
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bright green PhOLEDs using cyclometalated diiridium(III) complexes with bridging oxamidato ligands as phosphorescent dopants†
Ahmed
M’hamedi
a,
Mark A.
Fox
 b,
Andrei S.
Batsanov
b,
Hameed A.
Al-Attar
c,
Andrew P.
Monkman
c and
Martin R.
Bryce
*b
b,
Andrei S.
Batsanov
b,
Hameed A.
Al-Attar
c,
Andrew P.
Monkman
c and
Martin R.
Bryce
*b
aLaboratory of Structure Determination, Development, and Application of Molecular Material, Department of Chemistry, University of Abdelhamid Ibn Badis BP 227, Mostaganem 27000, Algeria
bDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
cDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK
First published on 28th June 2017
Abstract
In contrast to monoiridium complexes, the study of diiridium complexes as dopants in phosphorescent organic light-emitting devices (PhOLEDs) is largely unexplored. We now describe the syntheses, detailed NMR analyses, X-ray crystal structures and optoelectronic properties of the new cyclometalated diiridium complexes 5 and 6 in which the iridium centres are bridged by oxamidato ligands. These complexes contain diastereomers – the meso form (ΔΛ) and the racemic form consisting of two enantiomers (ΔΔ and ΛΛ) – with anti-oxamidato bridges. The precursor μ-dichloro-bridged complex 4 is very weakly emissive in solution, whereas the oxamidato bridged complexes 5 and 6 are highly emissive (ΦPL 73% and 63%) with short excited state lifetimes of τP 0.84 and 1.16 μs, respectively. Cyclic voltammetry studies demonstrate that the oxamidato bridging ligand plays a role in mediating intramolecular interactions between the iridium centres. Density functional theory (DFT) calculations and time dependent-DFT (TD-DFT) calculations provide further insights into the structural, electronic, and photophysical properties of the complexes in their ground and excited states. Phosphorescent organic light-emitting diodes (PhOLEDs) using complexes 5 and 6 as the emissive dopants in a simple architecture using a solution-processed active layer give bright green electroluminescence with remarkably high luminance (Lmax > 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2) for diiridium complexes.
000 cd m−2) for diiridium complexes.
Introduction
The rich photofunctional properties of Ir(III) complexes1–3 are currently exploited in applications such as photocatalysis,4 biological labelling and bioimaging5,6 and sensing.7 Most importantly, since the pioneering work of Baldo et al.8 Ir(III) complexes have been especially developed as emitters in phosphorescent organic light-emitting diodes (PhOLEDs) for displays,9 solid-state lighting10 and light-emitting electrochemical cells (LECs).11,12 Efficient spin–orbit coupling, induced by the significant contribution of metal orbitals to the excited state, enhances the formally forbidden intersystem crossing transitions and facilitates the harvesting of all the electro-generated excitons. Consequently, electrophosphorescence which derives from both singlet and triplet excited states theoretically achieves 100% internal quantum efficiency. Ir(III) complexes are synthetically versatile and they possess many favourable properties, such as high quantum efficiency (Φ), short phosphorescence lifetimes (τp) and reversible electrochemistry. The metal-ligand-based luminescent properties of Ir(III) complexes enable colour tuning across a broad range of the visible spectrum through systematic structural variation of the ligands.Cyclometalated diiridium complexes with a μ-dichloro-bridge13 are the standard precursors to homoleptic and heteroleptic monoiridium complexes.14 However, the μ-dichloro-bridged dimers are generally very poorly emissive15,16 (although there are a few exceptions).17,18 Most other diiridium systems also have low luminescence quantum yields19–24 which has focussed the vast majority of research onto the more strongly emissive monoiridium complexes.1–3 However, the bridging ligands within diiridium complexes provide unique potential for additional structural and electronic variations compared to monoiridium systems. High efficiency phosphorescence typically results from preventing non-radiative deactivation by rigidification of the emitter framework25 which can be enhanced by judicious choice of the bridging ligand. Additionally, the presence of a second metal might introduce a larger spin–orbit coupling effect than that associated with a single metal centre.
A few diiridium complexes, especially those with conjugated bridging ligands, are known to possess efficient photoluminescence (ΦPL typically ∼30–60% in solution).26–32 There are even examples of solution photoluminescence quantum yields (PLQYs) up to 100% for diiridium complexes with a bis-N^C-coordinating diarylpyrimidinyl bridging ligand.33 The use of diiridium complexes as dopants in phosphorescent organic light-emitting devices (PhOLEDs) is largely unexplored. However, solution-processed PhOLEDs with external quantum efficiencies (EQEs) of >10% have recently been fabricated by our group (complex 1, Fig. 1)34 and by Yang et al., using a 2-(4-tert-butylphenyl)pyrimidine bridging ligand.35 A few other examples of neutral diiridium PhOLEDs with lower efficiencies and low brightnesses have also been reported.36 A low-efficiency PhOLED using a cationic diiridium emitter has also been demonstrated.37 Therefore, it is important to explore diiridium systems in the search for new families of OLED materials.
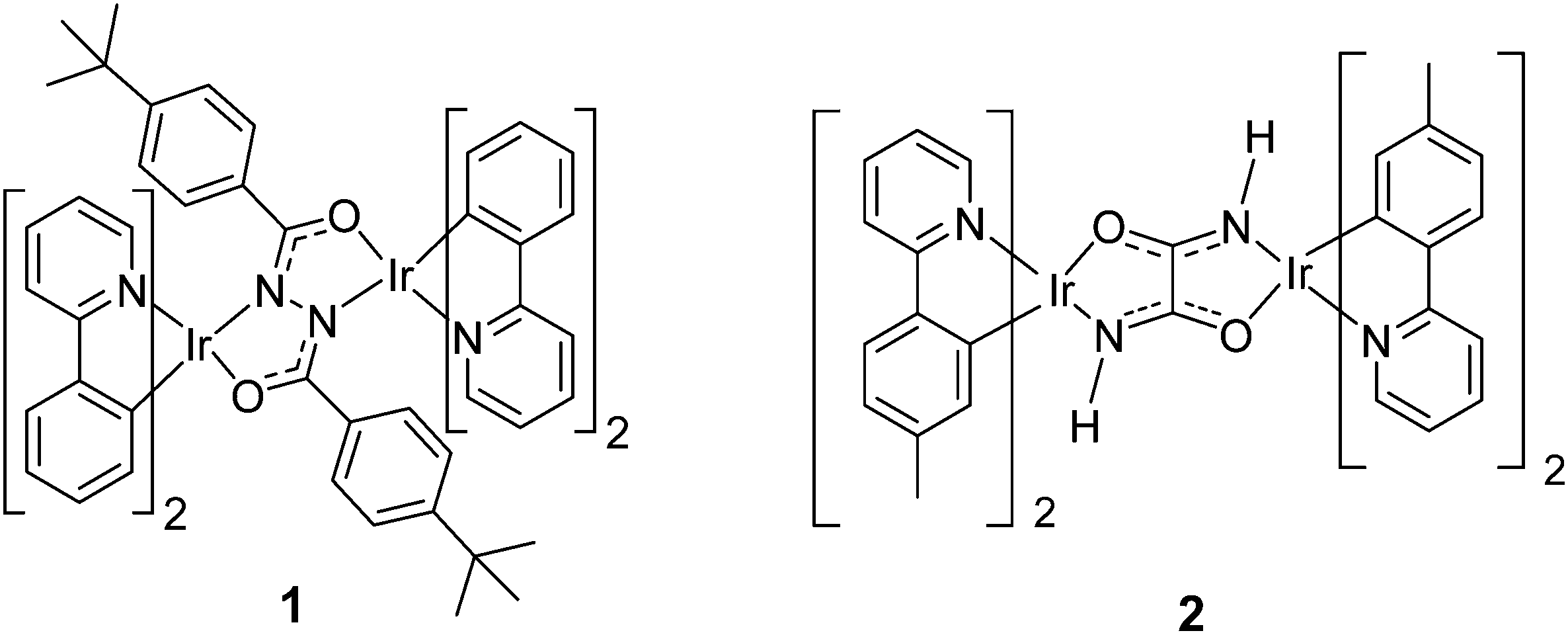
The motivation for the present work is to develop the optoelectronic properties and PhOLED applications of diiridium complexes with bridging oxamidato ligands, noting that their C2N2O2 structure is isomeric with the diarylhydrazido bridge of our previous complex 1.34 We reasoned that the rigidity of the bis-coordinated oxamidato ligands could lead to high photo- and electro-luminescence quantum yields. Indeed, Sünkel and co-workers recently showed that complex 2 is a green emitter with ΦPL 60% in degassed dichloromethane solution, although no OLEDs were reported in that work.32 We have now synthesised complexes 5 and 6 and discuss their single-crystal X-ray structures, NMR spectra, electrochemical and photophysical properties. Notably, complex 6 is the first reported example with any substituent (other than hydrogens) on the oxamidato bridge. Density functional theory (DFT) and time dependent DFT (TD-DFT) calculations provide further insights into the structural, electronic, and photophysical properties in the ground and excited states. Phosphorescent organic light-emitting diodes (PhOLEDs) using complex 5 or 6 as the emissive dopant in a solution-processed active layer give bright electroluminescence with luminance values as high as >25000 cd m−2. The present work, therefore, represents the first thorough investigation of the properties of diiridium complexes with bridging oxamidato ligands, including new structural modifications. To our knowledge these data strongly compete with the brightest reported PhOLEDs using bimetallic complexes as the emitter.34,35
Results and discussion
The synthesis of the diiridium complexes is shown in Scheme 1. Ligand 3 and the derived complexes 4–6 incorporate a mesityl substituent at C4 of each of the ppy ligands as there is precedent that a mesityl group at this position significantly improves the solubility of iridium complexes in organic solvents thereby facilitating device fabrication by solution processing, while having minimal effect on the emission colour.38–43 The μ-dichloro-bridged species 443 was obtained in 66% yield using the standard procedure44 by reacting 3 with IrCl3·3H2O in 2-ethoxyethanol. Reaction of 4 with oxamide or with N,N′-di-t-butylphenyloxamide in the presence of sodium methoxide gave the target oxamido-bridged complexes 5 and 6 as yellow-orange solids in ca. 60% yields after purification by column chromatography and recrystallisation.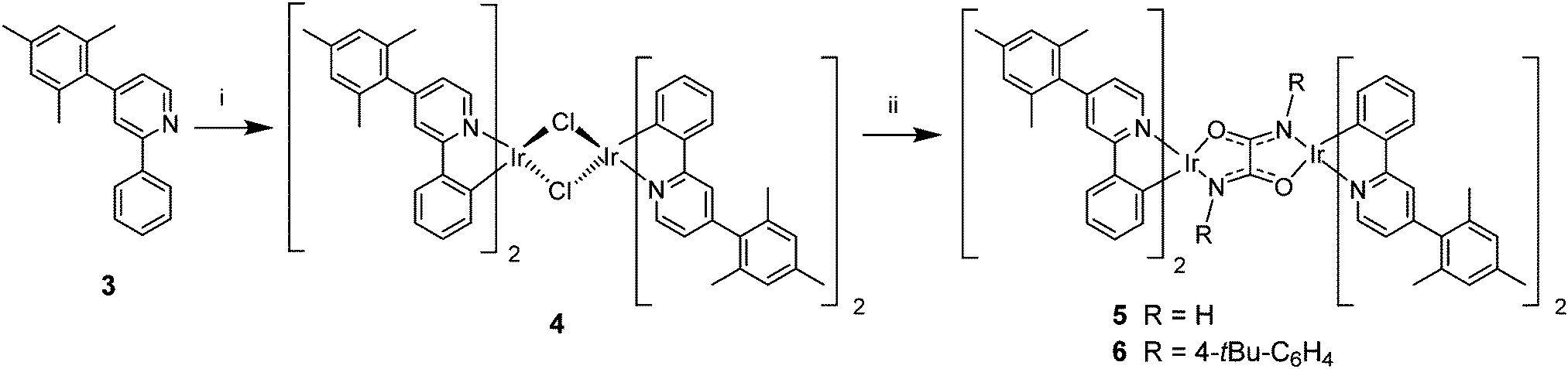 | ||
| Scheme 1 Reagents and conditions: (i) IrCl3·3H2O, 2-ethoxyethanol, 120 °C, 12 h; (ii) oxamide or N,N′-di-t-butylphenyloxamide, NaOMe, MeOH, 20 °C, 24 h. | ||
The structures of the dinuclear complexes were confirmed by MALDI mass spectra (Fig. S6, S7 in ESI†), elemental analysis and X-ray crystallography (for 5 and 6). The 1H and 13C NMR spectra of 5 and 6 (see the ESI†) exhibit many peaks, as noted previously for other diiridium complexes,31 due to the presence of isomers – the meso form (ΔΛ) and the racemic form consisting of two enantiomers (ΔΔ and ΛΛ). Furthermore, as noted by Sünkel for complex 2,32 both the syn- and anti-oxamidato bridge structures are feasible, although the anti geometry with N^O coordination at each iridium centre appears more reasonable, as observed unambiguously in the crystal structure of complex 6 (see below) and favoured in the crystal structure of 5. It is well known to be often very laborious, and sometimes not possible, to isolate the isomers of diiridium complexes in their pure form.33,45 We were unable to separate the isomers of 5 or 6 by recrystallisation or chromatography, including HPLC.
Table 1 lists all 13C NMR peak assignments corresponding to the ppy ligands for complexes 4–6. NMR data for complex 2 assigned elsewhere32 where two stereoisomers were observed in a 3:1 ratio, are also listed. The peaks for 4, 5 and 6 were assigned here with the aid of 2D 1H–1H COSY and 1H–13C correlation (HSQC and HMBC) NMR spectra. The dichloro-bridged complex 4 has only the racemic form (ΔΔ and ΛΛ) due to steric effects preventing the formation of the meso form (ΔΛ) and the higher symmetry of the dichloro-bridge results in one set of 13C NMR peaks for a ppy ligand observed here.
:1 for 2 and 3:2 for 5 and 6
| C2 | C3 | C4 | C5 | C6 | C7 | C8 | C9 | C10 | C11 | C12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | |||||||||||
| Major | 169.25 | 118.63 | 137.34 | 121.50 | 149.55 | 142.23 | 124.28 | 122.12 | 139.80 | 133.99 | 155.07 |
| Isomer | 168.72 | 118.43 | 136.47 | 121.44 | 148.34 | 142.20 | 124.18 | 121.84 | 139.50 | 133.36 | 145.73 |
| Difference | 0.53 | 0.20 | 0.87 | 0.06 | 1.21 | 0.03 | 0.10 | 0.28 | 0.30 | 0.63 | 9.34 |
| Minor | 169.17 | 118.54 | 137.16 | 121.50 | 149.81 | 142.26 | 124.22 | 122.05 | 139.74 | 133.99 | 155.11 |
| Isomer | 168.65 | 118.35 | 136.32 | 121.44 | 148.34 | 142.20 | 124.11 | 121.79 | 139.50 | 133.36 | 145.70 |
| Difference | 0.52 | 0.19 | 0.84 | 0.06 | 1.47 | 0.06 | 0.11 | 0.26 | 0.24 | 0.63 | 9.41 |
| 4 | |||||||||||
| 168.49 | 120.41 | 150.80 | 123.44 | 151.58 | 144.11 | 123.71 | 121.42 | 129.32 | 130.03 | 144.50 | |
| 5 | |||||||||||
| Major | 169.23 | 119.86 | 150.58 | 122.41 | 149.02 | 144.32 | 124.26 | 120.69 | 129.54 | 132.52 | 154.78 |
| Isomer | 168.98 | 119.42 | 149.88 | 122.38 | 147.76 | 144.30 | 123.87 | 120.36 | 129.34 | 132.11 | 144.78 |
| Difference | 0.25 | 0.44 | 0.70 | 0.03 | 1.26 | 0.02 | 0.39 | 0.33 | 0.20 | 0.41 | 10.00 |
| Minor | 169.20 | 119.76 | 150.48 | 122.51 | 149.23 | 144.35 | 124.29 | 120.72 | 129.50 | 132.66 | 154.71 |
| Isomer | 168.91 | 119.45 | 149.72 | 122.50 | 147.65 | 144.25 | 123.85 | 120.38 | 129.39 | 132.24 | 144.78 |
| Difference | 0.29 | 0.31 | 0.76 | 0.01 | 1.58 | 0.10 | 0.44 | 0.34 | 0.11 | 0.42 | 9.93 |
| 6 | |||||||||||
| Major | 169.43 | 120.47 | 150.77 | 122.88 | 149.35 | 145.05 | 124.31 | 120.91 | 129.13 | 133.24 | 152.88 |
| Isomer | 168.39 | 119.31 | 150.52 | 122.20 | 147.50 | 143.47 | 123.52 | 119.73 | 128.86 | 131.75 | 148.45 |
| Difference | 1.04 | 1.16 | 0.25 | 0.68 | 1.85 | 1.58 | 0.79 | 1.18 | 0.27 | 1.49 | 4.43 |
| Minor | 169.45 | 120.19 | 150.77 | 122.83 | 149.23 | 145.00 | 124.26 | 120.78 | 129.23 | 133.09 | 152.96 |
| Isomer | 168.21 | 119.19 | 150.27 | 122.30 | 147.63 | 143.43 | 123.40 | 119.71 | 128.88 | 132.05 | 148.64 |
| Difference | 1.24 | 1.00 | 0.50 | 0.53 | 1.60 | 1.57 | 0.86 | 1.07 | 0.35 | 1.04 | 4.32 |
The NMR data for complexes 5 and 6 indicate the presence of two isomers – the meso form (ΔΛ) and the racemic form (ΔΔ and ΛΛ). A 3:2 ratio of isomers is observed in both cases but it is unfortunately not possible to assign one form to one set of NMR data corresponding to the major isomer. Similarly, the NMR data for 2 could not be assigned to the isomers, but Sünkel assumed the anti geometry with N^O coordination at each iridium centre for both isomers.32 The proposed anti geometries are indeed supported by X-ray data here. The remarkable similarities between the 13C NMR data for 2 and for 5 with the largest shift differences of 9–10 ppm found for the carbon atom attached to iridium (C12) (Fig. 2) confirm that complexes 2 and 5 contain mixtures of meso (ΔΛ) and racemic (ΔΔ and ΛΛ) anti geometries. The effect of the tert-butylphenyl groups at the nitrogens of the bridge in complex 6 on the 13C NMR shifts compared to those for 5 is obvious with shift differences of over 1 ppm for seven carbons (C2, C3, C6, C7, C9, C11, C12) of the ppy ligands in 6 compared to only two (C6, C12) in 5. Interestingly, the shift differences for C12 in 6 are about half the corresponding values for 2 and 5.
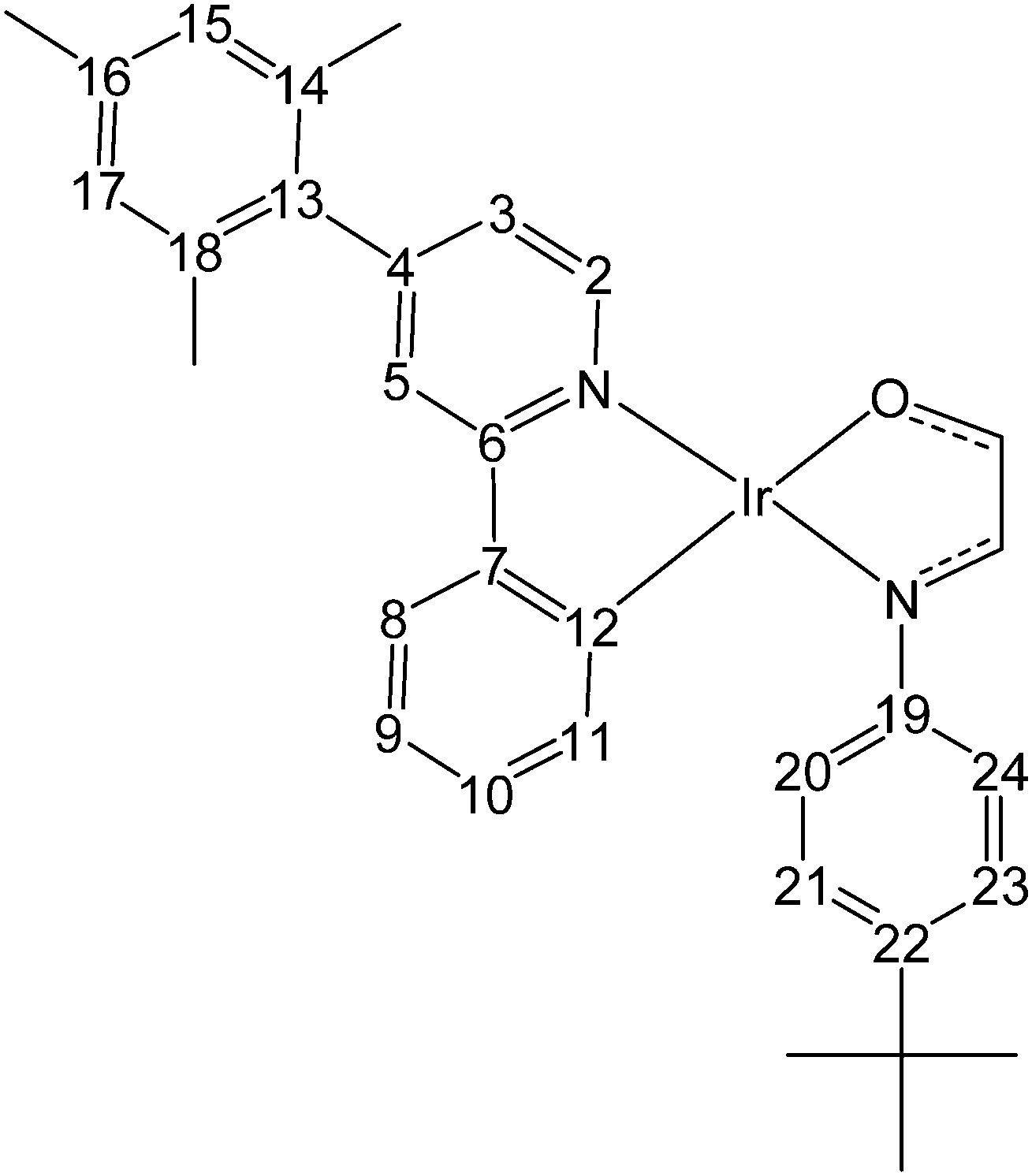 | ||
| Fig. 2 Carbon atom numbering used in the NMR peak assignments of 6 in Table 1. This numbering also applies to NMR data of diiridium complexes 2, 4 and 5. | ||
The thermal stabilities of the diiridium complexes 4–6 were evaluated using thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a nitrogen atmosphere. The 5% weight loss temperatures (Td) are > 370 °C for all complexes, suggesting the complexes should be thermally stable under the conditions of device operation. TGA and DSC traces for 5 and 6 are shown in Fig. S8–S11, ESI.†
Single crystal X-ray structures (Fig. 3, Table 2 and Fig. S1–S3, ESI†) were obtained for 5·hexane and 6·pentane·PhCl solvates, grown from mixtures of stereoisomers in solution. Molecule 5 lies on a crystallographic two-fold axis which is perpendicular to the bridge plane. As noted by Sünkel et al. for complex 2, the N(H) and O atoms are statistically mixed, hence the stereochemistry of the bridge cannot be established unequivocally; however, for 5 the observed electron density is best fit by an overlap of two alternative anti-configurations, with contributions of 0.64(3) [N trans to C(12)] and 0.36(3) [N trans to C(1)]. A similar (but 0.5:0.5) disorder has been observed in 2.32 Molecule 6 is the meso (ΔΛ) diastereomer, possessing crystallographic inversion symmetry. In both complexes, Ir atoms have distorted octahedral coordination with the pyridine N atoms trans to each other. The planarity of the Ir(O2C2N2)Ir system in 5 is distorted by a small (4.7°) twist around the C(23)–C(23′) bond. In 6 the O2C2N2 moiety is planar, the Ir atoms are tilted to opposite sides of its plane by 0.14 Å and the mean axes of the tolyl substituents by 19.4°, the bridging N(2) atom is planar-trigonal (sum of bond angles 358.7°). The anti geometry with N^O coordination at each iridium centre is observed unambiguously for the first time in the crystal structure of complex 6.
 | ||
| Fig. 3 X-ray molecular structures of 5 (major component) and 6. Displacement ellipsoids are drawn at 50% probability level. H atoms, disorder and solvent of crystallisation are omitted for clarity. Primed atoms are generated by twofold axis (5) or inversion centre (6). | ||
| 2 | 5 | 6 | |
|---|---|---|---|
| a N/O disorder in the bridge. | |||
| Ir–O(1) | 2.20(2)a | 2.194(13)a | 2.184(3) |
| Ir–N(1) | 2.18(2)a | 2.13(2)a | |
| Ir–N(2) | 2.132(15)a | 2.135(18)a | 2.147(3) |
| Ir–O(2) | 2.180(15)a | 2.19(2)a | |
| Ir–N(3) | 2.030(5) | 2.042(3) | 2.023(4) |
| Ir–N(4) | 2.029(5) | 2.032(3) | 2.040(4) |
| Ir–C(1) | 1.993(5) | 1.993(4) | 1.982(4) |
| Ir–C(12) | 2.008(6) | 2.013(3) | 2.013(4) |
| Ir⋯Ir′ | 5.718 | 5.688 | 5.726 |
All chelating phenylpyridine systems are slightly folded, with ph/py interplanar angles in 5 (10.9 and 12.5°) larger than in 6 (3.1 and 6.0°). The twists between pyridine rings and their mesityl substituents are much larger. This is expected due to steric effects involving the ortho-methyl substituents in the mesityl groups. These ortho-methyl groups in each mesityl group are non-equivalent in solution based on the observed solution-state NMR data of 4, 5 and 6 here. In 5, the mesityl group iv is twisted to the pyridine ring ii by 45.0°, whereas mesityl iii is disordered, in the ratio of 0.726(9) to 0.274(9), between two orientations inclined to pyridine ring i by 77.9° and 84.9°, respectively. In 6, the i/iii and ii/iv twists are 71.8° and 61.8°, respectively.
The redox properties of the diiridium complexes 4–6 were studied by cyclic voltammetry (CV) in dichloromethane (DCM) solution at 298 K (Fig. 4) and the data are tabulated along with those reported for 134 and [Ir(ppy)2Cl]217 for comparison in Table 3. CV data were not reported for complex 2 for comparison.32 All complexes display two quasi-reversible, one-electron oxidation waves which are assigned to sequential oxidation of the metal centred Ir3+/Ir4+ redox couples. The half-wave oxidation potentials for 4 are similar to that of [Ir(ppy)2Cl]217 as expected. The iridium atoms in 5 and 6 with first half-wave oxidation potentials of +0.38 to +0.40 V are more easily oxidised than for 4 at +0.56 V due to the stronger electron-withdrawing effect of the oxamidato bridge in 5 and 6 compared to the μ-dichloro bridge in 4. The two isomers of compound 1 have peak separations of 400–410 mV where the bridge plays a significant role in the photophysical properties of 1.34 Although complexes 1, 5 and 6 have similar Ir⋯Ir intramolecular distances, the peak separations observed for 5 and 6 are smaller than for 1 and suggest that oxamidato bridges play a minor role with respect to their photophysical properties. The larger peak separation (ΔE) of 220 mV between the two waves of 6 compared to ΔE 160 mV for 5 suggests the monocation species is more stable for 6 than for 5. This is consistent with more HOMO character on the bridge of 6 compared to 5 (see below). The tert-butylphenyl groups at the bridge must contribute to the increased stability of the monocation from 6 with respect to the monocation from 5.
 | ||
| Fig. 4 Cyclic voltammetry traces for 4, 5 and 6. | ||
| Complex |
E
ox(1)1/2a (V) |
E
ox(2)1/2a (V) |
ΔEa (mV) | Ir⋯Ir distance (Å) | HOMOb (eV) | HOMOc (eV) |
|---|---|---|---|---|---|---|
| a 0.1 M (nBu4NPF6) in DCM at 298 K, scan rate 100 mV s−1, referenced to the internal decamethylferrocene/decamethylferrocenium couple (Fc*Me/Fc*Me+) at −0.55 V with the ferrocenium/ferrocene (FcH/FcH+) couple as reference at 0.00 V. b HOMO levels calculated from CV potentials by HOMO = −4.8 + (−Eox1/2), using ferrocene as the standard. c HOMO energies calculated from optimised geometries at B3LYP/LANL2DZ:3-21G*. | ||||||
| 1 | +0.22 | +0.63 | 410 | 5.061 | −5.02 | −4.59 |
| 4 | +0.56 | +0.88 | 320 | 3.812 | −5.36 | −4.91 |
| 5 | +0.38 | +0.54 | 160 | 5.688 | −5.18 | −4.79 |
| 6 | +0.40 | +0.62 | 220 | 5.726 | −5.20 | −4.82 |
| Ir(ppy)2Cl2 | +0.53 | +0.79 | 260 | 3.771 | −5.33 | −4.98 |
The photophysical data for 4–6 are listed in Table 4, with data for [Ir(ppy)3] included for comparison. The absorption spectral profiles of 4–6 in 2-methylTHF are very similar (Fig. 5a). The high energy peaks are assigned to characteristic ligand-centred (LC) spin-allowed π → π* transitions.46 At longer wavelengths the complexes exhibit a series of weaker peaks between 350 and 520 nm which are attributed to singlet and triplet mixed metal-to-ligand charge transfer states (1MLCT and 3MLCT) with the lowest energy band at 492 nm for 4, and 500 nm for 5 and 6 assigned to the spin-forbidden triplet 3MLCT absorption.46,47 The PL spectra of 4 and 5 show well-defined peaks at λmax 518 and 529 nm, respectively, and like Ir(ppy)3,48 are broad and featureless which indicates a dominant 3MLCT contribution and negligible signature of a ligand centred triplet (3LC) contribution. The PL emission of 6 at λmax 522 nm also has a lower energy shoulder at 550 nm which indicates a higher contribution from the 3LC states for this complex as a result of the additional functionality on the bridge increasing the structural rigidity (Fig. 5b).
| Complex | λ Absmax /nm | λ PLmax /nm | PLQYcΦPL/% | τ p /μs |
|---|---|---|---|---|
| a Data obtained in <10 μM 2-methylTHF solutions at 20 °C. b Data obtained in degassed 2-methylTHF solution with excitation wavelength 340 nm. c Measured relative to Ir(ppy)3ΦPL = 0.97 (ref. 49) in degassed 2-methylTHF at 20 °C: estimated error ± 5%. d The phosphorescence decay lifetimes of thin films prepared by spin-coating on quartz substrate from chlorobenzene solutions in poly(methyl methacrylate): estimated error ± 5%. | ||||
| 4 | 341, 360, 410, 463, 492 | 518 | 5.6 | N/A |
| 5 | 315, 350, 415, 470, 500 | 529 | 73 | 0.84 |
| 6 | 315, 350, 415, 470, 500 | 522 | 63 | 1.164 |
| Ir(ppy)3 | 510 | 97 | 1.4 | |
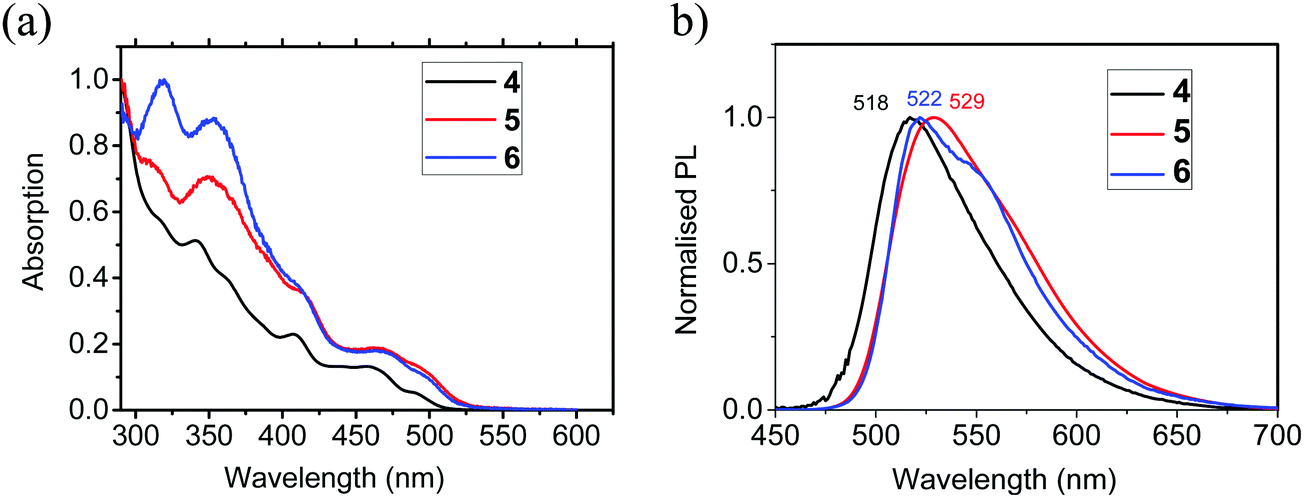 | ||
| Fig. 5 (a) Normalised absorption spectra of complexes 4–6 in 2-methylTHF. (b) Photoluminescence spectra of complexes 4–6 in 2-methylTHF at 20 °C. | ||
The photoluminescence quantum yields (PLQYs) of 4, 5 and 6 in deoxygenated 2-methylTHF solutions are 5.6%, 73% and 63%, respectively, compared with the reference complex Ir(ppy)3 at 97%.49 The very low QY for 4 is consistent with previously reported values for other μ-dichloro-bridged dimers.15,16 The considerably higher quantum yields for 5 and 6 demonstrate that enhancement is induced by the oxamidato bridging unit, and the values are consistent with that reported previously for complex 2 (ΦPL 60%).32
The phosphorescence decay lifetimes of thin films of the complexes 4–6 prepared by spin-coating on quartz substrate from chlorobenzene solutions in poly(methyl methacrylate) (PMMA) are shown in Fig. 6. The data for complex 4 deviate from single exponential characteristics, and this is ascribed to the low solubility and aggregation behaviour of this complex in solution which leads to a strong lifetime dispersion corresponding to different aggregated states. Complexes 5 and 6 both have short phosphorescence lifetimes of 0.84 and 1.164 μs, respectively, originating from 3MLCT which indicates strong spin-orbital coupling with the singlet states.
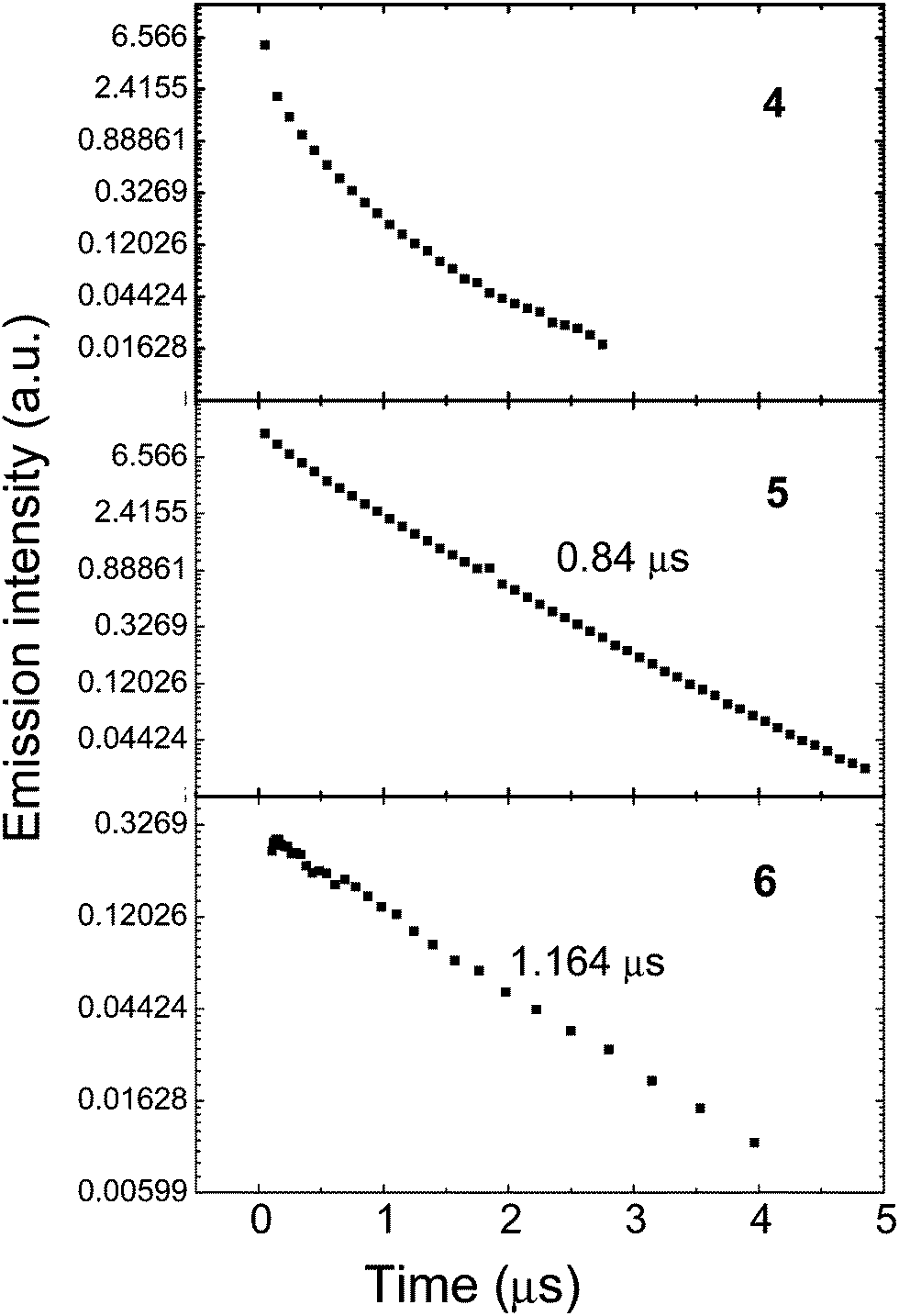 | ||
| Fig. 6 Phosphorescence decays of the complexes doped at 5 wt% in inert PMMA matrix on a log-linear scale. Excitation wavelength 355 nm. | ||
Computations
Optimised geometries of complexes, 2, 4, 5 and 6, in meso (ΔΛ) and racemic (ΔΔ/ΛΛ) forms were obtained by calculations using the hybrid DFT functional B3LYP. These geometries are in good agreement with their experimentally-determined molecular structures (Table S2, ESI†). The meso form of 4 (ΔΛ) is not known experimentally due to steric constraints, as previously reported34 for the parent derivative Ir(ppy)2Cl2, with the total energy of 4 (ΔΛ) higher than 4 (ΔΔ) by 9.0 kcal mol−1. The relative energy differences between meso (ΔΛ) and the more stable racemic (ΔΔ) forms for 5 and 6 are only 2.6 and 2.1 kcal mol−1 respectively thus both forms would be observed as expected here.Electronic structure calculations show that the frontier molecular orbitals for 4 resemble those reported17 for Ir(ppy)2Cl2 (Fig. 7). The HOMO has iridium and phenyl contributions whereas the LUMO contain pyridyl character (Table S3, ESI†). Similar orbital make ups are found for meso (ΔΛ) and racemic (ΔΔ) forms of 5 suggesting that the oxamidato bridges contribute little to the frontier orbitals (7–9% bridging character in HOMO, Fig. 7 and Fig. S4 and Tables S4, S5, ESI†). By contrast, the HOMO for complex 6 (ΔΛ) contains substantial oxamidato bridge character (35%) along with HOMO−1 and HOMO−2 being similar in energies to the HOMO (Fig. 8 and Table S6, ESI†). Complex 6 (ΔΔ) has the nature of HOMO, HOMO−1 and HOMO−2 in a different order while they all have similar energies (Fig. S5 and Table S7, ESI†). The greater bridge involvement in the properties of 6 with respect to 5 is supported by cyclic voltammetry and photophysical observations here.
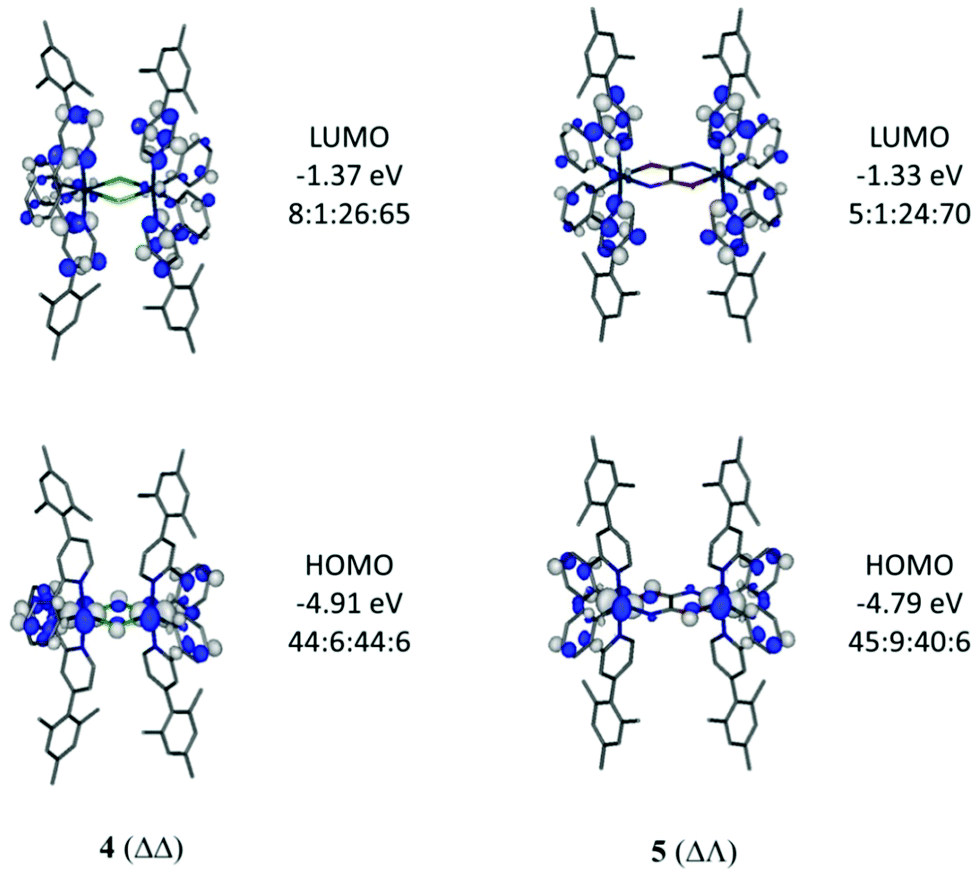 | ||
| Fig. 7 Frontier orbitals for 4 (ΔΔ) and 5 (ΔΛ). Contour values are plotted at ±0.035 e bohr−3. The Ir:bridge:phenyl:pyridyl ratios represent the atom/group MO contributions in percentages. | ||
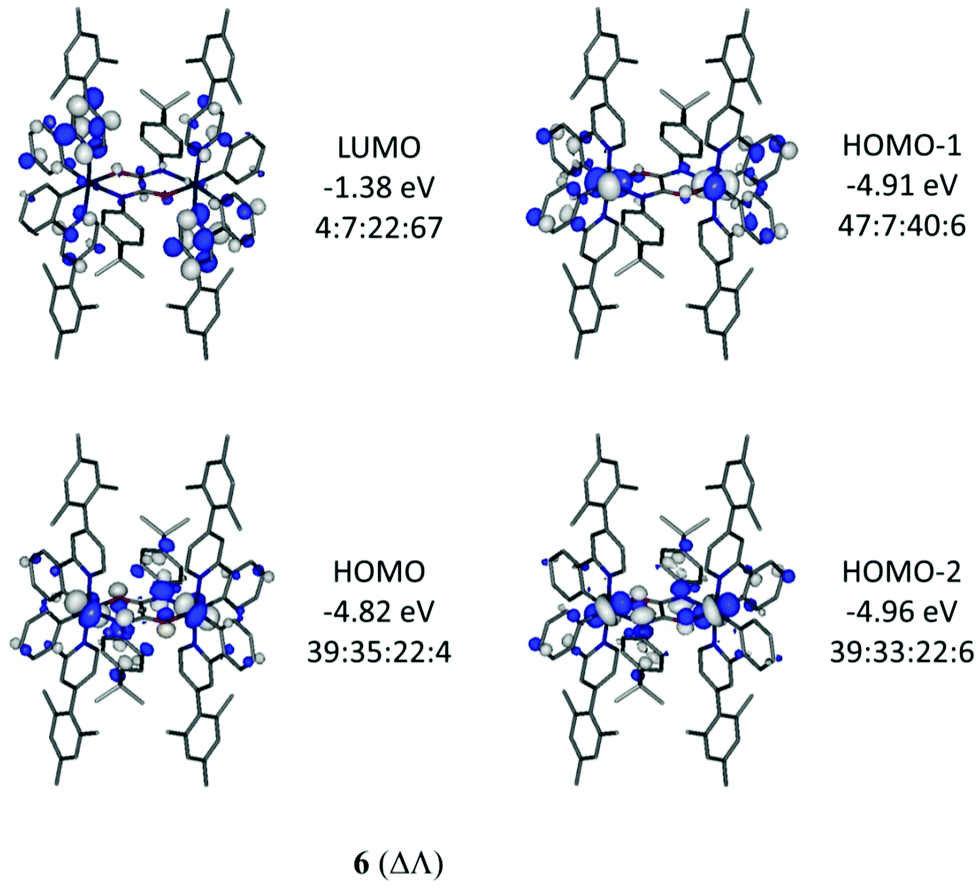 | ||
| Fig. 8 Important orbitals for 6 (ΔΛ). Contour values are plotted at ±0.035 e bohr−3. The Ir:bridge:phenyl:pyridyl ratios represent the atom/group MO contributions in percentages. | ||
The trend in the computed HOMO energies for 4, 5 and 6 is in good agreement with measured oxidation potentials (Table 3). Complex 4 has a considerably lower HOMO energy than the HOMO energies in 5 and 6; thus 4 has a higher oxidation potential than 5 and 6. The calculated HOMO–LUMO energy gaps are similar at 3.54, 3.46 and 3.44 eV for 4, 5 and 6, respectively, and are in accord with the trend in the observed emission energies. This agreement is supported further by TD-DFT data where the observed lowest absorption energies are in excellent agreement with predicted S0 → T1 energies (Table S8, ESI†).
Electrophosphorescent OLED characterisation
PhOLEDs were fabricated using complexes 5 or 6 as the dopant phosphor. Complex 4 was not studied because of its very low PLQY and very limited solubility. The device architecture comprised a simple single-emissive-layer which was a blend of poly(vinylcarbazole) (PVK) as the host material, OXD-7 (an electron-transporting material) and the Ir complex to give the architecture: ITO/PEDOT:PSS (45 nm)/PVK:OXD-7 (40%):Ir complex (5%) (70 nm)/TPBi (30 nm)/LiF/Al. An electron transporting layer of TPBi was incorporated adjacent to the cathode to optimize charge balance and confine excitons in the emitter layer.50 The current–voltage characteristics of the devices are shown in Fig. 9. Both complexes show similar device characteristics, with slightly higher external quantum efficiency and power efficiency for 6 (Table 5). The reason for the differences between 5 and 6 is not clear, but the data suggest that attaching substituents to the bridge (as in 6) may be a viable design strategy to further enhance the EL efficiency. The electroluminescence (EL) emission maxima of the devices from both 5 and 6 (λELmax 533 nm) are slightly red shifted compared to the corresponding photoluminescence maxima of the complexes in solution (529 and 522 nm, Table 1). The devices give green emission with a brightness Lmax > 25000 cd m−2 which is remarkably high for PhOLEDs fabricated using diiridium complexes. Compared to PhOLEDs of complex 1 under very similar conditions,34 the brightness of 5 and 6 is considerably higher, while the external quantum efficiency is lower. The moderate maximum external quantum efficiencies (EQE) for 5 and 6 (ηextca. 3–3.5%) indicate a significant dark current conduction through the device layers. It is well known that using PVK as a host and hole transporting layer can lead to increased device operating voltage and hence lower device power efficiency.51 Attempts were made to improve the EQE with an additional electron blocking layer consisting of poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(4,4′-(N-(4-sec-butylphenyl)diphenylamine))] (TFB)52 which was spin-coated on the top of the PEDOT:PSS layer from toluene solution. This device structure required preparing and spinning the emissive layer using N,N-dimethylformamide (DMF) as solvent to avoid dissolving the TFB layer. Since the high molecular weight PVK used as the host in the initial devices is insoluble in DMF, low molecular weight PVK (Mw 90000) which dissolves in DMF was used instead. The device efficiency and brightness with low molecular weight PVK is extremely low (<0.1%). However, by including the TFB layer the EQEmax of the low molecular weight PVK device improved from 3.5% (Table 5) to ca. 4% (Fig S13, ESI†). We have not explored alternative more complex device structures, as our aim in this work is to use a simpler single-active-layer solution-processable structure which is applicable to low-cost, large-area devices and panels for lighting applications. It is notable that the data for 5 and 6 strongly compete with the brightest reported PhOLEDs using bimetallic complexes as the emitter. They are more than twice as bright, although with lower EQE, than comparable PhOLEDs using complex 1 (maximum brightness 11000 cd m−2; EQEmax 7%; green emission).34 Electroluminescence data for other diiridium PhOLEDs are as follows: (bridge; brightness; EQE; colour). (i) μ-Dichloro; 13000 cd m−2; 0.8%; green.16 (ii) μ-Dichloro; 3700 cd m−2; 3.2%; green.17 (iii) μ-Dichloro; 6000 cd m−2; 2.6%; green.36a (iv) Diarylpyrimidinyl; 18410 cd m−2; 14.4%; red.33 (v) Fluorenyl; 1040 cd m−2; near-white.36b For a cationic diiridium complex with a carbazolyl bridge; 1022 cd m−2; yellow-orange.37
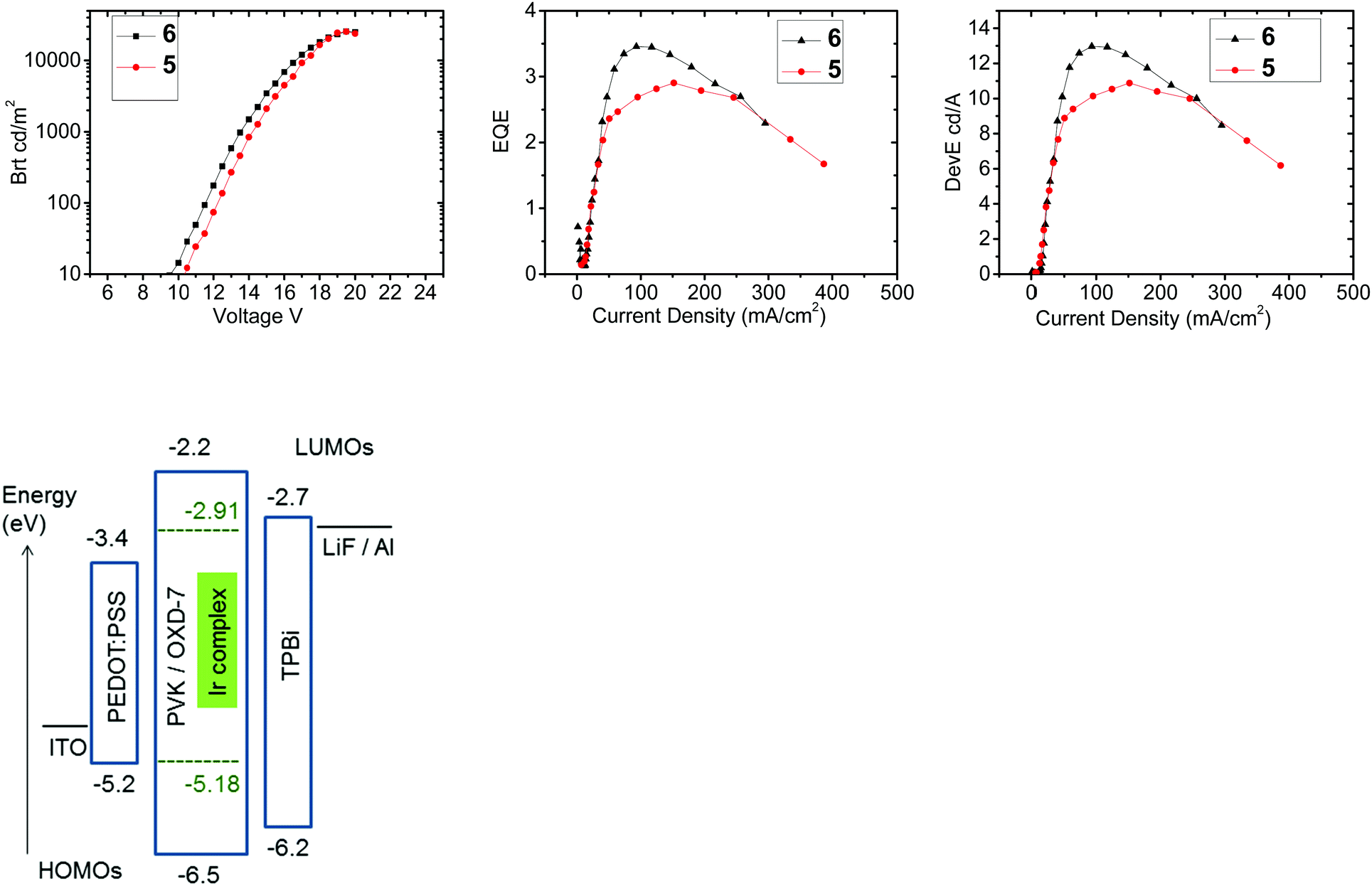 | ||
| Fig. 9 Efficiencies and brightness data for complexes 5 and 6 in the device architecture stated in Table 5, and an energy level diagram for the devices. | ||
The PhOLED data for 5 and 6 are, therefore, significantly different from data on other diiridium complexes studied in our laboratory,16,17,34 and elsewhere,33,36 demonstrating that properties can be tuned by varying the bridge structure. The high molecular weight of diiridium complexes generally precludes their fabrication into thin films by thermal evaporation, therefore, they are unlikely to achieve the very high EQEs which characterise PhOLEDs of mono-iridium complexes which are deposited by vacuum deposition.53 However, high brightness, as observed for 5 and 6, is widely recognised as an important figure-of-merit for PhOLED characterisation.52 Moreover, the solution-processability of the host-dopant systems is especially attractive for the construction of large-area electronics for relatively low-cost practical applications via spin-coating, roll-to-roll processing or ink-jet printing.54
Conclusions
Compared to the vast number of mononuclear iridium complexes used in PhOLEDs, the study of dinuclear complexes in this context is an emerging and very promising topic for further research. In this work we have described the synthesis, X-ray crystal structures and optoelectronic properties of the new cyclometalated diiridium complexes 5 and 6 bridged by oxamidato ligands. In particular, a novel structural feature is that complex 6 provides the first reported example with any substituent (other than hydrogens) on the oxamidato bridge. The complexes are highly emissive (ΦPL 73% and 63% in solution) with short excited state lifetimes of τP 0.84 and 1.16 μs, respectively. PhOLEDs using complexes 5 and 6 as the emissive dopants in a simple architecture using a solution-processed active layer give bright green electroluminescence with remarkably high luminance (Lmax > 25000 cd m−2) for diiridium complexes. These studies expand the range of diiridium complexes that are suitable as dopants for PhOLED applications and should stimulate further studies on chemically tailoring the structures of diiridium complexes to yield highly emissive and colour-tunable systems.
Experimental
General
All commercial chemicals were used without further purification unless otherwise stated. Solvents were dried through an HPLC column on an Innovative Technology Inc. solvent purification system. Column chromatography was carried out using 40–60 μm mesh silica. 1H, 13C{1H}, 2D 1H–1H COSY and 1H–13C correlation (HSQC and HMBC) NMR spectra were recorded on a solution-state Varian VNMRS-700 spectrometer. The proton chemical shifts are referenced to CHCl3 (7.25 ppm) or CHDCl2 (5.32 ppm) present in CDCl3 and CD2Cl2 solutions, respectively. The 13C chemical shifts are referenced to the deuterated solvent CDCl3 or CD2Cl2 at 77.00 and 53.38 ppm, respectively. Mass spectra were measured on a Waters Xevo OTof MS with an ASAP probe, a Thermoquest Trace or a Thermo-Finnigan DSQ. Elemental analyses were performed on a CE-400 Elemental Analyzer. Thermal analysis was performed in a helium atmosphere using a Perkin-Elmer Pyris 1 instrument.Synthesis
Anal. calcd for C80H72Cl2Ir2N4: C, 62.20; H, 4.70; N, 3.63. Found: C, 61.59; H, 4.56; N, 3.60%.
1H NMR (700 MHz, CD2Cl2) δ 9.67 (d, 3JHH = 5.5 Hz, 4H, C6H), 7.74 (s, 4H, C3H), 7.51 (d, 3JHH = 8.0 Hz, 4H, C8H), 7.04 (s, 4H, C15H), 7.01 (s, 4H, C17H), 6.83 (m, 8H, C5H and C9H), 6.68 (dd, 3JHH ∼ 7.5 Hz, 4H, C10H), 5.92 (d, 3JHH = 8.0 Hz, 4H, C11H), 2.39 (s, 12H, C16Me), 2.13 (s, 12H, C18Me), 2.12 (s, 12H, C14Me).
13C{1H} NMR (176 MHz, CD2Cl2) δ 168.49 (C2), 151.58 (C6), 150.80 (C4), 144.50 (C12), 144.11 (C7), 137.90 (C16), 135.33 (C13), 135.49 (C14), 134.62 (C18), 130.03 (C11), 129.32 (C10), 128.52 (C15), 128.23 (C17), 123.71 (C8), 123.44 (C5), 121.42 (C9), 120.41 (C3), 20.79 (C16Me), 20.46 (C18Me), 20.21 (C14Me). Complex 4 was very recently reported elsewhere and analysed by 1H NMR spectroscopy.40,41
1H NMR (700 MHz, CDCl3) δ major isomer 8.94 (d, 3JHH = 5.7 Hz, 2H, C6H), 8.82 (d, 3JHH = 5.6, 2H, C6′H), 7.64 (s, 2H, C3H), 7.61 (s, 2H, C3′H), 7.49 (d, 3JHH ∼ 7.6, 2H, C8H), 7.45 (d, 3JHH = 7.6, 2H, C8′H), 7.03 (dd, 3JHH = 5.7, JHH = 1.9 Hz, 2H, C5H), 7.02 (s, 2H, C15′H), 7.00 (s, 2H, C15H), 6.97 (s, 2H, C17H), 6.95 (s, 2H, C17′H), 6.85 (dd, 3JHH = 5.7, JHH = 1.9 Hz, 2H, C5′H), 6.81–6.75 (m, 4H, C9H, C9′H), 6.73–6.67 (m, 4H, C10H, C10′H), 6.35 (d, 3JHH ∼ 7.7, 2H, C11H), 6.30 (s, 2H, NH), 6.25 (d, 3JHH ∼ 7.7, 2H, C11′H), 2.39 (s, 6H, C16Me), 2.37 (s, 6H, C16′Me), 2.16 (s, 6H, C14Me), 2.14 (s, 6H, C14′Me), 2.02 (s, 6H, C18′Me), 1.99 (s, 6H, C18Me). Minor isomer 8.68 (d, 3JHH = 5.6, 2H, C6H), 8.63 (d, 3JHH = 5.7, 2H, C6′H), 7.68 (s, 2H, C3H), 7.65 (s, 2H, C3′H), 7.51 (d, 3JHH = 7.6, 2H, C8H), 7.48 (d, 3JHH ∼ 7.6, 2H, C8′H), 7.03 (s, 2H, C17H), 7.02 (s, 4H, C15H, C15′H), 6.98 (s, 2H, C17′H), 6.84 (dd, 3JHH = 5.7, JHH = 1.9, 2H, C5H), 6.81–6.75 (m, 4H, C9H, C9′H), 6.76 (dd, 3JHH ∼ 5.7, JHH = 1.9, 2H, C5′H), 6.73–6.67 (m, 2H, C10H, C10′H), 6.35 (d, 3JHH ∼ 7.7, 2H, C11H), 6.29 (s, 2H, NH), 6.25 (d, 3JHH ∼ 7.7, 2H, C11′H), 2.38 (s, 12H, C16Me, C16′Me), 2.16 (s, 6H, C14Me, C14′Me), 2.10 (s, 6H, C18Me), 2.07 (s, 6H, C18′Me).
13C{1H} NMR (176 MHz, CDCl3) δ major isomer 174.46 (CO). 169.23 (C2), 168.98 (C2′), 154.78 (C12), 150.58 (C4), 149.88 (C4′), 149.02 (C6), 147.76 (C6′), 144.78 (C12′), 144.32 (C7), 144.30 (C7′), 137.93 (C16), 137.89 (C16′), 135.82 (C13), 135.79 (C13′), 135.45 (C14), 135.33 (C14′), 135.13 (C18), 134.98 (C18′), 132.52 (C11), 132.11 (C11′), 129.54 (C10), 129.34 (C10′), 128.58 (C15), 128.56 (C15′), 128.48 (C17), 128.35 (C17′), 124.26 (C8), 123.87 (C8′), 122.41 (C5), 122.38 (C5′), 120.69 (C9), 120.36 (C9′), 119.86 (C3), 119.42 (C3′), 21.08 (C16Me), 21.05 (C16′Me), 20.85 (C18Me), 20.82 (C18′Me), 20.59 (C14Me), 20.56 (C14′Me). Minor isomer 174.35 (CO), 169.20 (C2), 168.91 (C2′), 154.71 (C12), 150.48 (C4), 149.72 (C4′), 149.23 (C6), 147.65 (C6′), 144.78 (C12′), 144.35 (C7), 144.25 (C7′), 137.83 (C16, C16′), 135.79 (C13, C13′), 135.36 (C14), 135.33 (C14′), 135.20 (C18), 135.01 (C18′), 132.66 (C11), 132.24 (C11′), 129.50 (C10), 129.39 (C10′), 128.53 (C15), 128.52 (C15′), 128.41 (C17, C17′), 124.29 (C8), 123.85 (C8′), 122.51 (C5), 122.50 (C5′), 120.72 (C9), 120.38 (C9′), 119.76 (C3), 119.45 (C3′), 21.08 (C16Me), 21.07 (C16′Me), 20.76 (C18Me), 20.61 (C18′Me), 20.53 (C14Me, C14′Me).
:hexane (1:1 v/v) (the DCM was saturated with K2CO3) yielded a light orange solid 6 (0.225 mg, 64%) which was recrystallised from a mixture of pentane and chlorobenzene.
MS(MALDI-TOF) m/z 1824.70 ([M + H, 191Ir, 193Ir], 100%). Anal. calcd For C102H98Ir2N6O2: C, 66.75; H, 5.32; N, 4.55. Found: C, 67.15; H, 5.41; N, 4.61%.
1H NMR (700 MHz, CD2Cl2) δ major isomer 9.03 (d, 3JHH = 5.7, 2H, C6H), 8.77 (d, 3JHH ∼ 5.7, 2H, C6′H), 7.76 (s, 2H, C3H), 7.59 (d, 3JHH ∼ 7.6, 2H, C8H), 7.45 (s, 2H, C3′H), 7.35 (s, 4H, C17H, C17′H), 7.10 (s, 4H, C15H, C15′H), 7.03 (dd, 2H, C5H), 7.02 (d, 2H, C8′H), 6.98 (dd, 2H, C5′H), 6.84 (dd, 3JHH ∼ 7.5, 2H, C9H), 6.74 (m, 2H, C10H), 6.72 (d, 3JHH = 8.6, 4H, C21/23H), 6.58 (dd, 2H, C9′H), 6.55 (m, 2H, C10′H), 6.49 (d, 3JHH = 7.8, 2H, C11H), 6.25 (d, 3JHH = 8.6, 4H, C20/24H), 6.15 (d, 3JHH = 7.8, 2H, C11′H), 2.38 (s, 12H, C16Me, C16′Me), 2.26 (s, 6H, C18′Me), 2.21 (s, 6H, C14Me), 2.19 (s, 6H, C18Me), 2.18 (s, 6H, C14′Me), 1.10 (s, CMe3). Minor isomer 9.25 (d, 3JHH = 5.6, 2H, C6H), 8.83 (d, 3JHH ∼ 5.6, 2H, C6′H), 7.76 (s, 2H, C3H), 7.60 (d, 3JHH ∼ 7.6, 2H, C8H), 7.40 (s, 2H, C3′H), 7.35 (s, 4H, C17H, C17′H), 7.10 (s, 4H, C15H, C15′H), 7.10 (dd, 3JHH = 7.2, JHH = 2.0, 2H, C5H), 7.04 (d, 2H, C8′H), 7.04 (dd, 2H, C5′H), 6.84 (dd, 2H, C9H), 6.74 (m, 2H, C10H), 6.68 (d, 3JHH = 8.6, 4H, C21/23H), 6.58 (dd, 2H, C9′H), 6.55 (m, 2H, C10′H), 6.41 (d, 3JHH = 7.7, 2H, C11H), 6.22 (d, 3JHH = 8.6, 4H, C20/24H), 6.27 (d, 3JHH = 7.8, 2H, C11′H), 2.41 (s, 6H, C16Me), 2.39 (s, 6H, C16′Me), 2.24 (s, 6H, C14Me), 2.15 (s, 6H, C14′Me), 2.02 (s, 6H, C18′Me), 1.97 (s, 6H, C18Me), 1.09 (s, CMe3).
13C{1H} NMR (176 MHz, CD2Cl2) δ major isomer 171.66 (CO), 169.43 (C2), 168.39 (C2′), 152.88 (C12), 150.77 (C4), 150.52 (C4′), 149.35 (C6), 148.45 (C12′), 147.50 (C6′), 145.87 (C22), 145.05 (C7), 143.47 (C7′), 141.79 (C19), 137.90 (C16), 137.88 (C16′), 135.92 (C13), 135.73 (C13′), 135.28 (C14), 135.27 (C14′), 134.86 (C18, C18′), 133.24 (C11), 131.75 (C11′), 129.13 (C10), 128.86 (C10′), 128.40 (C15, C15′, C17), 128.36 (C17′), 124.31 (C8), 124.02 (C21/23), 123.53 (C20/24), 123.52 (C8′), 122.88 (C5), 122.20 (C5′), 120.91 (C9), 120.47 (C3), 119.73 (C9′), 119.31 (C3′), 33.73 (C22CMe3), 30.99 (C22CMe3), 20.78 (C16Me, C16′Me), 20.58 (C18Me), 20.57 (C18′Me), 20.39 (C14Me), 20.31 (C14′Me). Minor isomer 171.62 (CO), 169.45 (C2), 168.21 (C2′), 152.96 (C12), 150.77 (C4), 150.27 (C4′), 149.23 (C6), 148.64 (C12′), 147.63 (C6′), 145.67 (C22), 145.00 (C7), 143.43 (C7′), 142.10 (C19), 137.92 (C16), 137.82 (C16′), 135.78 (C13), 135.73 (C13′), 135.28 (C14, C14′), 135.01 (C18, C18′), 133.09 (C11), 132.05 (C11′), 129.23 (C10), 128.88 (C10′), 128.40 (C15, C15′), 128.26 (C17), 128.19 (C17′), 124.26 (C8), 123.98 (C21/23), 123.64 (C20/24),123.40 (C8′), 122.83 (C5), 122.30 (C5′), 120.78 (C9), 120.19 (C3), 119.71 (C9′), 119.19 (C3′), 33.71 (C22![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) Me3), 30.97 (C22CMe3), 20.81 (C16Me), 20.78 (C16′Me), 20.55 (C18Me), 20.49 (C18′Me), 20.37 (C14Me), 20.27 (C14′Me).
Me3), 30.97 (C22CMe3), 20.81 (C16Me), 20.78 (C16′Me), 20.55 (C18Me), 20.49 (C18′Me), 20.37 (C14Me), 20.27 (C14′Me).
X-ray crystallography
The data for 5 was collected on a Rigaku Crystal Logic 4-circle κ-diffractometer with a Saturn 724+ CCD area detector at Station I19 of the Diamond Light Source synchrotron (undulator radiation, double-crystal Si monochromator, λ = 0.6889 Å, ω-scans, 1.0° per frame), and for 6 on a Bruker 3-circle CCD diffractometer D8 Venture with a PHOTON 100 CMOS area detector, using Mo-Kα radiation (λ = 0.71073 Å) from an Incoatec IμS microsource with focusing mirrors (Table S1, ESI†). Absorption corrections were carried out by empirical methods based on multiple scans and Laue equivalents (5) or numerical integration based on crystal face-indexing (6), using SADABS software.56 The structures were solved by direct (5) and Patterson (6) methods and refined by full matrix least squares using SHELXL57 and OLEX258 software. The unit cell of 5 contains 4 closed solvent-accessible voids of 214 Å3 each, presumably containing a disordered hexane molecule, whose contribution was removed using PLATON SQUEEZE solvent-masking procedure.59 The asymmetric unit of 6 (= half the formula unit) contains half a chlorobenzene and half a pentane molecule sharing the same void, which were refined at atomic resolution.Electrochemistry
Cyclic voltammograms were recorded at a scan rate of 100 mV s−1 at room temperature using an air-tight single-compartment three-electrode cell equipped with a Pt disk working electrode, Pt wire counter electrode, and Pt wire pseudo-reference electrode. The cell was connected to a computer-controlled Autolab PG-STAT 30 potentiostat. The solutions contained the complex and n-Bu4NPF6 (0.1 M) as the supporting electrolyte in dichloromethane (DCM). All potentials were determined with the decamethylferrocene/decamethylferrocenium couple as an internal reference in DCM at −0.55 V for the usual reference standard of the ferrocene/ferrocenium couple (FcH/FcH+) in DCM at 0.0 V.Photophysical measurements
UV-Vis and photoluminescence spectra for all samples were obtained with the Shimadzu UV-3600 spectrometer and Jobin Yvon Luminescence spectrometer (FluoroMax-3). Phosphorescence decay lifetime data were measured by time-resolved emission spectroscopy (TRES) using a pulsed Nd-yttrium aluminium garnet (Nd:YAG) laser (λ = 355 nm), repetition rate 1–10 Hz, FWHM 120 ps and maximum pulse energy of 7 mJ. The light emitted by the samples was dispersed through a spectrograph (TRIAX 180, Jobin Yvon-Spex) and the separated spectra were subsequently detected by a gated intensified charge coupled device (CCD) camera (4 pico, Stanford Computer Optics). The PL decay transients were obtained using exponentially increasing decay and integration times as previously described.60Computations
All calculations were carried out with the Gaussian 09 package.61 All optimised S0 geometries of 2, 4, 5 and 6 were carried out using B3LYP62 with the pseudopotential (LANL2DZ)63 for iridium and 3-21G* basis set for all other atoms.64 This model chemistry was selected on the basis of computational studies on Ir(ppy)2Cl2,17 Ir(ppy)365 and for direct comparison with the reported data for 1.34 All S0 geometries were found to be true minima based on no imaginary frequencies found. Electronic structure and TD-DFT calculations were also carried out on the optimised geometries at B3LYP/LANL2DZ:3-21G*. The MO diagrams and orbital contributions were generated with the aid of Gabedit66 and GaussSum67 packages, respectively. The time-dependent DFT (TD-DFT) method does not include spin-orbit couplings thus no oscillation strengths for the triplet excited states are given and also does not compute mixed singlet–triplet excited states (Table S8, ESI†). Predicted emission data from optimised triplet excited state (T1) geometries by TD-DFT for related iridium complexes are not reliable,17,62 thus T1 geometries are not examined here. Given that the calculated Stokes shifts are small (840–1100 cm−1, 0.10–0.14 eV, Table S8, ESI†), it is assumed that the T1 geometries are not significantly different to their corresponding S0 geometries in 2, 4, 5 and 6.Device fabrication
OLED devices were fabricated using precleaned indium tin oxide (ITO) coated glass substrate patterned to form four pixels of 4 × 4 mm in 24 × 24 mm samples, purchased from Kinetic. The ITO thickness was around 120 nm with a sheet resistance of 15 Ω square−1. The cleaned samples were exposed to UV-ozone for 10 min and purged with dry nitrogen. A hole injection layer of poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) (4083) of thickness 40 nm was spin coated at 5000 rpm for 1 min and then baked on a hotplate at 180 °C for 6 min to remove any remaining moisture. A chlorobenzene solution of 20 mg ml−1 of poly(9-vinylcarbazole) (PVK) of molecular weight 1000000 was both the host material and a hole-transport material. The PVK solution was doped with 40% w/w of (1,3-phenylene)bis[5-(4-tert-butylphenyl)-1,3,4-oxadiazole] (OXD-7) as an electron transport material for balancing charge carrier transport.
Complexes 5 and 6 readily dissolved in chlorobenzene without any aggregated particulates. Complex 4 has low solubility which is reflected in its photophysical properties. Therefore, its device characteristics were not measured. A mixture of PVK (20 mg):OXD-7 (40 wt%) and the complex (5 wt%) was spin-coated at 3000 rpm for 1 min on the top of the PEDOT:PSS layer and baked for 10 min at 110 °C. Each sample was shadow masked to produce four identical devices of area 4 × 4 mm; the samples were then introduced into a nitrogen glove box, where 2,2′,2′′-(1,3,5-benzenetriyl)tris-[1-phenyl-1H-benzimidazole] (TPBi) was evaporated as an electron injection/hole blocking layer at a rate of ∼1 Å s−1 under vacuum at a pressure of ca. 1 × 10−6 Torr, followed by 0.7 nm LiF and 100 nm Al cathodes at a rate of 0.2 and 1 Å s−1, respectively, under the same vacuum conditions. The devices were then encapsulated with DELO UV curable epoxy (Katiobond) and a 12 × 12 mm glass cover slide. The final device structure was ITO/PEDOT:PSS (40 nm)/[PVK:OXD-7 (40 wt%):Ir complex (5 wt%)] TPBi (30 nm)/LiF (0.7 nm)/Al (100 nm). For devices with an additional electron blocking layer of poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(4,4′-(N-(4-sec-butylphenyl)diphenylamine))] (TFB), this layer was spin-coated from toluene solution on the top of the PEDOT:PSS layer. The complex 6 (5 wt%) and OXD-7 (40 wt%) in PVK (Mw 90000) were spin coated from DMF. For all devices the current–voltage (I–V) characteristics and the emission intensities were measured in a calibrated Labsphere LMS-100 integrating sphere and the data acquisition was controlled using a home-written NI LabView program that controlled an Agilent Technologies 6632B power supply. The EL spectra were measured using an Ocean Optics USB 4000 CCD spectrometer supplied with a 400 μm UV-Vis fibre optic cable.
Acknowledgements
We thank EPSRC grant EP/K039423/1 for funding. The Diamond Light Source (RAL) is thanked for the award of instrument time on Station I19 (MT 11145) and the instrument scientists for their kind support. A. M. thanks the Algerian Ministry of Higher Education.References
- Z. Q. Chen, Z. Q. Bian and C. H. Huang, Adv. Mater., 2010, 22, 1534–1539 CrossRef CAS PubMed.
- T. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061–7084 RSC.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- K. K. W. Lo, K. H. K. Tsang, K. S. Sze, C. K. Chung, T. K. M. Lee, K. Y. Zhang, W. K. Hui, C. K. Li, J. S. Y. Lau, D. C. M. Ng and N. Zhu, Coord. Chem. Rev., 2007, 251, 2292–2310 CrossRef CAS.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Struct. Bonding, 2015, 165, 205–256 CrossRef CAS.
- G. Li, W. Guan, S. Du, D. Zhu, G. Shan, X. Zhu, L. Yan, Z. Su, M. R. Bryce and A. P. Monkman, Chem. Commun., 2015, 51, 16924–16927 RSC.
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- (a) W. C. H. Choy, W. K. Chan and Y. Yuan, Adv. Mater., 2014, 26, 5368–5398 CrossRef CAS PubMed; (b) L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 232, 926–952 CrossRef PubMed; (c) X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC; (d) W.-Y. Wong and C.-L. Ho, Coord. Chem. Rev., 2009, 253, 1709–1758 CrossRef CAS; (e) C.-L. Ho, L.-C. Chi, W.-Y. Hung, W.-J. Chen, Y.-C. Lin, H. Wu, E. Mondal, G.-J. Zhou, K.-T. Wong and W.-Y. Wong, J. Mater. Chem., 2012, 22, 215–224 RSC; (f) C.-L. Ho and W.-Y. Wong, New J. Chem., 2013, 37, 1665–1683 RSC; (g) G. Zhou, W.-Y. Wong and X. Yang, Chem. – Asian J., 2011, 6, 1706–1727 CrossRef CAS PubMed; (h) X. Xu, X. Yang, J. Zhao, G. Zhou and W.-Y. Wong, Asian J. Org. Chem., 2015, 4, 394–429 CrossRef CAS; (i) C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2013, 257, 1614–1649 CrossRef CAS.
- (a) K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS PubMed; (b) G. Zhou, W.-Y. Wong and S. Sou, J. Photochem. Photobiol., C, 2010, 11, 133–156 CrossRef CAS; (c) X. Yang, G. Zhou and W.-Y. Wong, J. Mater. Chem. C, 2014, 2, 1760–1778 RSC; (d) L. Ying, C.-L. Ho, H. Wu, Y. Cao and W.-Y. Wong, Adv. Mater., 2014, 26, 2459–2473 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2985–3007 CrossRef CAS.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767–768 CrossRef CAS.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. R. Thompson, J. Am. Chem. Soc., 2003, 125, 7377–7387 CrossRef CAS PubMed.
- G. A. Carlson, P. I. Djurovich and R. J. Watts, Inorg. Chem., 1993, 32, 4483–4484 CrossRef CAS.
- S. Bettington, M. Tavasli, M. R. Bryce, A. S. Batsanov, A. L. Thompson, H. A. Al Attar, F. B. Dias and A. P. Monkman, J. Mater. Chem., 2006, 16, 1046–1052 RSC.
- A. M’hamedi, A. S. Batsanov, M. A. Fox, M. R. Bryce, K. Abdullah, H. A. Al-Attar and A. P. Monkman, J. Mater. Chem., 2012, 22, 13529–13540 RSC.
- M. Y. Wong, G. Xie, C. Tourbillon, M. Sandroni, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Dalton Trans., 2015, 44, 8419–8432 RSC.
- X. Yuan, S. Zhang and Y. Ding, Inorg. Chem. Commun., 2012, 17, 26–29 CrossRef CAS.
- A. Tsuboyama, T. Takiguchi, S. Okada, M. Osawa, M. Hoshino and K. Ueno, Dalton Trans., 2004, 1115–1116 RSC.
- E. A. Plummer, W. Hofstraat and L. De Cola, Dalton Trans., 2003, 2080–2084 RSC.
- L. Donato, C. E. McCusker, F. N. Castellano and E. Zysman-Colman, Inorg. Chem., 2013, 52, 8495–8504 CrossRef CAS PubMed.
- E. S. Andreiadis, D. Imbert, J. Pécaut, A. Calborean, I. Ciofini, C. Adamo, R. Demadrille and M. Mazzanti, Inorg. Chem., 2011, 50, 8197–8206 CrossRef CAS PubMed.
- J. Fernández-Cestau, N. Giménez, E. Lalinde, P. Montaño, M. T. Moreno and S. Sánchez, Organometallics, 2015, 34, 1766–1778 CrossRef.
- F. M. Hwang, H. Y. Chen, P. S. Chen, C. S. Liu, Y. Chi, C. F. Shu, F. I. Wu, P. T. Chou, S. M. Peng and G. H. Lee, Inorg. Chem., 2005, 44, 1344–1353 CrossRef CAS PubMed.
- T. Hajra, J. K. Bera and V. Chandrasekhar, Aust. J. Chem., 2011, 64, 561–566 CAS.
- A. M. Prokhorov, A. Santoro, J. A. G. Williams and D. W. Bruce, Angew. Chem., Int. Ed., 2012, 51, 95–98 CrossRef CAS PubMed.
- G. Li, Y. Wu, G. Shan, W. Che, D. Zhu, B. Song, L. Yan, Z. Su and M. R. Bryce, Chem. Commun., 2014, 50, 6977–6980 RSC.
- P.-H. Lanoë, C. M. Tong, R. W. Harrington, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2014, 50, 6831–6834 RSC.
- M. Graf, K. Sünkel, R. Czerwieniec and H.-C. Böttcher, J. Organomet. Chem., 2013, 745–746, 341–346 CrossRef CAS.
- V. Chandrasekhar, T. Hajra, J. K. Bera, S. M. W. Rahaman, N. Satumtira, O. Elbjeirami and M. A. Omary, Inorg. Chem., 2012, 51, 1319–1329 CrossRef CAS PubMed.
- M. Graf, R. Czerwieniec and K. Sünkel, Z. Anorg. Allg. Chem., 2013, 639, 1090–1094 CrossRef CAS.
- R. E. Daniels, S. Culham, M. Hunter, M. C. Durrant, R. M. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Dalton Trans., 2016, 45, 6949–6962 RSC.
- Y. Zheng, A. S. Batsanov, M. A. Fox, H. A. Al-Attar, K. Abdullah, V. Jankus, M. R. Bryce and A. P. Monkman, Angew. Chem., Int. Ed., 2014, 53, 11616–11619 CrossRef CAS PubMed.
- X. Yang, X. Xu, J. Dang, G. Zhou, C.-L. Ho and W.-Y. Wong, Inorg. Chem., 2016, 55, 1720–1727 CrossRef CAS PubMed.
- (a) M. Y. Wong, G. Xie, C. Tourbillon, M. Sandroni, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Dalton Trans., 2015, 44, 8419–8432 RSC; (b) K. He, X. Wang, J. Yu, H. Jiang, G. Xie, H. Tan, Y. Liu, D. Ma, Y. Wang and W. Zhu, Org. Electrochem., 2014, 15, 2942–2949 CrossRef CAS.
- G. Nasr, A. Guerlin, F. Dumur, L. Beouch, E. Dumas, G. Clavier, F. Miomandre, F. Goubard, D. Gigmes, D. Bertin, G. Wantz and C. R. Mayer, Chem. Commun., 2011, 47, 10698–10700 RSC.
- V. N. Kozhevnikov, Y. Zheng, M. Clough, H. A. Al-Attar, G. C. Griffiths, K. Abdullah, S. Raisys, V. Jankus, M. R. Bryce and A. P. Monkman, Chem. Mater., 2013, 25, 2352–2358 CrossRef CAS.
- A. F. Henwood, A. K. Bansal, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, J. Mater. Chem. C, 2016, 4, 3726–3737 RSC.
- D. Rota Martir, A. K. Bansal, V. Di Mascio, D. B. Cordes, A. F. Henwood, A. M. Z. Slawin, P. C. J. Kamer, L. Martinez-Sarti, A. Pertegas, H. J. Bolink, I. D. W. Samuel and E. Zysman-Colman, Inorg. Chem. Front., 2016, 3, 218–235 RSC.
- H. Benjamin, Y. Zheng, A. S. Batsanov, M. A. Fox, H. A. Al-Attar, A. P. Monkman and M. R. Bryce, Inorg. Chem., 2016, 55, 8612–8627 CrossRef CAS PubMed.
- D. Rota Martir, G. J. Hedley, D. B. Cordes, A. M. Z. Slawin, D. Escudero, D. Jacquemin, T. Kosikova, D. Philip, D. M. Dawson, S. E. Ashbrook, I. D. W. Samuel and E. Zysman-Colman, Dalton Trans., 2016, 45, 17195–17205 RSC.
- D. Rota Martir, C. Momblona, A. Pertegás, D. B. Cordes, A. M. Z. Slawin, H. J. Bolink and E. Zysman-Colman, ACS Appl. Mater. Interfaces, 2016, 8, 33907–33915 Search PubMed.
- S. Sprouse, K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1984, 105, 6647–6653 CrossRef.
- A. Auffrant, A. Barbieri, F. Barigelletti, J. Lacour, P. Mobian, J.-P. Collin, J.-P. Sauvage and B. Ventura, Inorg. Chem., 2007, 46, 6911–6919 CrossRef CAS PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, C. Adachi, P. E. Burrows, S. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634–1641 CrossRef CAS.
- W. J. Finkenzeller and H. Yersin, Chem. Phys. Lett., 2003, 377, 299–305 CrossRef CAS.
- T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard III and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed.
- (a) J.-H. Jou, W.-B. Wang, S.-M. Shen, S. Kumar, I.-M. Lai, J.-J. Shyue, S. Lengvinaite, R. Zostautiene, J. V. Grazulevicius, S. Grigalevicius, S.-Z. Chen and C.-C. Wu, J. Mater. Chem., 2011, 21, 9546–9552 RSC; (b) M. Song, J. S. Park, Y.-S. Gal, S. Kang, J. Y. Lee, J. W. Lee and S.-H. Jin, J. Phys. Chem. C, 2012, 116, 7526–7533 CrossRef CAS.
- (a) H. A. Al-Attar and A. P. Monkman, J. Appl. Phys., 2011, 109, 074516 CrossRef; (b) F. Dumur, Org. Electrochem., 2015, 25, 345–361 CrossRef CAS.
- R. Jin, P. A. Levermore, J. Huang, X. Wang, D. D. C. Bradley and J. C. deMello, Phys. Chem. Chem. Phys., 2009, 11, 3455–3462 RSC.
- J.-W. Jou, S. Kumar, A. Agrawal, T.-H. Li and S. Sahoo, J. Mater. Chem. C, 2015, 3, 2974–3002 RSC.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- I. Welterlich, O. Charov and B. Tieke, Macromolecules, 2012, 45, 4511–4519 CrossRef CAS.
- G. M. Sheldrick, SADABS version 2014/5, Bruker AXS, Madison, USA Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. Rothe and A. P. Monkman, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 075208 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- (a) T. H. Dunning, Jr and P. J. Hay in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, vol. 3, 1976 Search PubMed; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (c) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS; (d) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- (a) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS; (b) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- M. Tavasli, T. N. Moore, Y. Zheng, M. R. Bryce, M. A. Fox, G. C. Griffiths, V. Jankus, H. A. Al-Attar and A. P. Monkman, J. Mater. Chem., 2012, 22, 6419–6428 RSC.
- A. R. Allouche, J. Comput. Chem., 2011, 32, 174–182 CrossRef CAS PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1521969 and 1521970. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7tc00628d |
| This journal is © The Royal Society of Chemistry 2017 |