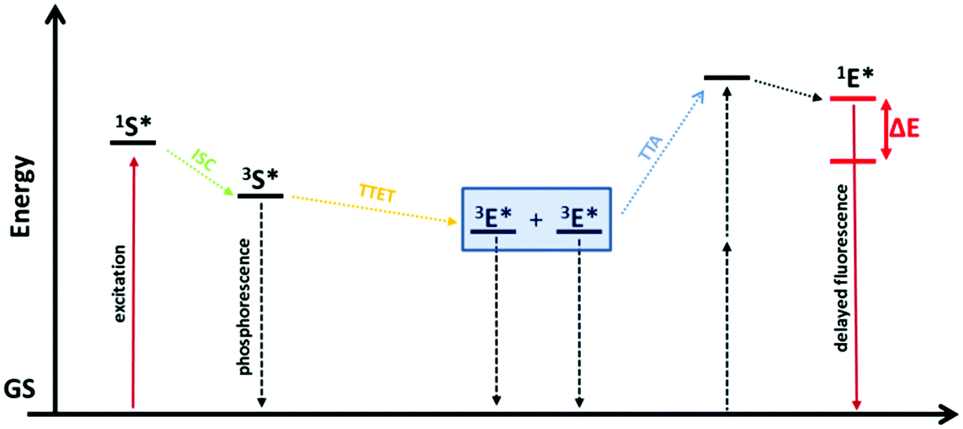 | ||
| Fig. 1 Jablonski energy level-diagram of TTA up-conversion. S stands for sensitizer and E for emitter. Coloured solid lines represent the intended radiative processes (absorption and emission). Vertical dashed lines represent undesired non radiative and radiative decay pathways. Slanted dotted arrows represent cascading energy transfer processes. GS denotes the ground states (for simplicity drawn at the same energy level) and ΔE is the energy difference between incident and emitted light.1 | ||
Regarding the chromophores, numerous chromophore combinations resulting in efficient TTA-UC have been established.10,11 The most commonly used dyes are perylenes, anthracenes, rubrene derivatives or borodipyrroles as emitters and porphyrin derivatives, supramolecular chromophores, phthalocyanines or cyclometallated Pt(II) complexes as sensitizers to name just a few.12–17 To obtain photon up-conversion, a chromophore pair, where the excited triplet state of the sensitizer is higher than the one of the emitter, has to be found. This is crucial for an efficient triplet–triplet energy transfer (TTET).
Although TTA is known in solution since over 50 years18 it has only recently been possible to realize this effect in solid polymeric films. The first examples were prepared in 2007 by the working group of Castellano et al. and they used blends of Pd 2,3,7,8,12,13,17,18-octaethylporphyrin (PdOEP) as sensitizer and diphenylanthracene (DPA) as emitter in an ethylene oxide/epichlorohydrin copolymer to obtain a rubber like matrix.19 Recent studies show that this effect is also possible with active polymeric structures,20–25 liposomes,26,27 ionogels,28 oil in water micro emulsions,29–31 micelles,17 dendrimers,32 nanocapsules,33–35 and many more.1,7,36 The translational and rotational mobility of the chromophores is essential for an efficient photon light up-converting process, due to the fact that mobility influences all the implemented energy transfer processes. Hence, there are only a few examples where covalently bound dye molecules are involved for efficient TTA quantum yields. Nevertheless, a close distance and a defined position of sensitizer and emitter to each other are at least equally important to obtain a high TTA photon emission. There are a few examples of a direct covalent coupling of both dyes.37–40 Only very recent studies investigated the covalent binding of one of the dyes to a polymeric matrix41–43 or even both dyes2,44 considering the fact that the sensitizer and emitter need to be in close proximity in order to efficiently undergo short-range interactions such as TTET and TTA (Fig. 1). Following this idea, the influence of different emitter to sensitizer ratios on the TTA efficiency was studied in methacrylate based materials synthesised by free radical polymerisation.2 Alternatively, the group of Ghiggino prepared star polymers with the sensitizer in the centre and emitter functionalized arms (2 and 6 arms) using RAFT as controlled polymerisation technique45 demonstrating that the placement of dye molecules at distinct places using controlled polymerisation techniques or post-functionalisation approaches is a strategy to study the TTA-process in polymeric materials in detail. A straightforward synthetic pathway to prepare such dye functionalized polymers with a defined structure is ring opening metathesis polymerisation (ROMP). A big advantage of ROMP is that it is a powerful method for the synthesis of novel materials with well-defined structures such as statistically distributed copolymers, block copolymers, alternating copolymers, crosslinked copolymers, end-group functionalized polymers or graft copolymers to name just a few.46 It is also known for its versatility, functional group tolerance and for the preparation of special polymers.47 In addition, ROMP offers different possibilities for the preparation of dye functionalized polymers for different applications in sensing, bio-imaging and other optoelectronic applications.48 The easiest way for this is to use dye-functionalized monomers, e.g. as shown by us in the synthesis of fluorescent polymeric materials using naphthalimide49 or xanthene50 containing norbornenes.
In this paper, we will investigate how the polymer architecture will influence the TTA efficiency using ROMP as polymerisation technique. In contrast to the work of Boutin,44 we will introduce both the sensitizer and the emitter via functionalized monomers. This allows the precise placement of both chromophore types in combination with a matrix monomer into defined terpolymer structures. As TTA-chromophore pair we chose the system: Pt(II) meso-tetraphenyltetra(tert-butyl)benzoporphyrin (TPTBTBP Pt) in combination with diisobutyl 3,9-perylene dicarboxylate, Solvent Green 5,51 due to a very high molar absorption coefficient of the former and matching energies of the triplet states of both dyes. As TPTBTBP Pt shows reduced solubility due to π–π stacking of the porphyrin rings,52 the benzo moieties were equipped with additional tert-butyl groups to increase solubility and this structure was chosen as precursor for the synthesis of the corresponding sensitizer monomer in this study. Both monomers are depicted in Fig. 2.
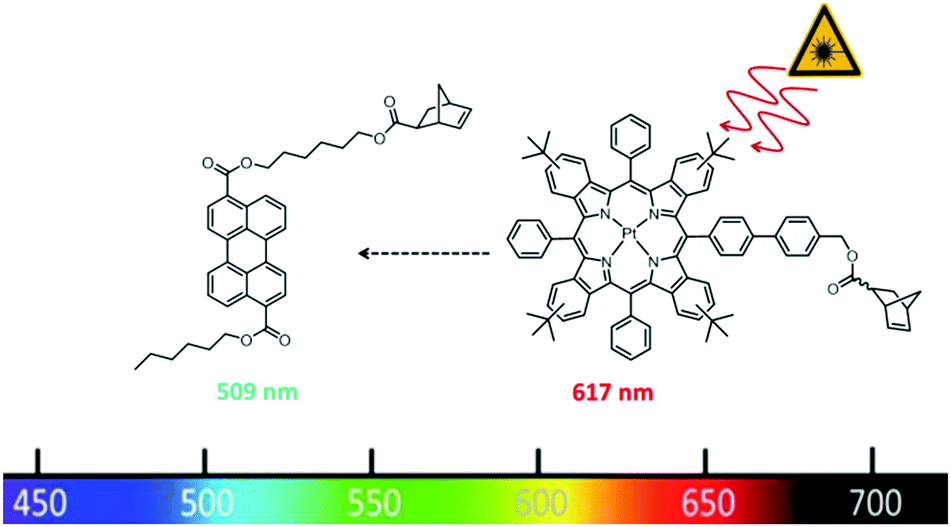 | ||
| Fig. 2 Perylene dicarboxylate and Pt(II) meso-tetraphenyltetra(tert-butyl)benzoporphyrin functionalized monomers used in this study. | ||
Results and discussion
Synthesis of emitter monomer
To ensure a covalent attachment of the chromophores to the polymer backbone, a norbornene moiety has to be linked to the dyes (cf.Fig. 2).The synthesis scheme for the preparation of the desired perylene emitter monomer is depicted in Fig. 3. The first reaction step was the saponification of diisobutyl 3,9-perylene dicarboxylate with potassium hydroxide to obtain potassium dicarboxylate (PDC). The second step was the unsymmetrical esterification with 1-bromobutane and 6-bromo-1-hexanol in water and tetra-n-butylammonium bromide as a phase transfer catalyst. Due to the differences in polarity of educts and products, the reaction products precipitate out of the reaction mixture and can be isolated by a simple filtration. The unsymmetrical perylene diester (PDE) can be separated from the two symmetrical esters via column chromatography. For the last step, PDE was added slowly to a solution of 5-norbornene-2-carbonyl chloride, which was prepared beforehand in the same flask by an Diels–Alder reaction of cyclopenta-1,3-diene with 2-propenoyl chloride. The emitter monomer PDEmon was finally isolated using column chromatography. The photophysical properties of PDEmon will be discussed later.
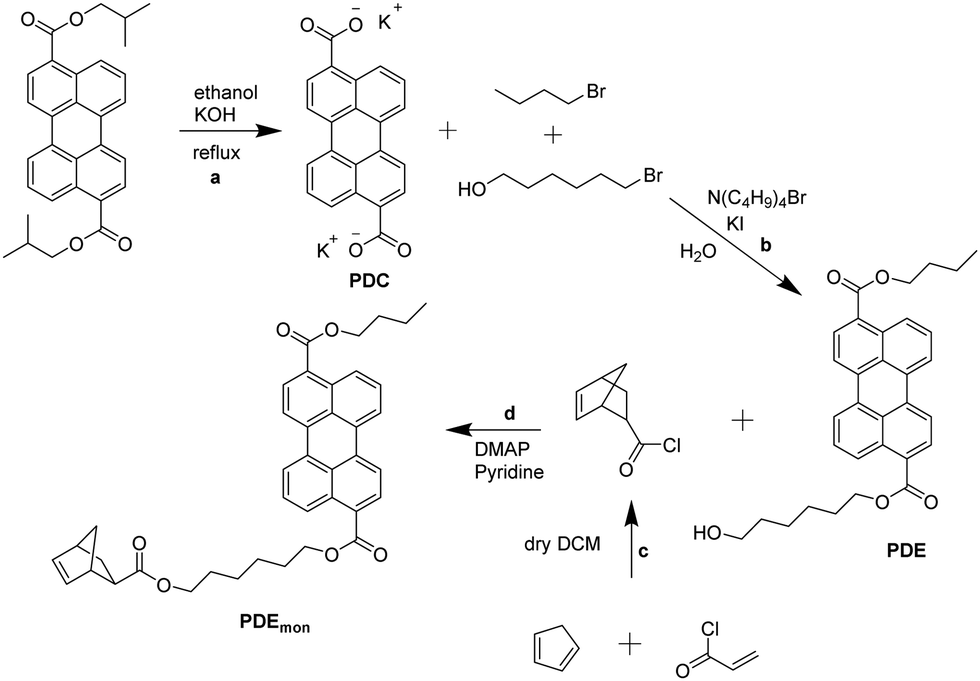 | ||
| Fig. 3 Synthetic pathway of PDEmon: (a) saponification, (b) esterification in water with a phase transfer catalyst, (c) Diels–Alder reaction, (d) Einhorn variation of the Schotten–Baumann reaction. | ||
Synthesis of sensitizer monomer
The overall reaction scheme for the sensitizer monomer TPTBTBP Ptmon is shown in Fig. 4. There are many techniques for the preparation of porphyrin ligands, e.g. the original synthesis by Rothemund53 or the often used route by Adler and Longo.54 For benzoporphyrins, the Lindsey method became popular.55–57 Alternatively, the template condensation allows a preparation of tetrabenzoporphyrins in a single step starting from phthalimide and phenylacetic acid.58 Recently, Hutter et al.59 demonstrated that substitution of phthalimide with 1,2-dicyanobenzene results in analytically pure benzoporpyhrins. This synthetic methodology was adapted to prepare mono-bromo-substituted benzoporphyrin for further modification. Therefore, phenyl acetic acid, zinc 4-bromophenylacetate and 4-(tert-butyl)phthalonitrile were melted together to obtain the mono bromo functionalized meso-tetraphenyl tetra(tert-butyl)benzoporphyrin complex with Zn(II). The introduction of tert-butyl moieties in the benzo core is highly advantageous, since these big sterically demanding moieties prevent noncovalent interactions of the aromatic macrocycles between each other (π–π stacking) which would lead to poor solubility. However, due to the formation of different side products with similar physical properties, i.e. multiple substituted bromo derivatives, laborious work-up steps, such as multiple precipitations and column chromatography are required. The moderate yield of only 6.4% is rather good considering that the complete mono-functionalized benzoporphyrin ring system is formed in this single step reaction. Further advantages are the low costs of starting materials and simplicity of the procedure. Although Zn(II) benzoporpyhrins can be used as sensitizers for TTA up-conversion,60 they neither possess high molar absorption coefficients for the Q-band nor show good energy match of the triplet excited state with that of the perylene emitter. Therefore, the Zn(II) complex was converted into the Pt(II) complex (BrTPTBTBP Pt) in two steps. First, demetallation of BrTPTBTBP Zn is conducted in acidic solution, followed by the metallation with Pt(C6H5CN)Cl2 in the second step. For this reaction step extreme caution is required; as during the metallation the evolving HCl can protonate the remaining free ligand which then precipitates and is removed from the reaction due to low solubility in cumene. Therefore, the emerging HCl was removed via an inert gas flow through the reaction mixture. The norbornene moiety was introduced via two further reaction steps. A Suzuki-cross coupling of BrTPTBTBP Pt with 4-(hydroxymethyl)phenylboronic acid was performed, followed by an Steglich esterification with the corresponding norbornene carboxylic acid in dichloromethane. The Steglich esterification was chosen because of its very mild reaction conditions.61 The original Zn porphyrine derivative differs significantly in the photophysical properties from the pure ligand and also from the Pt-containing ring, thus the demetallation and platination steps can be easily followed by measuring the absorption spectra, as shown in Fig. 4, right image. The sensitizer monomer TPTBTBP Ptmon shows optical characteristics similar to previously published TPTBP Pt59 and has identical absorption characteristics as the two platinated precursors (BrTPTPTBP Pt and HMP TPTPTBP Pt) but displays a hypsochromic shift compared to the free ligand BrTPTPTBP and the zinc derivative BrTPTPTBP Zn. The desired monomer exhibits a global absorption maximum at 429 nm (Soret-band), while the Q-bands appear at 567 and 618 nm. The molar absorption coefficient for the Soret-band is 155![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1. Due to the heavy atom effect, Pt-metalloporphyrins allow efficient intersystem crossing into excited triplet states leading to phosphorescence emission. The phosphorescence emission for this compound is in the near infrared area (λmax 775 nm, quantum yield ϕ = 63%). A comparison of the absorption and emission properties of TPTBTBP Ptmon with those of PDEmon is displayed in Fig. 5. The perylene monomer shows a global absorption maximum at 464 nm, a blue shifted local maximum at 438 nm and a slight shoulder at 413 nm with half of the intensity of the global maximum. The molar absorption coefficient for its global maximum is 29000 M−1 cm−1. The compound is highly fluorescent, emitting green light (λmax 509 nm, quantum yield ϕ = 97%). Furthermore, it has to be noted that the emission peak area is quite broad. Two shoulders are found at 476 nm and at 540 nm with half of the intensity of the global maximum.
500 M−1 cm−1. Due to the heavy atom effect, Pt-metalloporphyrins allow efficient intersystem crossing into excited triplet states leading to phosphorescence emission. The phosphorescence emission for this compound is in the near infrared area (λmax 775 nm, quantum yield ϕ = 63%). A comparison of the absorption and emission properties of TPTBTBP Ptmon with those of PDEmon is displayed in Fig. 5. The perylene monomer shows a global absorption maximum at 464 nm, a blue shifted local maximum at 438 nm and a slight shoulder at 413 nm with half of the intensity of the global maximum. The molar absorption coefficient for its global maximum is 29000 M−1 cm−1. The compound is highly fluorescent, emitting green light (λmax 509 nm, quantum yield ϕ = 97%). Furthermore, it has to be noted that the emission peak area is quite broad. Two shoulders are found at 476 nm and at 540 nm with half of the intensity of the global maximum.
 | ||
| Fig. 4 Left: Synthetic pathway of TPTBTBP Ptmon (a) melting process, (b) demetallation, (c) platination, (d) Suzuki-cross coupling, (e) Steglich esterification; right: normalized absorption spectra of porphyrin derivatives. The monomer (TPTBTBP Ptmon) exhibits the absorption peak maximum at 429 nm with a molar absorption coefficient of 155500 M−1 cm−1. | ||
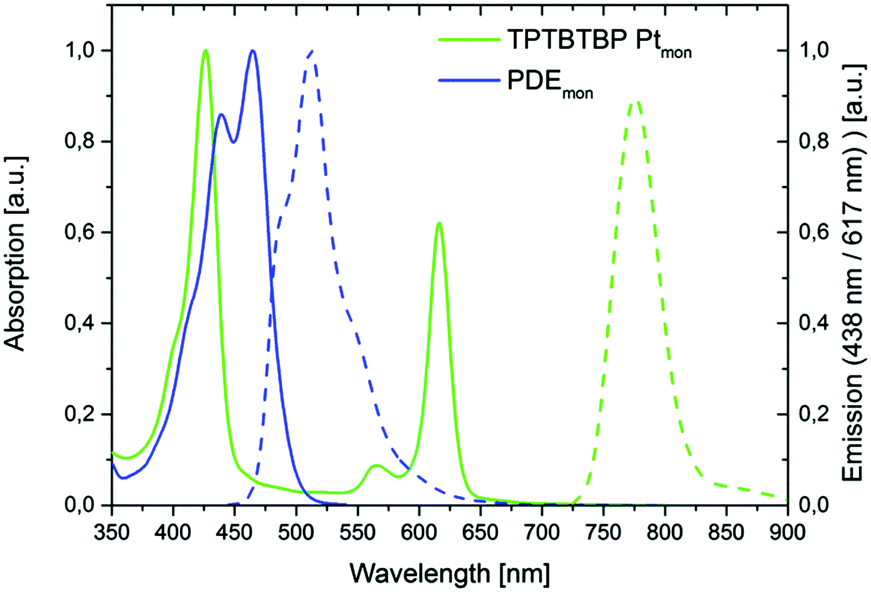 | ||
| Fig. 5 Normalized absorption (solid lines) and emission (dashed lines) spectra of PDEmon and TPTBTBP Ptmon PDEmon: absorption peak maximum exhibits at 464 nm with a molar absorption coefficient of 29200 M−1 cm−1TPTBTBP Ptmon: absorption peak maximum of the monomer exhibits at 429 nm with a molar absorption coefficient of 155500 M−1 cm−1. | ||
Polymers
Five different polymer architectures have been prepared with these two monomers and dimethyl-5-norbornene-2,3-dicarboxylate (N-DME) as matrix monomer in order to investigate the influence of the polymer architecture on the triplet–triplet annihilation induced photon up-conversion (Fig. 6). Due to its stability and wide functional group tolerance [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene] dichloro (3-phenyl-1H-inden-1-ylidene) (pyridyl) ruthenium(II) (also known as M31) was used for all polymerisations.62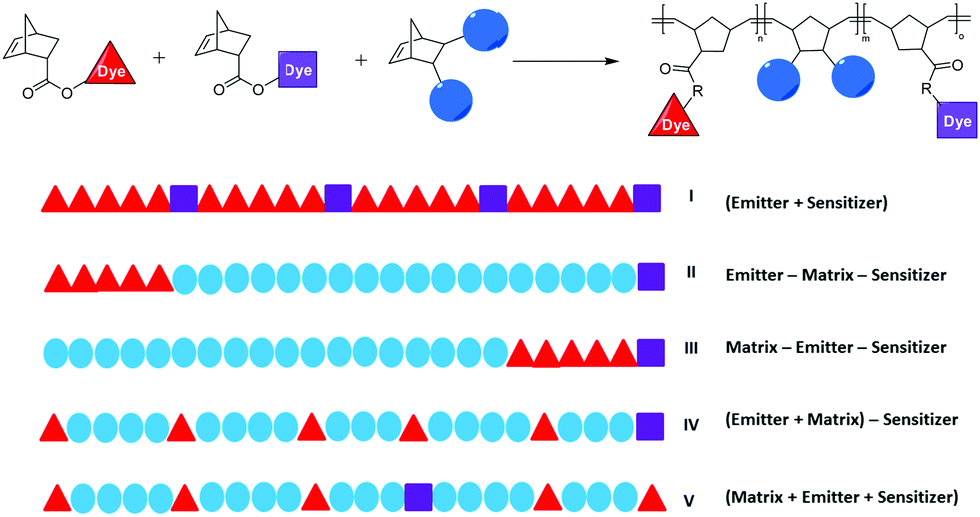 | ||
| Fig. 6 General synthetic pathway for the preparation of polymers II–V (top) and overview of different polymer architectures (red triangles = emitter, purple squares = sensitizer, blue spheres = matrix) (bottom). | ||
The ratio of the emitter to sensitizer was fixed to 5:1, in accordance to the up-conversion study reported for this chromophore pair (10:1 or 5:1).51 The following polymers were prepared: First a random copolymer of the emitter and sensitizer monomers in the ratio PDEmon:TPTBTBP Ptmon of 500:100 was prepared (polymer I). In addition, terpolymers (polymers II–V) were prepared with N-DME as matrix monomer and the chromophore system in a ratio N-DME:PDEmon:TPTBTBP Ptmon of 500:5:1. Polymer II is a triblock copolymer where the matrix block is separating the sensitizer and emitter. Polymer III is also a triblock copolymer, but now, emitter and sensitizer are directly linked to each other. Polymer IV is a diblock copolymer, where the emitter is randomly distributed in the matrix block and the sensitizer is added as the second “block”. It has to be noted that in our series, the average block length of the sensitizer is only 1, thus, some of the macromolecules will contain more than one but others contain no sensitizer molecule. Finally, polymer V is a random terpolymer of all three monomers. The GPC and DSC data of all polymers are summarized in Table 1.
Polymer I exhibits a melting temperature of 117.1 °C. The polydispersity index (Mw/Mn) of polymer I is quite high with a value of 1.6 due to sterical hindrance of the porphyrine side groups during polymerisation. Polymers II–V show glass transition temperatures between 89.2 and 91.9 °C which are in the range of the pure matrix polymer (Tg approx. 90.5 °C.63,64) Thus the concentration of the chromophores was too low to have a significant influence on the glass transition. The PDI of these polymers have values below 1.23 and polymers II–V exhibit very similar molar masses of approx. 81 kg mol−1.
TTA-UC
For the characterization of up-conversion properties of the chromophore functionalized terpolymers the concentration of acceptor and annihilator were adjusted to the values used previously for this dye system in solution.51 The chosen concentration of PDE was 5 × 10−4 M (and thus 1 × 10−4 M for TPTBTBP Pt) resulting in a polymer concentration of 0.46 g L−1 for polymer I and of 10.5 g L−1 for polymers II–V. Additionally, a solution of the unbound dye monomers PDEmon and TPTBTBP Ptmon were also analysed for comparison. All measurements were carried out in 1,4-dioxane as solvent. To ensure complete dissolution of the polymer, the mixtures were left in the ultrasonic bath for 30 minutes. Before the measurements were started, deoxygenation with argon for 10 minutes is essential to prevent quenching of the triplet states of the sensitizer and annihilator by molecular oxygen. The polymers have been excited with a 450 W Xe lamp (244 μmol s−1 m−2) at 617 nm and a 635 nm laser diode (36 200 μmol s−1 m−2). All TTA spectra show a broad phosphorescence signal with a peak maximum at 775 nm, which means that the triplet–triplet energy transfer can be improved further.The characteristic TTA up-conversion signal is very broad and occurs between 480 to 550 nm approximately, the maximum appears at 504 nm. The TTA spectrum of polymer V is illustrated below (Fig. 7, for the other polymers see ESI†). For laser excitation of polymers IV and V, the up-converted green fluorescence from TTA can be observed with the naked eye. For illustration purposes polymer II, IV and V have been excited with a laser diode at 635 nm (36 200 μmol s−1 m−2) which is shown below in Fig. 8. For the estimation of the quantum yields of the delayed up-converted fluorescence, a comparison of the peak area of the emission of a polymerized matrix:sensitizer system (ratio of matrix to sensitizer: 500:1) with the TTA polymers I to V was done. The measurements have been carried out in solution under room temperature and using a laser (λ = 635 nm) for excitation.
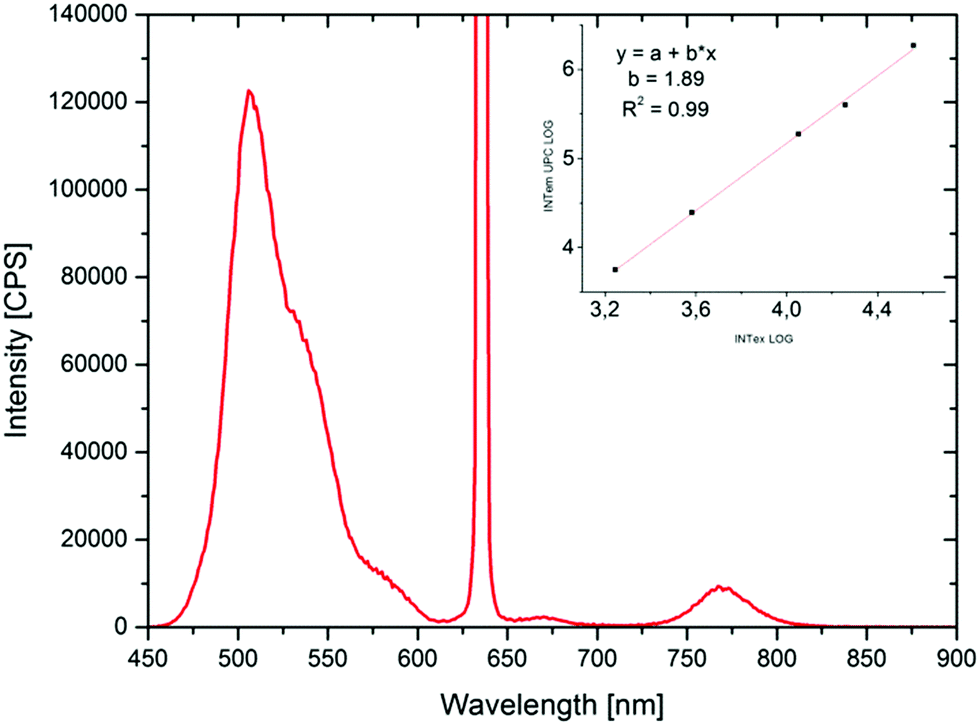 | ||
| Fig. 7 TTA emission spectrum of polymer V. The graph shows up-converted fluorescence where the peak maximum was detected at 509 nm and phosphorescence of the sensitizer which appears at 775 nm. The polymer was dissolved in 1,4-dioxane with a concentration of 10.5 g L−1 (Cemitter = 5 × 10−4 M, Csensitizer = 1 × 10−4 M). Excitation wavelength was set at 635 nm with a power density of 36 200 μmol s−1 m−2. Light irradiance dependent measurements are shown in the additional window on the right top. The logarithmic plot exhibits a slope of 1.89. | ||
 | ||
| Fig. 8 Photographic images of solutions of polymer II, IV and V (left to right). Polymers have been dissolved in deoxygenated 1,4-dioxane and were excited with a red laser at 635 nm. | ||
The TTA-UC quantum yield of the reference system (unbound dyes) exhibits a value of 8.7% (see ESI,† Fig. S14). The covalent attachment of the dye systems to the different polymer structures leads in all cases to a reduction in the TTA-UC quantum efficiency. The quantum yield of polymer I shows the lowest value (0.06%) compared to the other polymers. Due to very short distances between the chromophores it is very likely that the chromophores tend to aggregate which results in self-quenching. The block copolymer structures, polymer II and III exhibit slightly higher quantum yields (ϕ = 0.16% and ϕ = 0.10%). Also in these structures the emitter concentration is very high within one block. This correlates with the investigations of Xinjun Yu and co-workers.42 They reported a sharp maximum of the TTA-UC efficiency as a function of the emitter concentration and the inter-chromophore distance. Furthermore, they also suggested that self-quenching of the excited singlet state becomes significant at high dye concentrations. The UC emission is a little bit lower for polymer III, where both chromophores are next to each other. By increasing the distance between the emitter molecules on average as realized in polymer IV with a statistical distribution of the emitter molecules along the whole polymer chain, the up-conversion efficiency increases significantly (ϕ = 0.52%), by avoiding the previously described undesired effects. The best up-conversion by far was achieved with a statistical distribution of all three monomers in polymer V (ϕ = 2.95%). As in both polymers IV and V, one could argue that effects like aggregation or self-quenching of the emitter have been avoided or at least have the same extent in both structures, there is still a big difference in terms of quantum yield which is somehow surprising. The main difference between these two architectures is that the sensitizer is fixed to the end of the polymer chain in polymer IV instead of being distributed along the chain in polymer V. This leads obviously to a significant decrease of the TTA up-conversion quantum yield. At the same time the phosphorescence signal of polymer IV is much higher compared to the TTA signal of polymer V (see ESI,† Fig. S7 and S12). Therefore, it is obvious that the energy transfer from the sensitizer to the emitter is less effective in this structure, which can be rationalized by the fact that the mean distance between sensitizer and emitter is much larger.
Furthermore, an ideal polymer IV should have exactly one sensitizer monomer at the end of the polymer chain. However, in reality due to statistics there will be also polymer chains containing either zero, or even two and three sensitizer units at the end. Additionally, this will lead to self-aggregation and self-quenching effects of the sensitizer, which also decrease the quantum yield considerably. An overview of the observed ϕ values is shown in Table 1. Also for polymer V, it might also happen that one polymer chain contains more than one sensitizer, however, in this case the dye molecules will be distributed along the chain and all of them will be active in TTA. An indication, that aggregation of the emitter dye occurs, stems from the analysis of the PL spectra of the polymers (see Fig. S15 in the ESI†) showing a red-shift and an enhanced broadening of the emission signal especially in polymers II and III under selective excitation of the perylene unit (λEx = 430 nm). This behaviour is typically found upon aggregation of perylene ring systems.65 Further investigations of excitation light irradiance dependent up-conversion intensity (stepwise reduction of excitation light intensity using transmission filters: 50%, 30%, 10% and 5%) show a quadratic dependence (see Fig. 7 and ESI,† Fig. S4–S13).
This is typical for a nonlinear process such as TTA-based up-conversion. The logarithmic plot, for that measurement, exhibits a slope of two. This indicates that the triplet decay pathway is (quasi) first order (phosphorescence, quenching and intersystem crossing).66 Furthermore, this means that all measured ϕ values were below saturation. Hence, all values can be higher if higher excitation irradiance is used. Furthermore, a two-fold increase of the concentration of the polymers did not have much influence on the quantum yields (see ESI,† Table S3). All presented ϕ values were obtained with diode laser excitation at 635 nm (at power density of 36200 μmol s−1 m−2); in addition we tested the system with a Xe lamp at 617 nm with a two orders of magnitude lower power density compared to the laser. As expected, a tremendous decrease of the up-conversion emission was detected. The logarithmic plot of power density to UC emission still exhibits a slope of two (see ESI†). It can be summarized that the investigated terpolymer systems with covalently bound sensitizer and emitter chromophores can emit up-converted delayed fluorescence by TTA even upon irradiation with relatively low intensity excitation sources.
Finally, films of polymer V have been prepared via drop casting in order to examine the TTA up-conversion behaviour in the solid state. The results of the TTA experiments are shown in Fig. 9. On a first glance, the emission spectrum is dominated by the intense phosphorescence of the platinum benzoporphyrin system. However, by zooming into the low wavelength range, a small signal due to the up-converted emission can be identified. The weakness of this signal is not surprising considering the strongly restricted mobility of the chromophores and relatively high Tg of the polymers. So, for achieving solid state TTA up-conversion further optimization of the polymer structure by e.g. adjusting the dye concentrations and using other matrix monomers exhibiting lower glass transition temperatures can be pursued.
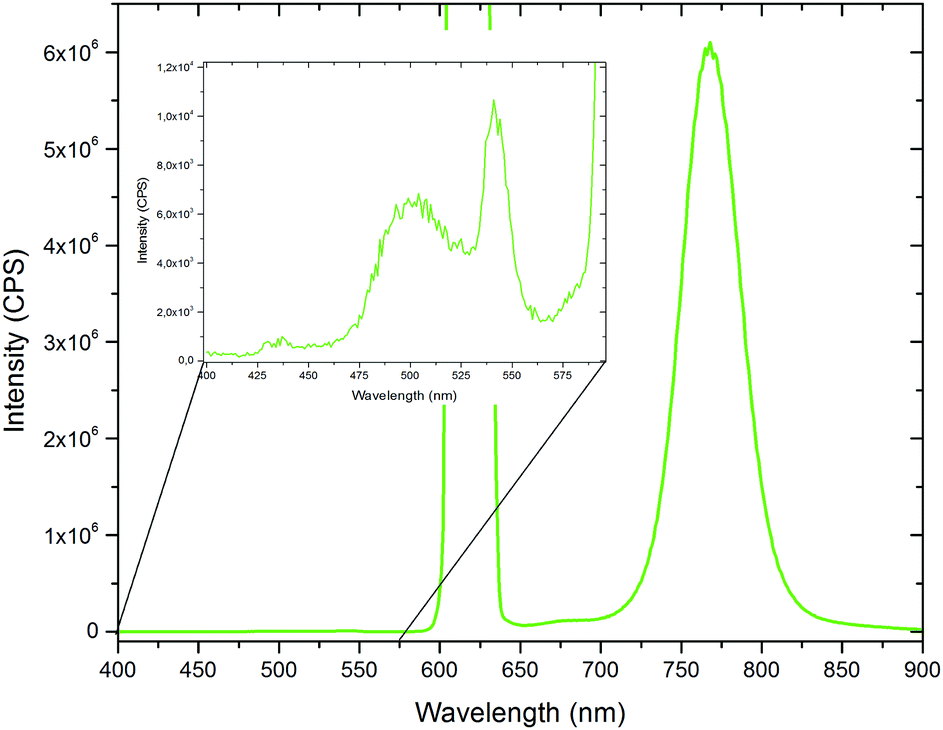 | ||
| Fig. 9 TTA-UC emission spectrum of the drop casted polymer V and its magnified spectrum between 400 and 600 nm (inset) (excitation with a 450 W Xe lamp: 244 μmol s−1 m−2). | ||
Conclusion
We presented the preparation of the first ROMP based dye functionalized polymer which is able to undergo triplet–triplet annihilation. The highest quantum yield was achieved in solution of 1,4-dioxane for the macromolecule having statistically distributed sensitizer and emitter (polymer V), with a value of ϕ ≈ 3%, after excitation with a red laser diode (635 nm). In this case up-conversion was detectable with the naked eye. The structural studies of the copolymers showed that an addition of a matrix monomer is essential for an efficient up-conversion; otherwise the chromophores are in too close proximity which results in self-quenching and aggregation, and consequently very low up-conversion efficiency. The same effect is observed for the prepared block copolymers. Up-conversion for all polymers was below saturation under the given experimental conditions which means that TTA up-conversion efficiency could be further enhanced if a higher light irradiance is used. Due to coiling of the matrix polymer, influences of different polymer matrices have to be investigated.Such polymer architectures capable of up-converted light emission and consisting of fully covalently bound chromophores are highly attractive if the functional polymers are used in solution e.g. for certain fluorescence microscopy and imaging techniques. The class of polymeric materials investigated here could e.g. be highly promising for applications in bioimaging (cell imaging, in vivo imaging, etc.). Therefore they could be easily modified by using water soluble matrix monomers in the ROMP process.
Finally it was also possible to prepare a polymer film which showed solid state up-conversion, albeit with very low efficiency.
Experimental
Materials and methods
All reagents and solvents (except dry CH2Cl2) were purchased from commercial sources (ABCR or Sigma Aldrich) with reagent grade quality and used as received. The dry CH2Cl2 was obtained through distillation over a drying agent (CaH2) and degassed with nitrogen. Zinc-4bromophenylacetate was prepared according to Ritveld et al.67 Complex M31 [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro-(3-phenyl-1H-inden-1ylidene) (pyridyl) ruthenium(II) was obtained from UMICORE AG Co. KG. Pt(C6H5CN)Cl2 was obtained according to ref. 59 by stirring PtCl2 in boiling benzonitrile for one hour and precipitating the resulted product with hexane. The yellow product was filtrated, washed with hexane and dried at 60 °C.NMR spectroscopy (1H, 13C, APT, COSY, HSQC) was performed on a Bruker Avance 300 MHz spectrometer. Deuterated solvents (chloroform-d, DMSO-d6, D2O) were obtained from Cambridge Isotope Laboratories Inc. and remaining solvent peaks were referenced according to literature.68 Peak shapes are specified as follows: s (singlet), bs (broad singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quadruplet) and m (multiplet). Silica gel 60 F254 and aluminium oxide 60 F254 (both from Merck) on aluminium sheets were used for thin layer chromatography. Visualization was done under UV light or by dipping into an aqueous solution of KMnO4 (0.1 wt%). MALDI-TOF mass spectrometry was performed on Micromass TofSpec 2E time-of-flight mass spectrometer. The instrument was equipped with a nitrogen laser (λ = 337 nm, operated at a frequency of 5 Hz) and a time lag focusing unit. Ions were generated just above the threshold laser power. Positive ion spectra were recorded in reflection mode with an accelerating voltage of 20 kV. The spectra were externally calibrated with a polyethylene glycol standard. Analysis of data was done with MassLynx-Software V3.5 (Micromass/Waters, Manchester, UK). The best ten shots were averaged to a spectrum. Samples were dissolved in acetone or CH2Cl2 (c = 1 mg mL−1). Solutions were mixed in the cap of a microtube in the ratio of 1 μL:10 μL. The resulting mixture (0.5 μL) was spotted onto the target and allowed to air-dry. The matrix was trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB). Absorption spectra were recorded on a Shimadzu spectrophotometer UV-1800. The emission was measured on a Hitachi F-7000 fluorescence spectrometer equipped with a red-sensitive photomultiplier R928 from Hamamatsu. For the TTA-measurements a Horiba Fluorolog-3 luminescence spectrometer was used. Polymers have been measured in solutions of 1,4-dioxane and excitation was done either with a 635 nm-laser diode or with a 450 W Xenon lamp at 617 nm. The laser diode was purchased from Roithner Lasertechnik (LDM 635/5LJM, 635 nm, 5 mW, focusable, 3–5 V, ∅ 12 × 30.5 mm). Relative luminescence quantum yields were determined according to Crosby and Demas69 using platinum(II) meso-tetraphenyltetra(tert-butyl)benzoporphyrin (ϕ = 0.51)70 as reference compound. Gel permeation chromatography (GPC) was used to determine molecular weights and the polydispersity index (PDI). These measurements were carried out, with chloroform as solvent, with the following instrument set up: Merck Hitachi L6000 (pump); Polymer Standards Service, 5 μm grade size (separation columns); Wyatt Technology (refractive index detector). Glass transition temperatures (Tg) were measured on a Perkin Elmer Differential Scanning Calorimeter (Hyper DSC 8500) under a nitrogen flow of 20 mL min−1. The scanning speed for cooling and heating was 20 °C min−1, the second heating run was used for determination of the Tg.
Synthetic procedures
:MeOH, 20:1). Yield: 59.2%. 1H-NMR (δ, 20 °C, CDCl3, 300 MHz): 8.94–8.91 (d, 3JHH = 8.4 Hz, 1H, Hpery), 8.85–8.83 (d, 3JHH = 8.4 Hz, 1H, Hpery), 8.31–8. 16 (m, 6H, Hpery), 7.67–7.61 (m, 2H, Hpery), 4.45–4.41 (t, 3JHH = 6.5 Hz, 4H, –COO–CH2–), 3.71–3.67 (t, 3JHH = 6.3 Hz, 2H, –CH2–OH) 1.87–1.43 (m, 12H, Halkyl) 1.05–1.01 (t, 3JHH = 7.3 Hz, 3H, –CH2–CH3). UV-Vis (CH2Cl2) λmax, nm (rel. in.): 437 (0.84), 464 (1).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 138.06, 135.78, 130.85, 130.47, 128.24, 122.10, 121.33, 120.40 (Cpery), 65.12, 64.37 (–COO–CH2–), 46.64, 46.39, 43.23, 41.65, 30.87, 30.36, 28.68, 25.89, 25.76 (Calkyl, Cnb), 19.42 (–CH2–CH3), 13.83 (–CH3). MALDI: m/z [M+] calc. for C40H40O6Na: 639.2723; found, 639.2746. UV-Vis (CH2Cl2) λmax, nm (rel. in.): 438 (0.86), 464 (1). λemission nm: 509.
:1, 100 mL) and dried with Na2SO4. The solvent was removed under reduced pressure to obtain a green solid. Yield: 99%. 1H-NMR (δ, 20 °C, CDCl3, 300 MHz): 8.41–6.90 (m, 31H, HPorphyrin, HAr), 1.36–1.13 (m, 36H, (CH3)3), −1.4 (bs, 2H, –NH–). UV-Vis (acetone) λmax, nm (rel. in.): 464 (1), 592 (0.05), 642 (0.17), 696 (0.06).
:CH2Cl2 3:1) and dried in vacuo to obtain a dark green solid. Yield: 49.5%. 1H-NMR (δ, 20 °C, CDCl3, 300 MHz): 8.45–6.80 (m, 31H, HPorphyrin, HAr), 1.19–1.01 (m, 36H, (CH3)3). UV-Vis (acetone) λmax, nm (rel. in.): 426 (1), 564 (0.09), 616 (0.62).
:EtOAc, 10:1) Yield: 37.5%. 1H-NMR (δ, 20 °C, DMSO-d6, 300 MHz): 8.35–6.81 (m, 35H, HPorphyrin, HAr), 5.34 (dd, 3JHH = 8.9, 5.1 Hz, 1H, –OH), 4.62 (d, 3JHH = 5.4 Hz, 2H, CH2–OH), 1.20–1.01 (m, 36H, (CH3)3). MALDI: m/z [M+] calc. for C83H74N4OPt: 1338.5535; found, 1338.6215. UV-Vis (acetone) λmax, nm (rel. in.): 426 (1), 564 (0.09), 616 (0.62).
:CH2Cl2 1:1) and dried in vacuo. Yield: 88%. 1H-NMR of the endo isomer (endo to exo: 80 to 20) (δ, 20 °C CDCl3, 300 MHz): 8.46–6.90 (m, 35H, HPorphyrin, HAr), 6.28 (dd, 3JHH = 5.1, 3.0 Hz, 1H, Hnb5), 6.06–5.98 (m, 1H, Hnb6), 5.25 (s, 2H, –O–CH2), 3.35 (bs, 1H, Hnb2), 3.11 (dd, 3JHH = 9.2, 3.7 Hz, 1H, Hnb1), 2.99 (bs, 1H, Hnb4), 2.11–1.88 (m, 2H, Hnb3), 1.62–1.11 (m, 38H, Hnb7, –(CH3)3). MALDI: m/z [M+] calc. for C91H82N4O2Pt: 1458.6111; found, 1458.769. UV-Vis (acetone) λmax, nm (rel. in.): 426 (1), 564 (0.09), 616 (0.62).
O), 138.06, 135.78, 130.85, 130.47, 128.24, 122.10, 121.33, 120.40 (Cpery), 65.12, 64.37 (–COO–CH2–), 46.64, 46.39, 43.23, 41.65, 30.87, 30.36, 28.68, 25.89, 25.76 (Calkyl, Cnb), 19.42 (–CH2–CH3), 13.83 (–CH3). MALDI: m/z [M+] calc. for C40H40O6Na: 639.2723; found, 639.2746. UV-Vis (CH2Cl2) λmax, nm (rel. in.): 438 (0.86), 464 (1). λemission nm: 509.
:1, 100 mL) and dried with Na2SO4. The solvent was removed under reduced pressure to obtain a green solid. Yield: 99%. 1H-NMR (δ, 20 °C, CDCl3, 300 MHz): 8.41–6.90 (m, 31H, HPorphyrin, HAr), 1.36–1.13 (m, 36H, (CH3)3), −1.4 (bs, 2H, –NH–). UV-Vis (acetone) λmax, nm (rel. in.): 464 (1), 592 (0.05), 642 (0.17), 696 (0.06).
:CH2Cl2 3:1) and dried in vacuo to obtain a dark green solid. Yield: 49.5%. 1H-NMR (δ, 20 °C, CDCl3, 300 MHz): 8.45–6.80 (m, 31H, HPorphyrin, HAr), 1.19–1.01 (m, 36H, (CH3)3). UV-Vis (acetone) λmax, nm (rel. in.): 426 (1), 564 (0.09), 616 (0.62).
:EtOAc, 10:1) Yield: 37.5%. 1H-NMR (δ, 20 °C, DMSO-d6, 300 MHz): 8.35–6.81 (m, 35H, HPorphyrin, HAr), 5.34 (dd, 3JHH = 8.9, 5.1 Hz, 1H, –OH), 4.62 (d, 3JHH = 5.4 Hz, 2H, CH2–OH), 1.20–1.01 (m, 36H, (CH3)3). MALDI: m/z [M+] calc. for C83H74N4OPt: 1338.5535; found, 1338.6215. UV-Vis (acetone) λmax, nm (rel. in.): 426 (1), 564 (0.09), 616 (0.62).
:CH2Cl2 1:1) and dried in vacuo. Yield: 88%. 1H-NMR of the endo isomer (endo to exo: 80 to 20) (δ, 20 °C CDCl3, 300 MHz): 8.46–6.90 (m, 35H, HPorphyrin, HAr), 6.28 (dd, 3JHH = 5.1, 3.0 Hz, 1H, Hnb5), 6.06–5.98 (m, 1H, Hnb6), 5.25 (s, 2H, –O–CH2), 3.35 (bs, 1H, Hnb2), 3.11 (dd, 3JHH = 9.2, 3.7 Hz, 1H, Hnb1), 2.99 (bs, 1H, Hnb4), 2.11–1.88 (m, 2H, Hnb3), 1.62–1.11 (m, 38H, Hnb7, –(CH3)3). MALDI: m/z [M+] calc. for C91H82N4O2Pt: 1458.6111; found, 1458.769. UV-Vis (acetone) λmax, nm (rel. in.): 426 (1), 564 (0.09), 616 (0.62).
13C-NMR (δ, 20 °C, CDCl3, 75 MHz): 175.08 (CO), 173.85 (CO), 137.69, 135.27 (Cnb5,6), 52.16 (–CH3), 51.88 (–CH3), 47.94, 47.71, 47.40, 47.17, 45.70 (Cnb1–4, Cnb7).
Polymer preparation
To ensure a precise ratio of monomers and initiator, stock solutions in CH2Cl2 have been prepared (N-DME: 1.01 × 10−1 g L−1, PDEmon: 1.74 × 10−2 g L−1, TPTBTBP Ptmon: 1.04 × 10−2 g L−1, M31:2.10 × 10−3 g L−1). The stock solutions have been filled into evacuated 10 mL Schlenk flasks purged with nitrogen and equipped with a stirring bar, amounts of the used monomers are shown in Table 2. First all stock solutions have been degassed. For polymer I 1.153 mL PDEmon solution, 0.9096 mL TPTBRBP Ptmon solution and 0.023 mL M31 have been filled, all at once, into the flask. Amounts of stock solutions for polymers II–V are listed below: 0.9865 mL N-DME solution, 0.1666 mL PDEmon solution, 0.1335 mL TPTBTBP Ptmon solution and 0.339 mL M31. For the preparation of block copolymers II and III just one monomer was added to the initiator solution. The following monomers have been added not until full conversion of the prior added ones was shown by thin layer chromatography. Polymer IV was prepared by adding initiator, matrix and emitter at once and after full conversion of these monomers the sensitizer solution was added. The statistically distributed polymer V was prepared by adding the corresponding amount of the stock solutions at once. Yield ≈ 90%. 1H-NMR (δ, 20 °C, CDCl3, 300 MHz): 5.54–5.11 (m, 2H, CHCH), 3.68–3.59, 3.32–2.62 (m, 4H, Hcp1–4), 1.98 (bs, 1H, Hcp5a), 1.47 (bs, 1H, Hcp5b). 13C-NMR (δ, 20 °C, CDCl3, 75 MHz): 174.6–173.3 (CO), 133.3–129.0 (HCCH), 53.4–51.4 (Ccp1–5), 40.8 (–CH3). (PDEmon and TPTBTBP Ptmon could not be detected, due to the low concentration of chromophores) UV-Vis (CH2Cl2) λmax, nm (rel. in.): 428 (1), 567 (0.1), 616 (0.6) Polymers II–V: PDI: 1.14. Mn: 8.105 × 104 g mol−1. Tg: 92.2 °C (177.9 °C, 193.6 °C).
| Matrix | PDEmon | TPTBTBP Ptmon | M31 | ||
|---|---|---|---|---|---|
| Polymer I | m [mg] | — | 20 | 9.46 | 4.9 × 10−3 |
| n [mmol] | — | 3.24 × 10−2 | 6.49 × 10−3 | 6.55 × 10−5 | |
| Ratio | — | 500 | 100 | 1 | |
| Polymer II–V | m [mg] | 100 | 2.89 | 1.39 | 0.72 |
| n [mmol] | 0.476 | 4.76 × 10−3 | 9.52 × 10−4 | 9.52 × 10−4 | |
| Ratio | 500 | 5 | 1 | 1 | |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project “PoTTA” (FFG: 841153) is funded by the Austrian “Climate and Energy Fund” within the program Energy Emission Austria. The authors thank Umicore for providing catalyst M31.References
- Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817–20830 RSC.
- S. H. Lee, D. C. Thévenaz, C. Weder and Y. C. Simon, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1629–1639 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- S. Mattiello, A. Monguzzi, J. Pedrini, M. Sassi, C. Villa, Y. Torrente, R. Marotta, F. Meinardi and L. Beverina, Adv. Funct. Mater., 2016, 16, 8447–8454 CrossRef.
- W. Kaiser and C. G. B. Garrett, Phys. Rev. Lett., 1961, 7, 229–231 CrossRef CAS.
- C. Ye, L. Zhou, X. Wang and Z. Liang, Phys. Chem. Chem. Phys., 2016, 18, 10818–10835 RSC.
- A. Monguzzi, S. M. Borisov, J. Pedrini, I. Klimant, M. Salvalaggio, P. Biagini, F. Melchiorre, C. Lelii and F. Meinardi, Adv. Funct. Mater., 2015, 25, 5617–5624 CrossRef CAS.
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 CAS.
- J. Zhao, S. Jia and H. Guo, RSC Adv., 2011, 1, 937–950 RSC.
- P. Ceroni, Chem. – Eur. J., 2011, 17, 9560–9564 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2008, 112, 3550–3556 CrossRef CAS PubMed.
- K. Sripathy, R. W. MacQueen, J. R. Peterson, Y. Y. Cheng, M. Dvořák, D. R. McCamey, N. D. Treat, N. Stingelin and T. W. Schmidt, J. Mater. Chem. C, 2015, 3, 616–622 RSC.
- T. N. Singh-Rachford, A. Haefel, R. Ziessel and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 16164–16165 CrossRef CAS PubMed.
- W. Wu, W. Wu, S. Ji, H. Guo and J. Zhao, Dalton Trans., 2011, 40, 5953–5963 RSC.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776–3778 RSC.
- A. Turshatov, D. Busko, S. Baluschev, T. Miteva and K. Landfester, New J. Phys., 2011, 13, 083035 CrossRef.
- C. A. Parker, C. G. Hatchard and T. A. Joyce, Nature, 1965, 205, 1282–1284 CrossRef CAS.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652–12653 CrossRef CAS PubMed.
- T. Gatti, L. Brambilla, M. Tommasini, F. Villafiorita-Monteleone, C. Botta, V. Sarritzu, A. Mura, G. Bongiovanni and M. D. Zoppo, J. Phys. Chem. C, 2015, 119, 17495–17501 CAS.
- C. Ye, J. Wang, X. Wang, P. Ding, Z. Liang and X. Tao, Phys. Chem. Chem. Phys., 2016, 18, 3430–3437 RSC.
- H. Kouno, T. Ogawa, S. Amemori, P. Mahato, N. Yanai and N. Kimizuka, Chem. Sci., 2016, 7, 5224–5229 RSC.
- C. Wohnhaas, K. Friedemann, D. Busko, K. Landfester, S. Baluschev, D. Crespy and A. Turshatov, ACS Macro Lett., 2013, 2, 446–450 CrossRef CAS.
- P. Mahato, A. Monguzzi, N. Yanai, T. Yamada and N. Kimizuka, Nat. Mater., 2015, 14, 924–930 CrossRef CAS PubMed.
- J. Zimmermann, R. Mulet, G. D. Scholes, T. Wellens and A. Buchleitner, J. Chem. Phys., 2014, 141, 184104 CrossRef PubMed.
- S. H. C. Askes, N. L. Mora, R. Harkes, R. I. Koning, B. Koster, T. Schmidt, A. Kros and S. Bonnet, Chem. Commun., 2015, 51, 9137–9140 RSC.
- M. Poznik, U. Faltermeier, B. Dick and B. König, RSC Adv., 2016, 6, 41947–41950 RSC.
- Y. Murakami, T. Ito and A. Kawai, J. Phys. Chem. B, 2014, 118, 14442–14451 CrossRef CAS PubMed.
- M. Penconi, P. L. Gentili, G. Massaro, F. Elisei and F. Ortica, Photochem. Photobiol. Sci., 2014, 13, 48–61 CAS.
- C. Ye, B. Wang, R. Hao, X. Wang, P. Ding, X. Tao, Z. Chen, Z. Liang and Y. Zhou, J. Mater. Chem. C, 2014, 2, 8507–8514 RSC.
- Q. Liu, B. Yin, T. Yang, Y. Yang, Z. Shen, P. Yao and F. Li, J. Am. Chem. Soc., 2013, 135, 5029–5037 CrossRef CAS PubMed.
- K. Tanaka, K. Inafuku and Y. Chujo, Chem. Commun., 2010, 46, 4378–4380 RSC.
- A. J. Tilley, B. E. Robotham, R. P. Steer and K. P. Ghiggino, Chem. Phys. Lett., 2015, 618, 198–202 CrossRef CAS.
- K. Katta, D. Busko, Y. Avlasevich, R. Muñoz-Espí, S. Baluschev and K. Landfester, Macromol. Rapid Commun., 2015, 36, 1084–1088 CrossRef CAS PubMed.
- C. Wohnhaas, V. Mailänder, M. Dröge, M. A. Filatov, D. Busko, Y. Avlasevich, S. Baluschev, T. Miteva, K. Landfester and A. Turshatov, Macromol. Biosci., 2013, 13, 1422–1430 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- J.-H. Olivier, Y. Bai, H. Uh, H. Yoo, M. J. Therien and F. N. Castellano, J. Phys. Chem. A, 2015, 119, 5642–5649 CrossRef CAS PubMed.
- R. Andernach, H. Utzat, S. D. Dimitrov, I. McCulloch, M. Heeney, J. R. Durrant and H. Bronstein, J. Am. Chem. Soc., 2015, 137, 10383–10390 CrossRef CAS PubMed.
- J. Sun, J. Yang, C. Zhang, H. Wang, J. Li, S. Su, H. Xu, T. Zhang, Y. Wu, W.-Y. Wong and B. Xu, New J. Chem., 2015, 39, 5180–5188 RSC.
- S. Yu, Y. Zeng, J. Chen, T. Yu, X. Zhang, G. Yang and Y. Li, RSC Adv., 2015, 5, 70640–70648 RSC.
- D. C. Thévenaz, A. Monguzzi, D. Vanhecke, R. Vadrucci, F. Meinardi, Y. C. Simon and C. Weder, Mater. Horiz., 2016, 3, 602–607 RSC.
- X. Yu, X. Cao, X. Chen, N. Ayres and P. Zhang, Chem. Commun., 2015, 51, 588–591 RSC.
- C. Fan, W. Wu, J. J. Chruma, J. Zhao and C. Yang, J. Am. Chem. Soc., 2016, 138, 15405–15412 CrossRef CAS PubMed.
- P. C. Boutin, K. P. Ghiggino, T. L. Kelly and R. P. Steer, J. Phys. Chem. Lett., 2013, 4, 4113–4118 CrossRef CAS.
- A. J. Tilley, M. J. Kim, M. Chen and K. P. Ghiggino, Polymer, 2013, 54, 2865–2872 CrossRef CAS.
- E. Khosravi, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 3, pp. 72 and 255 Search PubMed.
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS.
- M. Hollauf, G. Trimmel and A. C. Knall, Monatsh. Chem., 2015, 146, 1063–1080 CrossRef CAS.
- M. Hollauf, M. Cajlakovič, M. Tscherner, S. Köstler, A. C. Knall and G. Trimmel, Monatsh. Chem., 2016, 148, 121–129 CrossRef PubMed.
- M. Sandholzer, A. Lex, G. Trimmel, R. Saf, F. Stelzer and C. Slugovc, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1336–1348 CrossRef CAS.
- S. M. Borisov, C. Larndorfer and I. Klimant, Adv. Funct. Mater., 2012, 22, 4360–4368 CrossRef CAS.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- P. Rothemund, J. Am. Chem. Soc., 1936, 58, 625–627 CrossRef CAS.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS.
- S. J. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836 CrossRef.
- O. S. Finikova, A. V. Cheprakov, I. P. Beletskaya, P. J. Carroll and S. A. Vinogradov, J. Org. Chem., 2004, 69, 522–535 CrossRef CAS PubMed.
- C. Borek, K. Hanson, P. I. Djurovich, M. E. Thompson, K. Aznavour, R. Bau, Y. Sun, S. R. Forrest, J. Brooks and L. Michalski, Angew. Chem., Int. Ed., 2007, 46, 1109–1112 CrossRef CAS PubMed.
- I. Kunihiro, S. Masako, M. Hisayuki, Y. Madoka, F. Masaru and O. Osamu, Inorg. Chim. Acta, 1991, 182, 83–86 CrossRef.
- L. H. Hutter, B. J. Müller, K. Koren, S. M. Borisov and I. Klimant, J. Mater. Chem. C, 2014, 2, 7589–7598 RSC.
- X. Cui, J. Zhao, P. Yang and J. Sun, Chem. Commun., 2013, 49, 10221–10223 RSC.
- B. Neises and W. Steglich, Org. Synth., 1985, 63, 183 CrossRef CAS.
- D. Burtscher, C. Lexer, K. Mereiter, R. Winde, R. Karch and C. Slugovc, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4630–4635 CrossRef CAS.
- K. Stubenrauch, M. Sandholzer, F. Niedermair, K. Waich, T. Mayr, I. Klimant, G. Trimmel and C. Slugovc, Eur. Polym. J., 2008, 44, 2558–2566 CrossRef CAS.
- K. Gallas, A. C. Knall, S. R. Scheicher, D. E. Fast, R. Saf and C. Slugovc, Macromol. Chem. Phys., 2014, 215, 76–81 CrossRef CAS.
- M. S. Glaz, J. D. Biberdorf, M. T. Nguyen, J. J. Travis, B. J. Holliday and D. A. Vanden Bout, J. Mater. Chem. C, 2013, 1, 8060–8065 RSC.
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, H. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC.
- I. B. Rietveld, E. Kim and S. A. Vinogradov, Tetrahedron, 2003, 59, 3821–3831 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- S. M. Borisov, G. Nuss, W. Haas, R. Saf, M. Schmuck and I. Klimant, J. Photochem. Photobiol., A, 2009, 201, 128–135 CrossRef CAS.
- R. Wang, Z. Shi, C. Zhang, A. Zhang, J. Chen, W. Guo and Z. Sun, Dyes Pigm., 2013, 98, 450–458 CrossRef CAS.
- B. Balaganesan, W. J. Shen and C. H. Chen, Tetrahedron Lett., 2003, 44, 5747–5750 CrossRef CAS.
- A. J. Lowe, G. A. Dyson and F. M. Pfeffer, Eur. J. Org. Chem., 2008, 1559–1567 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tc01639e |
| This journal is © The Royal Society of Chemistry 2017 |