
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDielectric elastomers with dual piezo-electrostatic response optimized through chemical design for electromechanical transducers†
C.
Tugui
a,
A.
Bele
a,
V.
Tiron
b,
E.
Hamciuc
a,
C. D.
Varganici
 a and
M.
Cazacu
*a
a and
M.
Cazacu
*a
aPetru Poni Institute of Macromolecular Chemistry Iasi, Grigore Ghica Voda Alley, no. 41A, 700487, Iasi, Romania. E-mail: mcazacu@icmpp.ro
bFaculty of Physics, Alexandru Ioan Cuza University of Iasi, Blvd. Carol I no. 11, Iaşi 700506, Romania
First published on 22nd December 2016
Abstract
A new class of elastomers that simultaneously shows sensing, actuation and energy conversion functionalities is synthesized to meet the current requirements for electroactive materials. These new materials consist of a silicone network (polydimethylsiloxane-α,ω-diol crosslinked through chain ends) semi-interpenetrated with different percentages (2, 5, and 10 wt%) of polyimide derivatives stepwise modified by different strategies to improve the compatibility with the silicone core network. By addressing the right chemical pathway, the resulting semi-interpenetrated structures (S-IPNs) show noticeable dielectric permittivity, eps′ (up to 11), and breakdown strength, Ebd (up to 88 μm V−1), improvements as compared with the starting polymers (silicone with eps′ = 2.9 and Ebd = 38 μm V−1 and our best polyimide with eps′ = 6.2 and Ebd = 23 μm V−1). The S-IPNs with 10 wt% polyimide are able to gain energy up to 132 mJ cm−3 at 100% strain and up to 164 mJ cm−3 at maximum strain to develop large actuation strain (up to 8.7%) and show very good piezo-response (up to 44 pm V−1), making them highly suitable for cutting-edge electromechanical applications. For a better evaluation, S-IPNs are compared with one of the commercially available dielectric elastomers, most often used for this purpose.
Introduction
Dielectric elastomer actuators (DEAs) are able to transform electrical energy into mechanical displacement, known as actuation strain.1 These devices are employed in a wide spectrum of applications, such as robotics,2,3 optical systems,4 sensors5–7 and energy harvesting.8,9 The simplest DEAs consist of a thin elastomeric film that is sandwiched between two compliant electrodes.10 Siloxanes, polyurethanes, acrylics and natural rubber represent the main materials that are used as dielectric elastomers. The electrostatic equation which describes the actuation strain in the thickness direction, sz = −εε0E2/Y (where ε and ε0 are the dielectric permittivity of the dielectric layer, and of the free space, respectively, while E represents the applied electric field and Y – Young modulus), reveals very clearly the main factors that influence the actuation efficiency. In soft dielectric elastomers, the permittivity may change considerably with the strain as a result of a strain-dependent polarization response under an imposed electric field. This phenomenon that generally accompanies the “Maxwell stress” is called electrostriction and is related to the change in the material dielectric constant.11–14 In this sense, many studies were focused to enhance the polymer’s ability, like dielectric permittivity,15,16 electrical breakdown strength,17,18 energy density19,20 or mechanical properties, so that they become more suitable for such applications.21,22 In order to improve these features, most studies have been focused on using various fillers23,24 and chemical modification of the polymer matrix.25–27 However, there are certain drawbacks for each of these strategies such as increase of the Young’s modulus by incorporation of different fillers or decrease of the dielectric strength by adding polar contents.28–30 Beside these possibilities, interpenetrating polymer networks represent a new category of materials that can provide high electromechanical properties. In the literature are reported several types of interpenetrating polymer networks (IPNs), mainly based on silicones, acrylics and polyurethanes.31–35 IPNs based on acrylics seem to provide the highest actuation strain, over 300%.36,37 However, to reach these results, the acrylic films need to be prestrained. Moreover, acrylics and polyurethanes exhibit high plastic deformation when they are subjected to repeated mechanical stress cycles.31 On the other hand, silicone elastomers possess high elasticity but require a high electric field to be actuated due to their lower dielectric permittivity.32 Therefore, finding new materials with desired properties represents a big challenge in terms of developing better actuators. Considering the above and continuing our previous work in the development of interpenetrated networks derived from silicones (full silicone21,33 or polyurethane–silicone31 interpenetrating networks), in this paper we used polyimides (PI) as a partner for polydimethylsiloxane (PDMS). Due to their excellent thermal, mechanical and dielectric properties, aromatic polyimides attract particular interest in the development of high temperature piezoelectric and pyroelectric sensors and microelectromechanical devices (MEMS).38 Their investigation was mainly motivated by NASA's interest for developing high performance piezoelectric polymers for a variety of aerospace applications.39 For this purpose, amorphous polyimides containing polar functional groups (mainly –CN) have been synthesized and investigated at Virginia Commonwealth University and NASA LaRC for their potential use as high temperature piezoelectric sensors.40 As a result, a new class of electroactive polymeric blend materials has been created that offers both sensing and actuation dual functionality.41 In principle, these materials consist of an electrostrictive graft elastomer (a flexible backbone elastomer) grafted with polymeric polarizable moieties, such as polyvinylidene difluoride (PVDF) and polarizable crystalline groups, and a poly(vinylidene fluoride-trifluoroethylene) copolymer.41,42 By carefully selecting the composition, there is potential to create a blend system with electromechanical properties that can be tailored for various conditions and applications.42 An actuator was also built comprising both piezoelectric and electrostrictive components in a monolithic construction material, useful as a component in electronic devices with a large and precise displacement for high reliability applications (precision machining, micropumps, optics, astronomy, fluid control, etc.).43 As it is well known, piezoelectricity is the ability of some dielectric materials to change their polarization state when receiving a mechanical impulse and inversely to mechanically deform when applying an electric field. Thus, such a material can be used both as a sensor and as an actuator.38,44 A stress applied on a piezoelectric material alters the separation between the positive and negative charge sites leading to a net polarization at the crystal surface. Similarly, a deformation of these materials is effected by placing them under an electric charge.45 Piezoelectricity in amorphous polymers is different from that in crystalline polymers or compounds, in that polarization is not in a steady state, rather in the quasi-stable state due to freezing molecular dipoles.38In our study, we combined all these properties in one highly sensitive material that was able to develop a large actuation strain. In a first approach, we used a polyimide functionalized with polar cyanide groups as a partner for silicone. In order to improve its compatibility with silicone, we prepared a siloxane–imide copolymer. For comparison, we also prepared materials by incorporating a high polar polyamic precursor without siloxane segments in the silicone matrix. At the other extreme, to ensure a better compatibility between components, we prepared and incorporated within the siloxane matrix a hexafluoroisopropylidene-containing polyimide–polydimethylsiloxane copolymer. The obtained materials were studied from morphological, thermal, mechanical and dielectric points of view. The electromechanical behaviour (actuation and piezoelectricity) was evaluated. The results were compared between them, in correlation with the material composition and reported to those obtained on a commercial elastomer, often used as a dielectric elastomer, Elastosil film produced by Wacker. All samples were measured under similar conditions. There are few examples of PDMS–PI interpenetrating networks reported in the literature, but none of them addresses these materials from this perspective.46,47
Results and discussion
Starting with one of the current requirements for electroactive materials (that is to exhibit high piezoelectric strain and large electric-field induced strain responses, thus conferring sensing and actuation dual functionality41,42), identified and outlined in the Introduction, the idea of the authors was to combine polysiloxane as the dielectric and polyimide, as a piezoelectric component, in a single elastomer. To this aim, a polydimethylsiloxane-α,ω-diol (PDMS) having an average molecular weight Mn = 230![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 was used as a dielectric component and as a matrix for the incorporation of the polyimides with different structures. First, polyamic acid (PAA-1) (Scheme 1a) derived from 4,4′-oxydiphthalic anhydride and 2,6-bis(3-aminophenoxy)benzonitrile in a 1:1 molar ratio was used to obtain series Si-1 (Table 1). As expected, the incompatibility of the two components, due to the large difference of their polarity, led to materials with mechanical properties inappropriate for future use. Therefore, in the second approach, a completely condensed derivative was used. Moreover, for its synthesis, 2,6-bis(3-aminophenoxy)benzonitrile was half replaced with a siloxane diamine (α,ω-bis(aminopropyl)oligodimethylsiloxane with Mn = 1120 g mol−1) thus resulting in a polyimide (PICOSI-1) containing long dimethylsiloxane sequences to ensure better compatibility with the silicone matrix (Scheme 1b). The PICOSI-1 product was used to build the Si-2 series (Table 1). To go to the other extreme, in the third approach a polyimide (PICOSI-2) was used for the preparation of which 4,4′-oxydiphthalic anhydride was replaced with a less polar one, 4,4′-(hexafluoroisopropylidene)diphthalic anhydride, and 2,6-bis(3-aminophenoxy)benzonitrile with 4,4′-bis(4-aminophenoxy)biphenyl, while the ratio between organic and siloxane diamine was increased to 3:2 and the average molecular weight of the latter fell to 750 g mol−1 (Scheme 1c). The obtained polyimide PICOSI-2 was used to prepare the network series Si-3. Each of the three types of polymers was incorporated in different weight percentages (2, 5, and 10%) into polydimethylsiloxane, coded as shown in Table 1, and the mixture was cast as a thin film, which has been stabilized by curing. This occurs as a result of crosslinking polydimethylsiloxane-α,ω-diol according to a well-known pathway,21 with tetraethylorthosilicate (TEOS) as a tetra-functional crosslinking agent in the presence of dibutyltin dilaurate (DBTDL) as a catalyst, with simultaneously evaporating the solvent and alcohol by-product of the condensation-crosslinking reaction. The latter is a slower process because it is controlled by the diffusion of the secondary product outside of the film. Therefore, prior to the investigation, the samples were allowed to age for stabilizing the properties. During this time, it is assumed that polyimide chains without groups able to participate in the crosslinking reaction remain tangled with dimethylsiloxane chains. The aromatic rings within structure permit their self-assembly by π–π stacking that can be associated in this case with physical crosslinking. However, according to the IPN definition, the material cannot be considered to be an interpenetrating network but rather a semi-interpenetrating one, S-IPN (Scheme 2).
000 g mol−1 was used as a dielectric component and as a matrix for the incorporation of the polyimides with different structures. First, polyamic acid (PAA-1) (Scheme 1a) derived from 4,4′-oxydiphthalic anhydride and 2,6-bis(3-aminophenoxy)benzonitrile in a 1:1 molar ratio was used to obtain series Si-1 (Table 1). As expected, the incompatibility of the two components, due to the large difference of their polarity, led to materials with mechanical properties inappropriate for future use. Therefore, in the second approach, a completely condensed derivative was used. Moreover, for its synthesis, 2,6-bis(3-aminophenoxy)benzonitrile was half replaced with a siloxane diamine (α,ω-bis(aminopropyl)oligodimethylsiloxane with Mn = 1120 g mol−1) thus resulting in a polyimide (PICOSI-1) containing long dimethylsiloxane sequences to ensure better compatibility with the silicone matrix (Scheme 1b). The PICOSI-1 product was used to build the Si-2 series (Table 1). To go to the other extreme, in the third approach a polyimide (PICOSI-2) was used for the preparation of which 4,4′-oxydiphthalic anhydride was replaced with a less polar one, 4,4′-(hexafluoroisopropylidene)diphthalic anhydride, and 2,6-bis(3-aminophenoxy)benzonitrile with 4,4′-bis(4-aminophenoxy)biphenyl, while the ratio between organic and siloxane diamine was increased to 3:2 and the average molecular weight of the latter fell to 750 g mol−1 (Scheme 1c). The obtained polyimide PICOSI-2 was used to prepare the network series Si-3. Each of the three types of polymers was incorporated in different weight percentages (2, 5, and 10%) into polydimethylsiloxane, coded as shown in Table 1, and the mixture was cast as a thin film, which has been stabilized by curing. This occurs as a result of crosslinking polydimethylsiloxane-α,ω-diol according to a well-known pathway,21 with tetraethylorthosilicate (TEOS) as a tetra-functional crosslinking agent in the presence of dibutyltin dilaurate (DBTDL) as a catalyst, with simultaneously evaporating the solvent and alcohol by-product of the condensation-crosslinking reaction. The latter is a slower process because it is controlled by the diffusion of the secondary product outside of the film. Therefore, prior to the investigation, the samples were allowed to age for stabilizing the properties. During this time, it is assumed that polyimide chains without groups able to participate in the crosslinking reaction remain tangled with dimethylsiloxane chains. The aromatic rings within structure permit their self-assembly by π–π stacking that can be associated in this case with physical crosslinking. However, according to the IPN definition, the material cannot be considered to be an interpenetrating network but rather a semi-interpenetrating one, S-IPN (Scheme 2).
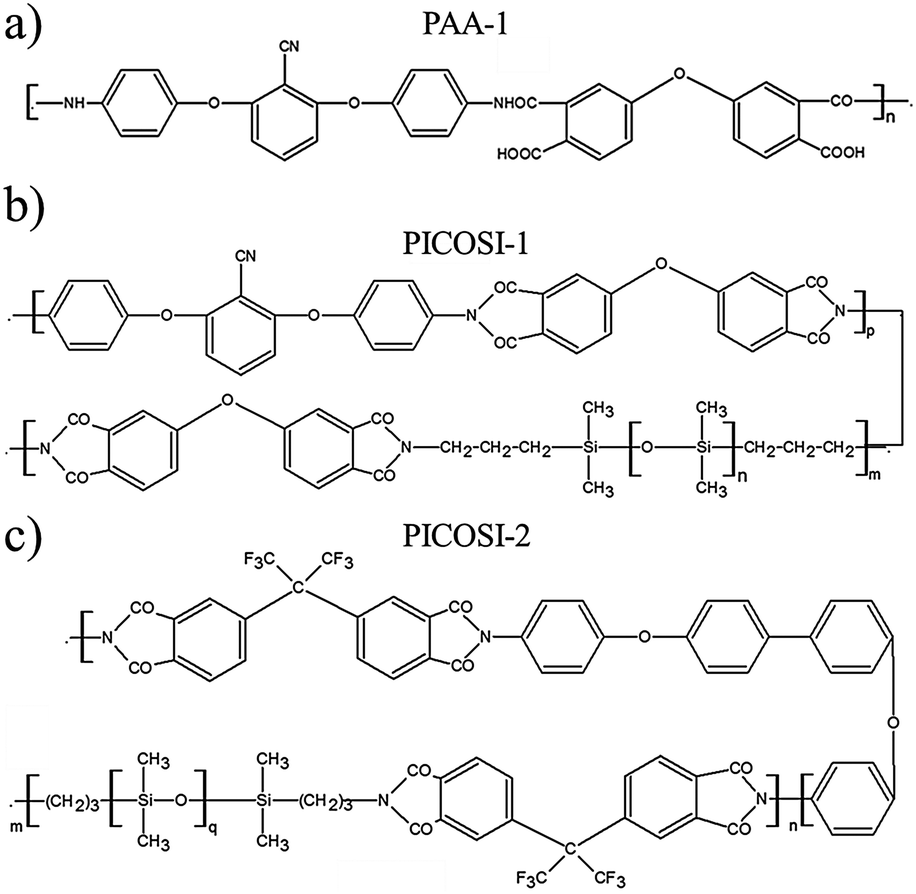 | ||
| Scheme 1 Chemical structures of: (a) polyamic acid, PAA-1 (b) poly(siloxane-imide) containing CN groups, PICOSI-1, and (c) poly(siloxane-imide) containing hexafluoropropylidene groups, PICOSi-2. | ||
| Samplea | PDMS, g | PAA-1, g | PICOSI-1, g | PICOSI-2, g | TEOS, mL |
|---|---|---|---|---|---|
| a 25 mL THF was used as a solvent and 0.01 mL of DBTDL as a catalyst. Meaning of sample codes: Si-X-Y% where, X represents the polyimide type and Y represents the percentage of polyimide reported to the PDMS weight. | |||||
| Si-Blank | 1 | 0 | 0 | 0 | 0.07 |
| Si-1-2% | 0.5 | 0.01 | 0 | 0 | 0.035 |
| Si-1-5% | 0.5 | 0.025 | 0 | 0 | 0.035 |
| Si-1-10% | 0.5 | 0.05 | 0 | 0 | 0.035 |
| Si-2-2% | 0.5 | 0 | 0.01 | 0 | 0.035 |
| Si-2-5% | 0.5 | 0 | 0.025 | 0 | 0.035 |
| Si-2-10% | 0.5 | 0 | 0.05 | 0 | 0.035 |
| Si-3-2% | 0.5 | 0 | 0 | 0.01 | 0.035 |
| Si-3-5% | 0.5 | 0 | 0 | 0.025 | 0.035 |
| Si-3-10% | 0.5 | 0 | 0 | 0.05 | 0.035 |
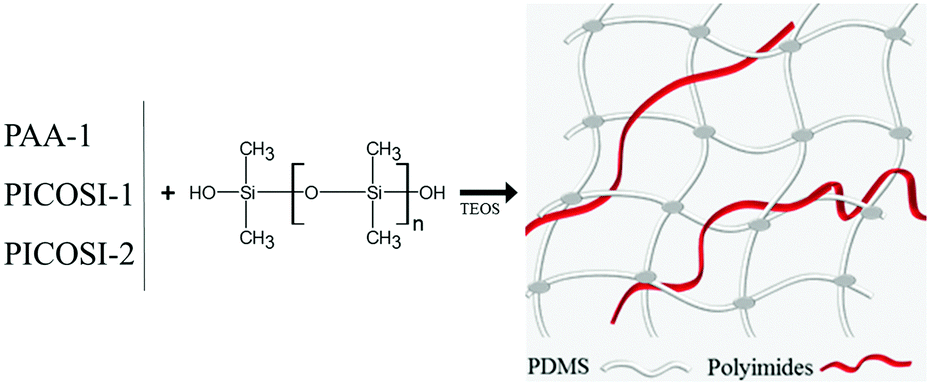 | ||
| Scheme 2 Schematic representation of S-IPN formation. | ||
Morphological and thermal characterization
The prepared films were investigated morphologically using scanning electron microscopy (SEM). The images indicate some structuring in the mixed network compared to the pure PDMS network, being more pronounced in samples of the series Si-1 (Fig. S1, ESI†). In fact, the network obtained by adding 10% of PAA-1 formed very frail films unable to be peeled off nicely from the substrate. This can be explained by the higher polarity of the polyamic acid compared with the other two polyimides. However, additional techniques do not highlight a real phase separation. Thus, energy-dispersive X-ray spectroscopy (EDX) analysis reveals a relatively uniform distribution of elements in the section (Fig. S2, ESI†), while the atomic force microscopy (AFM) phase contrast images (Fig. S3, ESI†) shows a variation of contrast (i.e., 1° for Si-2-10% and 4° for Si-3-10%) well below 10 degrees beyond which phase separation is considered.48For a better evaluation of this phenomenon, the samples were investigated using DSC, a useful analytical tool in assessing solid state interactions. In Fig. 1 are shown the second heating curves of individual linear components (Fig. 1a) and those of the network sets (Fig. 1b–d). On the first heating runs (not shown), the thermal history was removed (i.e. physical dehydration and thermal cyclization for PAA-1, endotherm at 192 °C).49 From Fig. 1a may be observed two glass transition temperature domains (Tg) for the copolymer PICOSI-1, one at −115 °C, corresponding to PDMS moieties, and the other one at 100 °C, describing the polyimide phase (Fig. 1a). In general, the latter is significantly lower than that of a polyimide, due to the presence of the highest PDMS block content. The increase in the Tg value of PDMS from −122 °C to −115 °C (Fig. 1b), as a block in the copolymer PICOSI-1, is explained by partial phase mixing.50 This effect is more pronounced in the case of PICOSI-2 with shorter silicone blocks and a different organic imide, when the Tg value of the former increases from −115 °C to −100 °C (Fig. 1a), this being in good agreement with reported literature data.51 Sample PAA-1 exhibited a Tg value of 212 °C, close to that previously reported for the corresponding imide structure.49
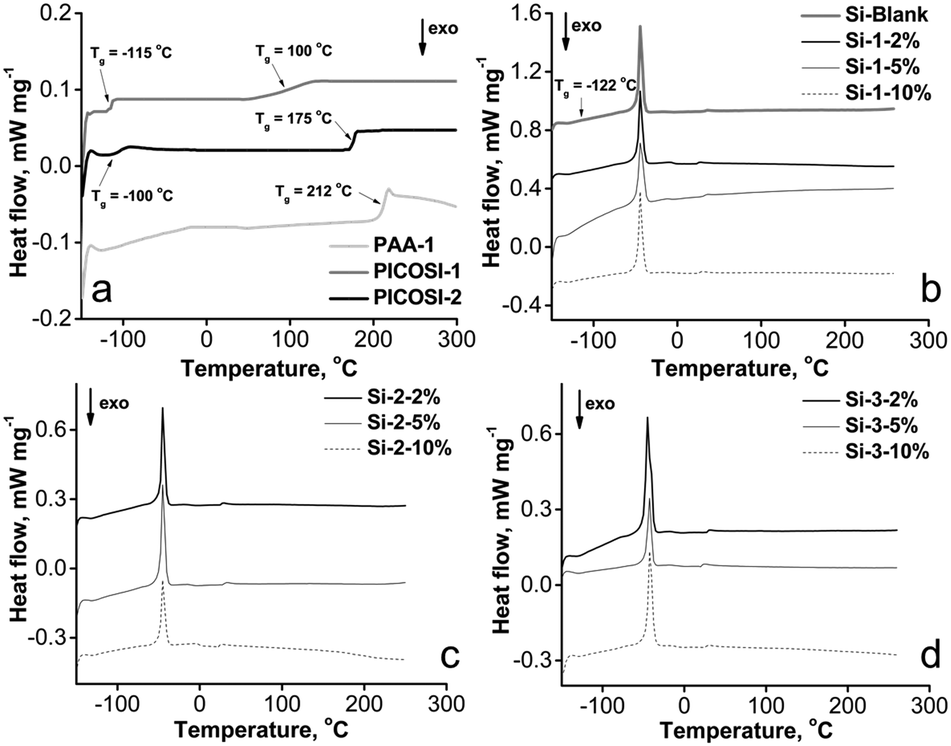 | ||
| Fig. 1 DSC curves of the polyimides and prepared S-IPNs. | ||
DSC measurements on polyimide derivatives revealed neither melting nor crystallization transitions, demonstrating an amorphous morphology. The DSC curves of the networks indicate only a weak Tg value in the range −125 to −122 °C, and a melting endotherm around −45 °C, which describes the PDMS component of the films (Fig. 1b–d).
The presence of a single Tg domain could be due to the low content of polyimide within the network but also to the good miscibility between individual comprising components,52–54 similar to that was found for highly miscible S-IPNs based on linear aromatic polyurethane and crosslinked epoxy resin.55 It is also known that during the synthesis of such network types there occurs a synergism of the individual component’s properties through a forced phase compatibilization,55 hence the improved properties of the films are also described by means of other characterization methods depicted in this article. The glass transition is particularly important for the thermal and dynamic properties of polymers56 and establishes the polarization process conditions.38 Piezoelectric response is determined by the orientation of molecular dipoles. The main factors influencing the piezoelectric response on such materials are: glass transition temperature, which determines the polarization process conditions, dipole concentration, their ability to orient and stability of their orientation.38 Therefore, it is important to identify this parameter by adequate sensitive techniques. Among the techniques suitable for this purpose, although DSC is the most commonly used method, dynamic mechanical analysis (DMA) is considered to be the most sensitive, while dielectric analysis (DEA) is less sensitive.57 In general, the values determined by different methods differ slightly.58 These values determined through DSC, DMA and DEA are, for example, −122, −120 and −115 °C, respectively (for the Si-Blank sample), and −125, −123 and −120 °C, respectively, for sample Si-2-5% (Fig. S4–S7, ESI†), these suggesting that in our case the values determined by DSC are quite close to those determined by DMA.
Mechanical and dielectric behaviour
The effects of the polyimide incorporation within a silicone matrix on the mechanical properties of the resulting IPNs were investigated by stress–strain measurements (Fig. 2 and Table 2). Due to the rigid nature of the polyimides, an increase in the modulus and a decrease in the elongation would have been expected.59 Overall, these trends take shape although in our case there are several parameters related to polyimide structure (nature of the anhydride and diamine monomers, presence/absence of siloxane segments, their proportion and length) that can influence the material properties, including mechanical ones. Thus, the three series of samples have in common the silicone matrix and differ in polyimide structure, whose polarity appears to decrease from PAA-1, a polyamic acid derived from 4,4′-oxydiphthalic anhydride and 2,6-bis(3-aminophenoxy)benzonitrile to PICOSI-1 due to polyamic acid cyclization and partial replacement of polar diamine, 2,6-bis(3-aminophenoxy)benzonitrile, with long, flexible, non-polar siloxane diamine and then to PICOSI-2 by further replacing 4,4′-oxydiphthalic anhydride with less polar anhydride, 4,4′-(hexafluoroisopropylidene)diphthalic anhydride. From another perspective, the three polyimides differ by flexibility growing in the order PAA-1 < PICOSI-2 < PICOSI-1 as a result of the incorporation of siloxane diamine which is the longest and is introduced in the highest percentage in polyimide PICOSI-1. The values found for the mechanical properties (elongation at break and Young's modulus) for the prepared samples could be logically interpreted as follows. In all three series, by incorporating 2 wt% polyimide, the modulus does not change (0.3 MPa in Si-2-2% and Si-3-2%) or change insignificantly (0.4 MPa in Si-1-2%) as compared to the Si-Blank sample (0.3 MPa), while elongation increases slightly in the first two series (1129% for Si-1-2% and 1106% for Si-2-2%) and significantly decreased (723%) in series Si-3.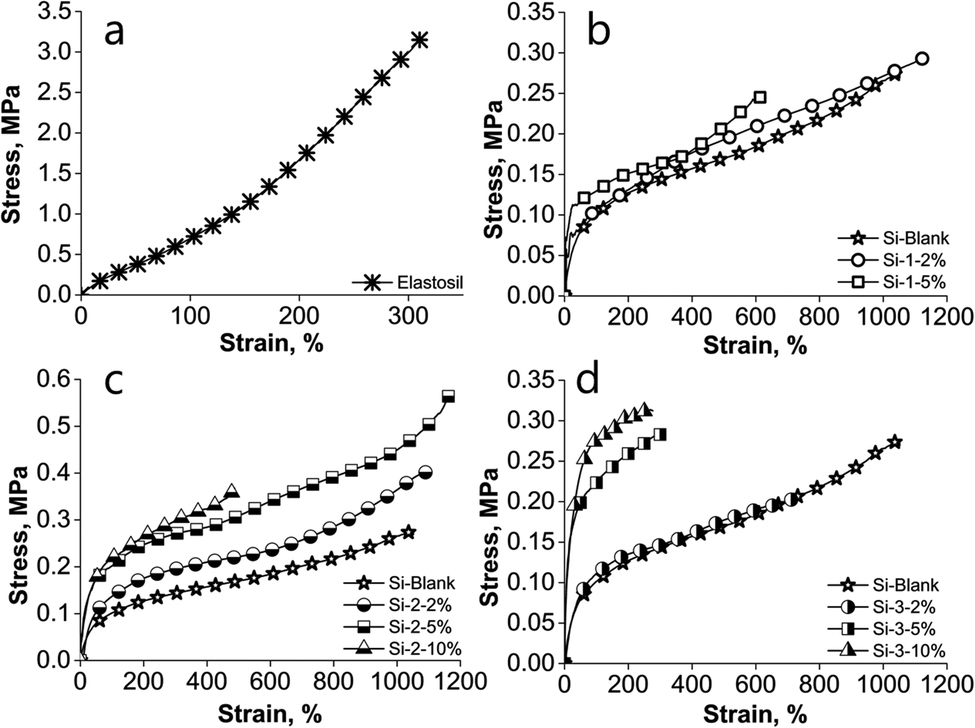 | ||
| Fig. 2 Stress–strain curves of: (a) Elastosil; (b) PAA-1 series; (c) PICOSI-1 series and (d) PICOSI-2 series. | ||
| Sample | Young’s modulus, MPaa | Strain@break, % | eps′@100, Hz | eps′@104, Hz | p , KPa | s x , % | s x , % | Ebde, V μm−1 | Ebdf, V μm−1 |
|---|---|---|---|---|---|---|---|---|---|
| a @20% strain. b Maxwell pressure@15 V μm−1. c Lateral actuation strain@15 V μm−1. d Lateral actuation strain@maximum electric field. e Electrical breakdown from actuation. f Electrical breakdown in the static regime. | |||||||||
| Elastosil | 1 | 314 | 2.6 | 2.6 | 5.1 | 0.3 | 22.5 | 75 | 105 |
| Si-Blank | 0.3 | 1052 | 2.9 | 2.8 | 5.7 | 1.1 | 3.2 | 25 | 38 |
| PAA-1 | — | — | 4.2 | 3.9 | — | — | — | — | 19 |
| PICOSI-1 | — | — | 2.4 | 2.3 | — | — | — | — | 20 |
| PICOSI-2 | — | — | 6.2 | 3.8 | — | — | — | — | 23 |
| Si-1-2% | 0.4 | 1129 | 4.4 | 3.4 | 8.8 | — | 0.4 | 5 | 38 |
| Si-1-5% | 0.5 | 623 | 10.2 | 4.2 | 20.3 | — | 0.2 | 3 | 34 |
| Si-1-10% | — | — | 9.6 | 6.8 | — | — | — | — | 33 |
| Si-2-2% | 0.3 | 1106 | 3.1 | 2.7 | 6.2 | 1.4 | 4.4 | 35 | 54 |
| Si-2-5% | 0.6 | 1176 | 3.3 | 2.3 | 6.6 | 1.4 | 5.5 | 40 | 71 |
| Si-2-10% | 0.6 | 487 | 3.9 | 2.6 | 7.8 | 1.7 | 8.7 | 50 | 88 |
| Si-3-2% | 0.3 | 725 | 4.6 | 3.1 | 9.2 | 1.6 | 1.6 | 15 | 48 |
| Si-3-5% | 0.8 | 313 | 6.5 | 2.5 | 12.9 | 2 | 2 | 15 | 53 |
| Si-3-10% | 0.8 | 277 | 11.1 | 4.1 | 22.1 | 2.2 | 4.3 | 25 | 60 |
This means that due to the incompatibility of the two components and low polyimide content, intermolecular interactions are lacking so that polyimide acts much like a plasticizer. Due to its less polar nature, polyimide PICOSI-2 is more compatible with silicones, its presence leading to lower elongation. Increasing the polyimide content to 5 wt% increases the possibility of the polyimide to form extensive physical intermolecular self-interactions which increase the modulus (0.5, 0.6 and 0.8 MPa for Si-1-5%, Si-2-5% and Si-3-5%, respectively) and elongation decreases dramatically (623% for Si-1-5% and 313% for Si-3-5%). The exception is the sample Si-2-5% for which elongation becomes higher (1176%). Responsible for this are the densely and long, flexible siloxane segments within the copolymer PICOSI-1, which allow easy sliding of chains to each other. Upon adding 10 wt% polyimide, the appropriate sample belonging to the series Si-1 becomes friable, unable to form freestanding films, while in the series Si-2 and Si-3 significant decreases in elongation are recorded (487% for Si-2 and 277% for Si-3). This is due to a structuring of the material as shown by SEM images. Overall, the obtained S-IPNs meet all mechanical requirements of a dielectric elastomer designed for actuation devices. When comparing with the commercial silicone sample (Elastosil), the Young's modulus of the prepared films is lower, while almost all samples show higher elongation values (Fig. 2 and Table 2).
Because the dielectric properties of the elastomers represent also an important aspect for electromechanical applications, the prepared films were investigated using dielectric spectroscopy. To get a complete picture of the influence of the polyimides on the dielectric properties, first the three derivatives PAA-1, PICOSI-1 and PICOSI-2 were analysed after being pressed in the pellet form (Fig. S8, ESI†). The lowest value of the dielectric permittivity, around 2.5, similar to the commercial silicone (Table 2), found in the case of PICOSI-1, is mainly attributed to the presence of high siloxane moieties from the polyimide backbone (Fig. 3a).
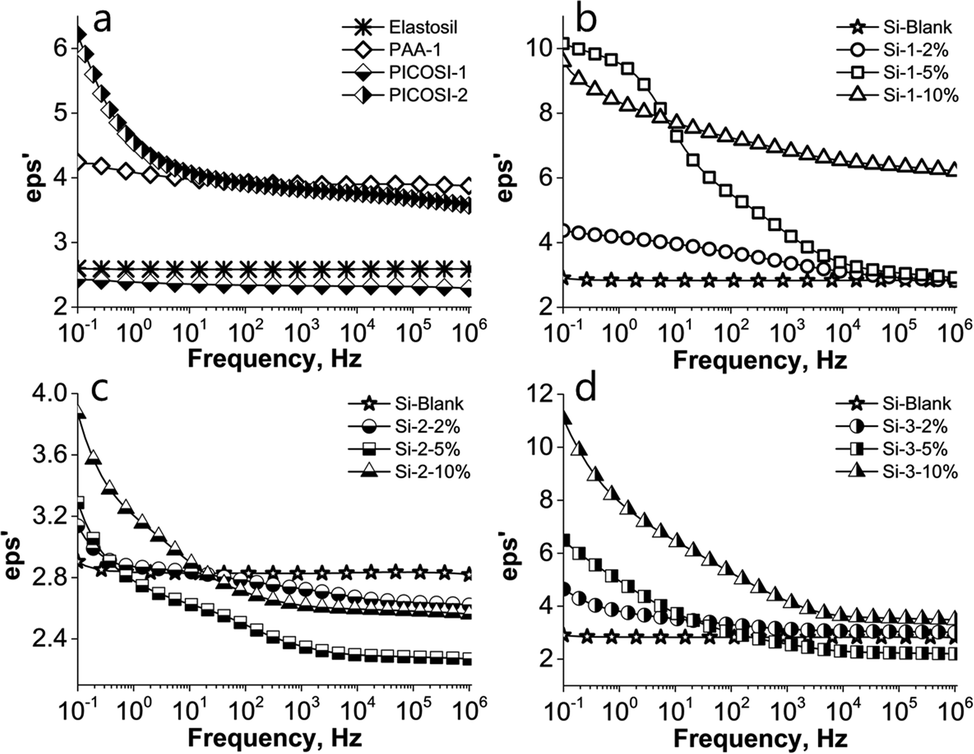 | ||
| Fig. 3 Dielectric permittivity dependence on the frequency of: (a) Elastosil; (b) PAA-1 series; (c) PICOSI-1 series and (d) PICOSI-2 series. | ||
The dielectric permittivity of the imidic copolymers decreases as the number of siloxane units increases. In addition, due to their relatively high mobility, the siloxane chains can increase the free volume, thus decreasing the dielectric permittivity.60,61 On the other side, the higher permittivity of the PICOSI-2 copolymer can be related to the reduced amount of siloxane from the main chain compared to the PICOSI-1 copolymer. In the cases of PICOSI-1, Elastosil and Si-Blank, due to their low polarity, the dielectric permittivity is constant over the entire range of frequency, these being only electronically polarized. In contrast, all the prepared S-IPNs show an increasing permittivity in the low frequency range suggesting dipole polarization (Fig. 3). The presence of the COOH and CF3 groups in S-IPNs with PAA-1 and PICOSI-2, respectively, increases the polarity, thus leading to an increase of dielectric permittivity in the low frequency range from 4.2 for PAA-1 to 9.6 for Si-1-10% and from 6.2 for PICOSI-2 to 11.1 for Si-3-10% (Fig. 3b and d). As expected, due to the low content of the polar component within PICOSI-1, the resulting IPNs show a very slight increase of permittivity, from 2.4 for PICOSI-1 to 3.9 for the derived network Si-2-10%. Dielectric losses of IPNs follow the same trend as the dielectric permittivity increases with increasing amounts of polyamic acid/polyimide (Fig. S9, ESI†). Excepting the S-IPNs that contain PICOSI-2, where the dielectric losses are up to 10 even at low concentrations of polyimide, in the other IPNs the dielectric losses are generally below 1. By interpenetrating the Si-Blank network with the three types of polymers (PAA-1, PICOSI-1 and PICOSI-2) we obtained an improvement of the dielectric features in almost all S-IPNs.
Electromechanical behaviour
Beyond mechanical and dielectric aspects, the prepared samples were electromechanically tested. Both electrical breakdown and actuation measurements were undertaken in accordance with the standards established by the EuroEAP society.62 The breakdown tests reveal that sample PAA-1 has the lowest breakdown value, 19 V μm−1, while the two polyimides, PICOSI-1 and PICOSI-2, show slightly higher values of 23 V μm−1 and 20 V μm−1, respectively (Fig. 4). Moreover, the breakdown strength values of the S-IPNs based on PDMS–PAA-1 decrease with the increasing amount of polyamic acid. By contrast, the electrical breakdown of S-IPN series based on PDMS–PICOSI-1 and PDMS–PICOSI-2 increases with the amount of polyimide starting from 54 V μm−1 for Si-2-2% to 88 V μm−1 for Si-2-10% and from 48 V μm−1 for Si-3-2% to 60 V μm−1 for Si-3-10%, respectively.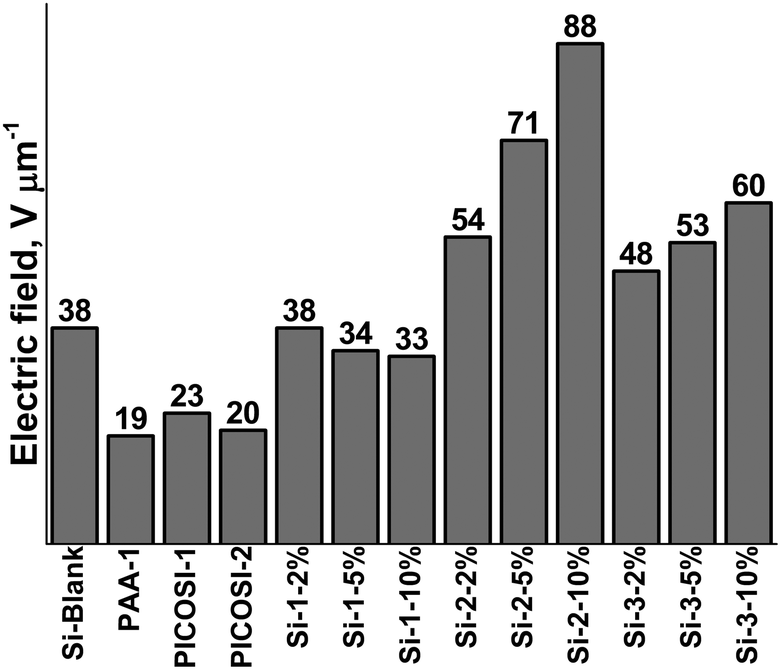 | ||
| Fig. 4 Comparative electrical breakdown values of the prepared samples. | ||
Furthermore, the breakdown values of these S-IPNs are higher than of PDMS network and polyimide, suggesting that there is a synergistic effect between the IPN components, as was also found in the case of the previously discussed thermal behaviour. The breakdown values determined during actuation measurements follow a similar trend as in the case of static measurements, increasing by enriching the polyimide content (Table 2).
The decrease in thickness during actuation measurements because of electrostatic attraction between electrodes leads to lower values for dielectric breakdown. By interpenetrating a PDMS network with 10% of polyimide based on a 1:1 molar ratio between organic and siloxane diamine (PCOSI-1) the dielectric strength was enhanced by more than 130%.
Further, all the obtained IPNs were evaluated in actuation tests. The electromechanical actuation measurements were performed on circular films 20% equiaxial prestrained by applying a symmetric rectangular signal and kept at the required voltage for 2 s (Fig. S10, ESI†). An AFG300 wave generator and a Trek 20/20C-HS power supply were used to provide the voltage signal and the high voltage needed for the actuation measurements. The voltage was stepwise increased to the required actuation voltage until the breakdown occurred (Fig. S11, ESI†).
The S-IPN based on PDMS-PAA-1 being very frail showed poor actuation displacement, the breakdown occurring at very low voltages (Fig. 5b). The commercial film Elastosil has the highest lateral strain of about 22%, but a very high electric field, nearly 80 V μm−1, was required to reach this value (Fig. 5a). Instead, although the actuation strain values of siloxane–polyimide semi-interpenetrating networks are lower, the electric field required to be actuated is much lower (Fig. 5c and d). For instance, at an electric field of 15 V μm−1, the lateral strain of the commercial film Elastosil is 0.3%, while for all S-IPNs the lateral strain is higher than 1% and even higher than 2% for sample Si-3-10% (Table 2). Likewise, the calculated electrostatic pressure, p, confirms the actuation results, having a similar tendency, to increase with the increasing amount of polyimide within IPNs (Table 2).
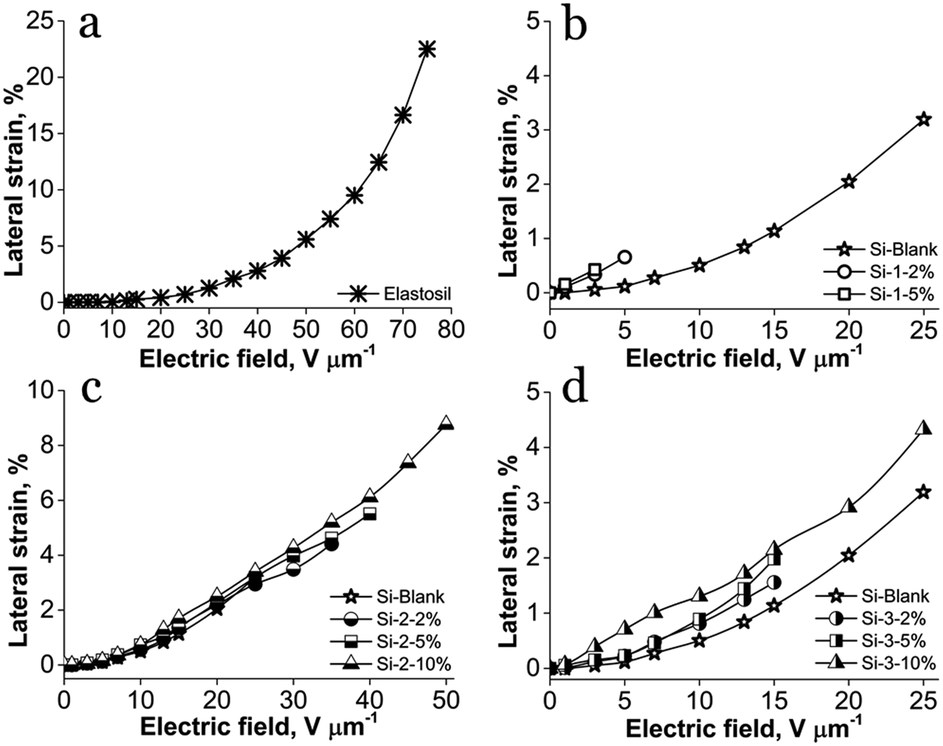 | ||
| Fig. 5 Lateral actuation strain of: (a) Elastosil; (b) PAA-1 series; (c) PICOSI-1 series and (d) PICOSI-2 series. | ||
Beside the electrical breakdown improvements, by interpenetrating the siloxane network with the two polyimides, PICOSI-1 and PICOSI-2, an improvement in actuation displacement was achieved as compared with the pure crosslinked PDMS used as a reference sample.
Considering that the dielectric elastomers can also be used as generators, by converting the mechanical work into electrical energy, the gain energy over a cycle of operation was calculated using eqn (1), assuming a linear deformation (Fig. 6).63,64 The maximum energy, ΔWmax, that can be converted is limited by the electrical breakdown Ebd2 and the maximum strain smax.
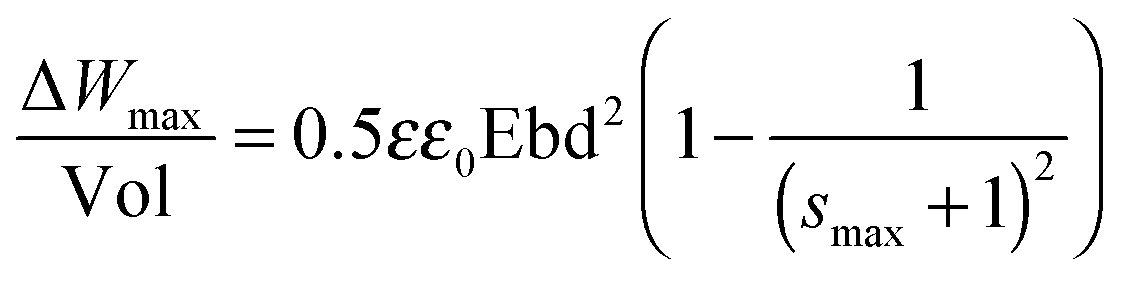 | (1) |
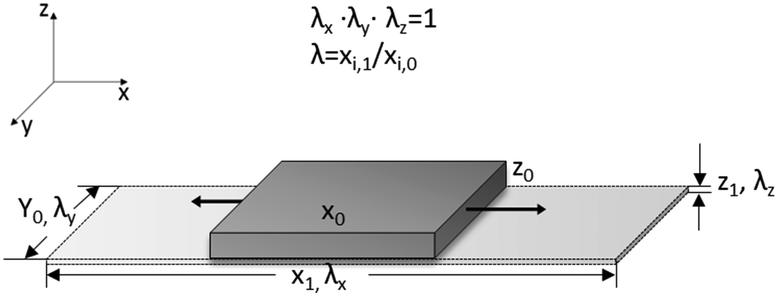 | ||
| Fig. 6 Dielectric elastomer uniaxially stretched. | ||
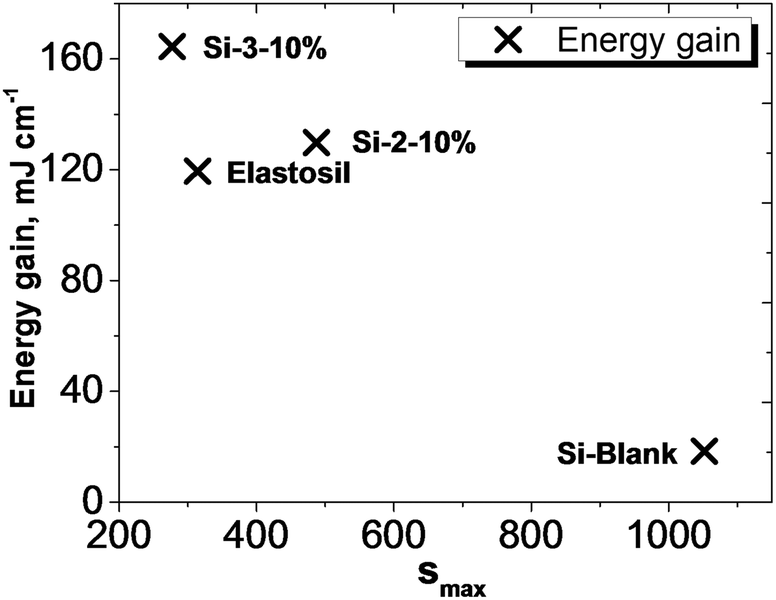 | ||
| Fig. 7 Comparative energy gain values at maximum strain for Si-Blank, Elastosil, Si-2-10% and Si-3-10%. | ||
Piezoelectric measurements
As is known, the electromechanical responses of the two components on which the new dielectric elastomers were built are based on different phenomena, electrostatic in the case of silicone, while the polyimides meet the conditions for piezoelectric (non-symmetric, polar and insulating).65,66 It has been demonstrated by modelling and experimentally that the dipole moment of the polar (e.g., nitrile) substituent as well as the dianhydride contribute significantly to the polarization and piezoelectric response of the polyimide.39 Thus, one can assume that the total strain response of the system is cumulative one being mainly due to the electrostatic compression (Maxwell stress effect) of the silicone network attributed to the force generated by the accumulated charges on the opposing surfaces of the film under an applied electric field but also to the piezoelectric effect. The surface topography and piezoelectricity were evaluated by piezoelectric force microscopy (PFM) measurements for the sample films Si-2-10% and Si-3-10% having the highest polyimide content (10 wt%) and compared with pure crosslinked silicone as a reference (Si-Blank), the results being presented in Fig. 8.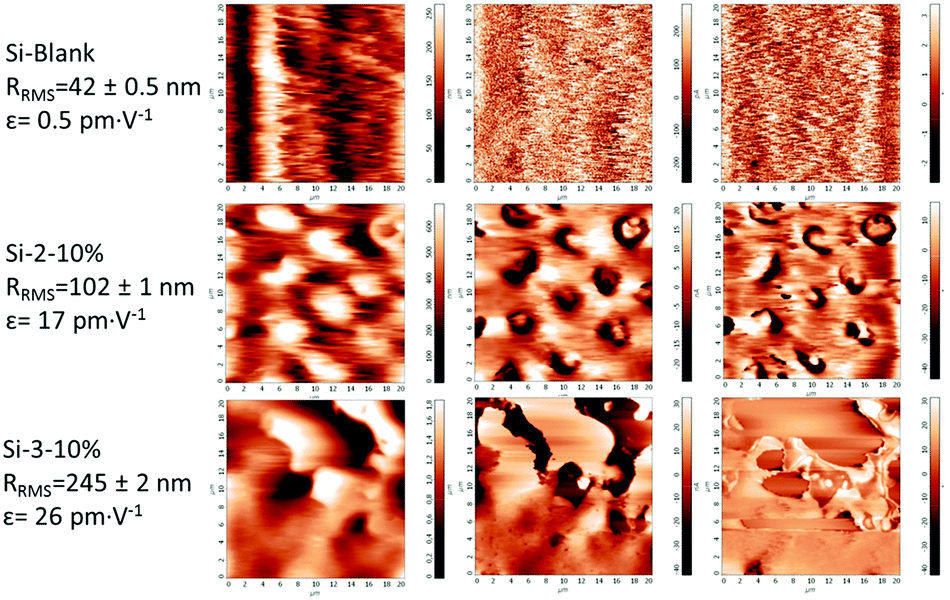 | ||
| Fig. 8 Piezoelectric force microscopy images of Si-Blank, Si-2-10% and Si-3-10%. | ||
In the case of pure crosslinked silicone, no noticeable piezo-response and phase contrast were found, while the magnitude and phase images of polysiloxane–polyimide film show a variety of contrasts implying widespread local d33 values and allow identifying domain structures with different orientations of polarization. The surface roughness as root mean square roughness, RRMS, was estimated from topography images and was found to be in the range of 100 to 240 nm. The dependence of the piezo-response (amplitude signal) versus the applied alternating current (AC) bias voltage was plotted for each of the three samples in 100 different points placed equidistantly on the surface of the film with an area of 20 × 20 μm2. From the slopes of the piezo-response curves, the average piezoelectric constant (d33) values were estimated.67
The polysiloxane–polyimide film Si-2-10% shows a piezoelectric constant of up to 30 pm V−1 with an average value of 17 ± 0.3 pm V−1, while the Si-3-10% sample shows a piezoelectric constant of up to 44 pm V−1 with an average value of 26 ± 0.6 pm V−1 (Fig. 8).
As can be seen, the average piezoelectric coefficient of S-IPNs increases drastically with the polyimide content from almost 0 pm V−1 for Si-Blank up to 44 pm V−1 for the S-IPN based on the polyimide poorest in siloxane and containing hexafluoropropylidene sequences, Si-3-10%. These values are lower than those reported for a highly pure piezoelectric such as PbZrxTi1−xO3 (PZT) (360 to 500 pm V−1),68 similar to a PC/PVDF/PZT (portland cement/polyvinylidene fluoride/lead zirconate titanate) composite (up to 24 pm V−1)69 and much larger (about 7.5 times) than values found in the case of Parylene C films.70
Experimental section
Materials
Polydimethylsiloxane-α,ω-diol (PDMS) having a molecular weight Mn = 230000 g mol−1 was synthesized through cation ring-opening polymerization of octamethylcyclotetrasiloxane in the presence of sulfuric acid according to ref. 71. Tetraethylorthosilicate (TEOS) was purchased from Merck, dibutyltin dilaurate (DBTDL) from Sigma-Aldrich, while the Elastosil film having 100 μm thickness was acquired from Wacker.
Polyamic acid, PAA-1 (Scheme 1a), was prepared according to the already reported procedure72 consisting in a polycondensation reaction of 2,6-bis(3-aminophenoxy)benzonitrile with 4,4′-oxydiphthalic anhydride in a 1:1 molar ratio, in solution (in N-methyl-2-pyrrolidone) at room temperature (Scheme S1, ESI†). The nitrile-containing polyimide–polydimethylsiloxane copolymer, PICOSI-1 (Scheme 1b), was prepared by the polycondensation reaction of 4,4′-oxydiphthalic anhydride with a mixture of an aromatic diamine, 2,6-bis(3-aminophenoxy)benzonitrile, and α,ω-bis(aminopropyl)oligodimethylsiloxane of molecular weight 1120 g mol−1, in a 1:0.5:0.5 molar ratio. The reaction was carried out in two stages, first in a tetrahydrofuran (THF)/N-methyl-2-pyrrolidone (NMP) mixture at room temperature when the polyamic acid is formed, and the second in a mixture of m-xylene/NMP at reflux when the cyclization of polyamic acid occurs with the formation of the appropriate polyimide (Scheme S2, ESI†).49 The hexafluoroisopropylidene-containing polyimide–polydimethylsiloxane copolymer, PICOSI-2 (Scheme 1c), was prepared by the reaction between 4,4′-(hexafluoroisopropylidene)diphthalic anhydride and 4,4′-bis(4-aminophenoxy)biphenyl and α,ω-bis(aminopropyl)oligodimethylsiloxane, in a 1:0.6:0.4 molar ratio, in solvent, temperature and time conditions similar to those in the case of the sample PICOSI-1, excepting the adding reactants algorithm that is described in ref. 50 (Scheme S3, ESI†). The molecular weight of the siloxane diamine was 750 g mol−1.
The three reaction products were isolated by precipitation in water and their structures were verified by IR (Fig. S12, ESI†) and 1H-NMR (Fig. S13–S15, ESI†) spectral analysis.
The specific IR absorption bands (KBr, cm−1): PAA-1 (3200–3700m (OH and NH from COOH and CONH stretching vibration), 1668s (amide I, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from CONH stretching vibration), 1543s (amide II, C–NH), 2231m (CN from the nitrile group)); PICOSI-1 (1778w (CO asymmetric stretching), 1720vs (CO symmetric stretching), 1371s (C–N stretching of imide), 744m (CO bending), 2962m (C–H from Si–CH3 asymmetric stretching), 1261vs (C–H deformation in Si–CH3), 1095–1026s (Si–O–Si stretching), 798vs (C–H from Si–CH3 stretching), 2233w (C
O from CONH stretching vibration), 1543s (amide II, C–NH), 2231m (CN from the nitrile group)); PICOSI-1 (1778w (CO asymmetric stretching), 1720vs (CO symmetric stretching), 1371s (C–N stretching of imide), 744m (CO bending), 2962m (C–H from Si–CH3 asymmetric stretching), 1261vs (C–H deformation in Si–CH3), 1095–1026s (Si–O–Si stretching), 798vs (C–H from Si–CH3 stretching), 2233w (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N from the nitrile group)) and PICOSI-2 (1782m (CO asymmetric stretching), 1726vs (CO symmetric stretching), 1379s (C–N stretching of imide), 746m (CO bending), 2962m (C–H from Si–CH3 asymmetric stretching), 1257vs (CH3 deformation in Si–CH3), 1095–1014s (Si–O–Si stretching), 800vs (CH3 from Si–CH3 stretching), 1211vs and 1164s (C–F from the CF3 group)).73–75
N from the nitrile group)) and PICOSI-2 (1782m (CO asymmetric stretching), 1726vs (CO symmetric stretching), 1379s (C–N stretching of imide), 746m (CO bending), 2962m (C–H from Si–CH3 asymmetric stretching), 1257vs (CH3 deformation in Si–CH3), 1095–1014s (Si–O–Si stretching), 800vs (CH3 from Si–CH3 stretching), 1211vs and 1164s (C–F from the CF3 group)).73–75
1H-NMR (DMSO-d6, ppm) of PAA-1 13.0 (COO![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) ), 10.6 (CON), and the signals between 6.3 and 8.0 (aromatic protons); PICOSI-1 6.8–8.0 (aromatic protons), 3.5 (–C2–CH2–CH2–Si–), 1.6 (–CH2–C2–CH2–Si–), 0.5 (–CH2–CH2–C2–Si–), 0 (C3–Si); PICOSI-2 6.6–8.2 (aromatic protons), 3.54 (–C2–CH2–CH2–Si–), 1.6 (–CH2–C2–CH2–Si–), 0.5 (–CH2–CH2–C2–Si–), 0.0 (C3–Si).
), 10.6 (CON), and the signals between 6.3 and 8.0 (aromatic protons); PICOSI-1 6.8–8.0 (aromatic protons), 3.5 (–C2–CH2–CH2–Si–), 1.6 (–CH2–C2–CH2–Si–), 0.5 (–CH2–CH2–C2–Si–), 0 (C3–Si); PICOSI-2 6.6–8.2 (aromatic protons), 3.54 (–C2–CH2–CH2–Si–), 1.6 (–CH2–C2–CH2–Si–), 0.5 (–CH2–CH2–C2–Si–), 0.0 (C3–Si).
Measurements
Infrared spectra were recorded on a FTIR Bruker Vertex 70 apparatus in transmission mode on KBr pellets at room temperature. 1H-NMR measurements were conducted in DMSO on a Bruker AVANCE III 400 MHz spectrometer. The molecular masses of the polymers were determined through gel permeation chromatography (GPC) on a PL-EMD 950 apparatus. SEM images were taken using a Quanta 200 scanning electron microscope in a low vacuum mode, while the energy-dispersive X-ray spectroscopy (EDX) system available on the equipment was used for elemental mapping. The surface morphology of the films was studied by atomic force microscopy (AFM) using a Scanning Probe Microscopy Solver PRO-M, NT-MDT equipment (Russia), in semi-contact mode. Differential scanning calorimetry (DSC) measurements were conducted on a DSC 200 F3 Maia device (Netzsch, Germany). 6 mg of each sample was heated in aluminum crucibles with pierced and shut lids. A heating rate of 10 °C min−1 was applied and nitrogen was used as an inert atmosphere (flow rate 50 mL min−1). The device was calibrated with indium, according to standard procedures. Dynamic mechanical analysis (DMA) measurements were conducted in tension mode under a nitrogen atmosphere, with a heating rate of 2 °C min−1 at 1 Hz, on a PerkinElmer apparatus. The size of the measured samples was 12 mm in length, 10 mm in width and 0.27 mm thickness for the PDMS sample and 11 mm in length, 0.96 mm in width and 0.06 mm thickness for Si-2-5%. Dielectric analysis (DEA) was performed on a Novocontrol setup operating in the frequency range of 10−1–106 Hz (Concept 40 GmbH) with an ALPHA analyzer and a Quatro temperature controller. The temperature effect over the dielectric properties was obtained using a heating rate of 5 °C min−1 between −150 and 150 °C. The mechanical stress–strain tests were conducted on a TIRA test 2161 instrument, Maschinenbau GmbH Ravenstein at an extension rate of 20 mm min−1 at ambient temperature. The breakdown voltage tests were performed on a home-made setup based on international standards. The polymer films were placed between two electrodes, a spherical electrode of 5 mm in diameter and a planar electrode (ground) of 150 mm in diameter (Image S1, ESI†).31 The applied voltage was stepwise increased with 500 V s−1 until breakdown occurs. Actuation measurements were made on circular films of 50 mm in diameter, 20% equiaxially prestrained (Image S2, ESI†), and fixed between two circular frames. Circular electrodes of carbon black powder (8 mm in diameter) were deposited concentrically on both sides of the polymeric film. The lateral actuation displacement was optically measured using a digital camera and a software in order to evaluate the electrode extension.21,76 Piezoelectric force microscopy (PFM) was employed to investigate the local electromechanical (piezoelectric) properties of the polysiloxane–polyimide films using a multimode AFM setup (NT-MDT SolvePro). Essentially, PFM technique is based on the detection of the local electromechanical vibration of the sample caused by an external alternating current (AC) voltage. The voltage is applied to a conductive (platinum covered) AFM probing tip, which is used as a movable top electrode. The external driving voltage with frequency ν generates a sample surface vibration with the same frequency due to the converse piezoelectric effect. The modulated deflection signal from the cantilever, which oscillates together with the sample, is detected using the lock-in technique. The amplitude of the first harmonic signal from the lock-in amplifier is a function of the magnitude of piezoelectric displacement and phase shift between the AC electric field and the cantilever displacement. Thus, during AFM scanning of the sample surface the magnitude of the AFM tip displacement is recorded simultaneously with the phase of the displacement (orientation of the piezoelectric response). This means that regions with opposite piezoelectric orientation will vibrate in the counter phase with respect to each other under the applied electric field. The amplitude of displacement allows estimating the magnitude of the piezoelectric response, while the phase image gives information on the sequence of the mechanical oscillation that is related to the orientation of the polarization, allowing the identification of domain structures. In this work, all images were taken during one imaging session with the same cantilever (nominal spring constant 0.1 N m−1 and free resonant frequency of 245.9 kHz) and laser position in order to allow for quantitative comparison across the samples investigated. PFM images (surface topography, magnitude and phase) were obtained in contact mode at a vertical deflection setpoint of 2 nm, drive voltage of 1 V, scan rate of 0.5 Hz, and surface area of 20 × 20 μm2.S-IPN preparation procedure
0.5 g of PDMS and the specific amount of polyimide were dissolved together in 25 mL THF and stirred approximately for 10 hours, enabling a complete dissolution of the polymers. Afterwards, 0.035 mL of TEOS and 0.01 mL of DBTDL were added and the obtained solutions were again mixed thoroughly for another 10 min and then cast as thin films. The formed films were left for 24 hours under normal conditions to allow the curing process. A reference sample based only on crosslinked PDMS was also prepared. All samples were left for one month under normal conditions before being analysed.Conclusions
The interpenetration of the flexible polysiloxane with more rigid polyimide chains, the latter modified appropriately to minimize the known incompatibility between the two polymer classes, lead in all cases, when percent greater than 2% PI was incorporated, to increasing Young’s modulus in the resulting products as compared with pure polysiloxane but remained in reasonable limits. Elongation is generally maintained at high or reasonably high values depending on the polyimide content. In almost all cases, significant improvements in dielectric properties (high permittivity and dielectric loss) of the mixed networks relative to those of the Si-Blank network were obtained. The breakdown values of the S-IPNs are much higher than both the PDMS network and cyclized polyimides, suggesting that there is a synergetic effect between the S-IPN components, as was also found in the case of thermal behaviour. A substantial improvement in actuation displacement of the S-IPNs films was also achieved as compared with the pure crosslinked PDMS used as the reference sample. These materials have also potential application as generators, calculated values for the energy increasing with the content of polyimide so at 10 wt% polyimide content these are higher than those for Elastosil, one of the preferred materials at this time for such applications. Besides the large deformation, up to 8.7%, mainly given by a flexible dielectric silicone component, the presence of the imide component confers the piezoelectric effect, as has been revealed and measured by piezoelectric force microscopy, the average values obtained (17–26 pm V−1) being of high interest. The characteristics obtained for the developed materials recommend them as active elements in sensors, actuators or combined functions for medical devices (i.e., controlling and stimulating functions of certain organs). The presence of PDMS with its high surface activity as the major component in these materials guarantees the biocompatibility of the material.Acknowledgements
Thanks to Dr Mariana Cristea for DMA measurements and to Dr Violeta Melinte for mechanical measurements. The work presented in this paper is developed in the context of the project PolyWEC (http://www.polywec.org, prj. ref. 309139), a FET-Energy project that is partially funded by the 7th Framework Programme of European Community.Notes and references
- R. E. Pelrine, R. D. Kornbluh and J. P. Joseph, Sens. Actuators, A, 1998, 64, 77–85 CrossRef CAS.
- H. R. Choi, K. M. Jung, J. C. Koo and J. D. Nam, in Electroactive Polymers for Robotic Applications, ed. K. J. Kwang and S. Tadokoro, Robotic Applications of Artificial Muscle Actuators, Springer-Verlag, London, 2007, ch. 3, pp. 49–96 Search PubMed.
- J. Shintake, S. Rosset, B. Schubert, D. Floreano and H. Shea, Adv. Mater., 2016, 28(2), 231–238 CrossRef CAS PubMed.
- F. Carpi, G. Frediani, S. Turco and D. De Rossi, Adv. Funct. Mater., 2011, 21(21), 4152–4158 CrossRef CAS.
- M. Kollosche, H. Stoyanov, S. Laflamme and G. Kofod, J. Mater. Chem., 2011, 21, 8292–8294 RSC.
- H. Zhang, M. Y. Wang, J. Li and J. Zhu, Smart Mater. Struct., 2016, 25(3), 035045 CrossRef.
- D. Vilela, A. Romeo and S. Sánchez, Lab Chip, 2016, 16, 402–408 RSC.
- S. Chiba, M. Waki, T. Wada, Y. Hirakawa, K. Masuda and T. Ikoma, Appl. Energy, 2013, 104, 497–502 CrossRef CAS.
- P. Brochu, H. Stoyanov, R. Chang, X. Niu, W. Hu and Q. Pei, Adv. Energy Mater., 2014, 4(3), 1300659 CrossRef.
- R. Pelrine, R. Kornbluh, J. Joseph, R. Heydt, Q. Pei and S. Chiba, Mater. Sci. Eng., C, 2000, 11, 89–100 CrossRef.
- M. Gei, S. Colonneli and R. Springhetti, Int. J. Solids Struct., 2014, 51, 848–860 CrossRef CAS.
- A. G. Bejenariu, M. Boll, M. R. Lotz, C. Vraa, A. L. Skov, in Proceedings of SPIE, ed. Y. Bar-Cohen and F. Carpi, 2011, p. 7976 Search PubMed.
- I. Krakovsky, T. Romijn and A. Posthuma de Boer, J. Appl. Phys., 1999, 85(1), 628 CrossRef CAS.
- B. Li, L. Liu, J. Wu, Z. Zhu, H. Chen, in Proceedings of SPIE, ed. Y. Bar-Cohen, 2010, p. 7642 Search PubMed.
- F. B. Madsen, A. E. Daugaard, S. Hvilsted, M. Y. Benslimane and A. L. Skov, Smart Mater. Struct., 2013, 22, 104002 CrossRef.
- D. M. Opris, M. Molberg, C. Walder, Y. S. Ko, B. Fischer and F. A. Nüesch, Adv. Funct. Mater., 2011, 21(18), 3531–3539 CrossRef CAS.
- J. Chon, S. Ye, K. J. Cha, S. C. Lee, Y. S. Koo, J. H. Jung and Y. K. Kwon, Chem. Mater., 2010, 22(19), 5445–5452 CrossRef CAS.
- C. Racles, A. Bele, M. Dascalu, V. E. Musteata, C. D. Varganici, D. Ionita, S. Vlad, M. Cazacu, S. J. Dünki and D. M. Opris, RSC Adv., 2015, 5(72), 58428–58438 RSC.
- H. Stoyanov, M. Kollosche, D. N. McCarthy and G. Kofod, J. Mater. Chem., 2010, 20, 7558–7564 RSC.
- X. Zhou, X. Zhao, Z. Suo, C. Zou, J. Runt, S. Liu, S. Zhang and Q. M. Zhang, Appl. Phys. Lett., 2009, 94, 162901 CrossRef.
- C. Tugui, G. Stiubianu, M. Iacob, C. Ursu, A. Bele, S. Vlad and M. Cazacu, J. Mater. Chem. C, 2015, 3, 8963–8969 RSC.
- P. H. Vargantwar, A. E. Özçam, T. K. Ghosh and R. J. Spontak, Adv. Funct. Mater., 2012, 22(10), 2100–2113 CrossRef CAS.
- M. Iacob, G. Stiubianu, C. Tugui, L. Ursu, M. Ignat, C. Turta and M. Cazacu, RSC Adv., 2015, 5(56), 45439–45445 RSC.
- J. E. Q. Quinsaat, M. Alexandru, F. A. Nüesch, H. Hofmann, A. Borqschulte and D. M. Opris, J. Mater. Chem. A, 2015, 3, 14675–14685 CAS.
- F. B. Madsen, I. Dimitrov, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polym. Chem., 2013, 4, 1700–1707 RSC.
- F. Carpi, G. Gallone, F. Galantini and D. De Rossi, Adv. Funct. Mater., 2008, 18, 235–241 CrossRef CAS.
- A. H. A. Razak, P. Szabo and A. L. Skov, RSC Adv., 2015, 5, 53054–53062 RSC.
- M. Dascalu, S. J. Dünki, J.-E. Q. Quinsaat, Y. S. Ko and D. M. Opris, RSC Adv., 2015, 5, 104516 RSC.
- M. Poikelispää, A. Shakun, A. Das and J. Vuorinen, J. Appl. Polym. Sci., 2016, 133(42), 1–9 CrossRef.
- G. Stiubianu, A. Soroceanu, C. D. Varganici, C. Tugui and M. Cazacu, Composites, Part B, 2016, 93, 236–243 CrossRef CAS.
- C. Tugui, S. Vlad, M. Iacob, C. D. Varganici, L. Pricop and M. Cazacu, Polym. Chem., 2016, 7, 2709–2719 RSC.
- F. B. Madsen, A. E. Daugaard, S. Hvilsted and A. L. Skov, Macromol. Rapid Commun., 2016, 37, 378–413 CrossRef CAS PubMed.
- C. Tugui, M. Cazacu, L. Sacarescu, A. Bele, G. Stiubianu, C. Ursu and C. Racles, Polymer, 2015, 77, 312–322 CrossRef CAS.
- P. Brochu, H. Stoyanov, X. Niu and Q. Pei, Smart Mater. Struct., 2013, 22(5), 055022 CrossRef.
- H. Zhang, L. Düring, G. Kovacs, W. Yuan, X. Niu and Q. Pei, Polym. Int., 2010, 59(3), 384–390 CrossRef CAS.
- S. M. Ha, W. Yuan, Q. Pei, R. Pelrine and S. Stanford, Smart Mater. Struct., 2007, 16(2), S280–S287 CrossRef CAS.
- S. M. Ha, W. Yuan, Q. Pei, R. Pelrine and S. Stanford, Adv. Mater., 2006, 18(7), 887–891 CrossRef CAS.
- A. Maceiras, C. M. Costa, A. C. Lopes, M. San Sebastián, J. M. Laza, J. L. Vilas, J. L. G. Ribelles, R. S. Serra, A. A. Balado, S. Lanceros-Méndez and L. M. León, Appl. Phys. A: Mater. Sci. Process., 2015, 120(2), 731–743 CrossRef CAS.
- Z. Ounaies, J. A. Young, J. O. Simpson and B. L. Farmer, Dielectric Properties of Piezoelectric Polyimides, in Materials Research Society Symposium Preceding, 1996, vol. 53, p. 459.
- G. M. Atkinson, R. E. Pearson, Z. Ounaies, C. Park, J. S. Harrison, S. Dogan and J. A. Midkiff, Novel Piezoelectric Polyimide MEMS, in The 12th International Conference on Solid State Sensors, Actuators and Microsystems, Boston US, 2003, vol. 1, pp. 782–781.
- T. L. St. Clair, J. S. Harrison, J. Su and Z. Ounaies, Polymeric Blends for Sensor and Actuation Dual Functionality, US6689288 B2, 2004 Search PubMed.
- J. Su, Z. Ounaies, J. S. Harrison, Y. Bar-Cohen and S. P. Leary, Proc. SPIE, 2000, 3987 Search PubMed.
- A. Safari and P. Ngernchuklin, Piezoelectric Electrostrictive Composition Apparatus and Method, US2011/0043081 A1, 2011 Search PubMed.
- Y. Bar-Cohen, Electroactive Polymers as actuators, in Advanced Piezoelectric Materials: Science and Technology, ed. K. Uchino, Woodhead Publishing, Cambridge, UK, 2010, ch. 8, pp. 287–317 Search PubMed.
- http://SensorWiki.org, http://www.sensorwiki.org/doku.php/actuators/piezoelectric, accesed: october, 2016.
- P. Garg, R. P. Singh and V. Choudhary, Purif. Technol., 2011, 76(3), 407–418 CrossRef CAS.
- F. Abbasi, H. Mirzadeh and A. A. Katbab, Polym. Int., 2001, 50(12), 1279–1287 CrossRef CAS.
- M. D. Damaceanu, C. P. Constantin, M. Bruma and N. M. Belomonia, Polymer, 2014, 55(17), 4488–4497 CrossRef CAS.
- E. Hamciuc, M. Bruma, C. Hamciuc and R. Lungu, Rev. Roum. Chim., 2006, 51, 765–771 CAS.
- E. Hamciuc, C. Hamciuc, M. Cazacu, M. Ignat and G. Zarnescu, Eur. Polym. J., 2009, 45, 182–190 CrossRef CAS.
- E. Hamciuc, C. Hamciuc, M. Cazacu, G. Lisa and L. Okrasa, J. Macromol. Sci., Part A: Pure Appl.Chem., 2007, 44, 1069–1078 CrossRef CAS.
- I. R. Harrison and J. Runt, J. Polym. Sci., Polym. Phys. Ed., 1980, 18(11), 2257–2261 CrossRef CAS.
- M. Aubin, Y. Bédard, M. F. Morrissette and R. E. Prud'homme, J. Polym. Sci., Polym. Phys. Ed., 1983, 21(2), 233–240 CrossRef CAS.
- G. Stiubianu, A. Nicolescu, A. Nistor, M. Cazacu, C. Varganici and B. C. Simionescu, Polym. Int., 2012, 61(7), 1115–1126 CrossRef CAS.
- C. D. Varganici, L. Rosu, D. Rosu and B. C. Simionescu, Composites, Part B, 2013, 50, 273–278 CrossRef CAS.
- Z. Lei, W. Xing, J. Wu, G. Huang, X. Wang and L. Zhao, J. Therm. Anal. Calorim., 2014, 116(1), 447–453 CrossRef CAS.
- J. Foreman, S. R. Sauerbrunn and C. L. Marcozzi, Exploring the Sensitivity of Thermal Analysis Techniques to Glass Transition, TA Instruments, Inc., New Castle, UK, 1993 Search PubMed.
- M. Alexandru, M. Cazacu, M. Cristea, A. Nistor, C. Grigoras and B. C. Simionescu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49(7), 1708–1718 CrossRef CAS.
- C. E. Sroog, Films, Moldings and other Applications, in Polyimides, ed. D. Wilson, H. D. Stezenberger and P. M. Hergenrother, Springer Science+Business Media, New York, US, 1990, ch. 9, pp. 252–284 Search PubMed.
- T. L. St. Clair, Structure–property relationships in linear aromatic polyimides, in Polyimides, ed. D. Wilson, H. D. Stezenberger and P. M. Hergenrother, Springer Science+Business Media, New York, US, 1990, ch. 3, pp. 58–78 Search PubMed.
- Z. Ahmad, in Dielectric material, ed. M. A. Silaghi, InTech, 2012, ch. 1, pp. 1–26 Search PubMed.
- F. Carpi, I. Anderson, S. Bauer, G. Frediani, G. Gallone, M. Gei, C. Graaf, C. Jean-Mistral, W. Kaal, G. Kofod, R. Kornbluh, B. Lassen, M. Matysek, S. Michel, S. Nowak, B. O'Brien, Q. Pei, R. Pelrine, B. Rechenbach, S. Rosset and H. Shea, Smart Mater. Struct., 2015, 24(10), 105025 CrossRef.
- C. Racles, V. E. Musteata, A. Bele, M. Dascalu, C. Tugui and A. L. Matricala, RSC Adv., 2015, 5, 102599 RSC.
- M. Molberg, D. Crespy, P. Rupper, F. Nüesch, J. A. E. Månson, C. Löwe and D. M. Opris, Adv. Funct. Mater., 2010, 20(19), 3280–3291 CrossRef CAS.
- J. Su, J. S. Harrison and T. St Clair, Proceedings of The 12th IEEE International Symposium on Application of Ferroelectrics, 2000, vol. 2, pp. 811–814.
- F. Carpi, C. Salaris and D. De Rossi, Smart Mater. Struct., 2007, 16(2), S300–S305 CrossRef CAS.
- H. J. Fan, W. Lee, R. Hauschild, M. Alexe, G. Le Rhun, R. Scholz, A. Dadgar, K. Nielsch, H. Kalt, A. Krost, M. Zacharias and U. Gösele, Small, 2006, 2(4), 561–568 CrossRef CAS PubMed.
- K. S. Ramadan, D. Sameoto and S. Evoy, Smart Mater. Struct., 2014, 23(3), 033001 CrossRef.
- N. Jaitanong, R. Yimnirun, H. R. Zeng, G. R. Li, Q. R. Yin and A. Chaipanich, Mater. Lett., 2014, 130, 146–149 CrossRef CAS.
- J. Y. H. Kim, PhD thesis, California Institute of Technology, Pasadena CA, 2012.
- M. Cazacu, M. Antohi, C. Racles, A. Vlad and N. Forna, J. Compos. Mater., 2009, 43, 2045–2055 CrossRef CAS.
- E. Hamciuc, C. Hamciuc and M. Ignat, High Perform. Polym., 2010, 22, 225–236 CrossRef CAS.
- Q. Li, X. Yang, W. Chen, C. Yi and Z. Xu, Macromol. Symp., 2008, 261, 148–156 CrossRef CAS.
- A. Kumar, S. Tateyama, K. Yasaki, M. A. Ali, N. Takaya, R. Singh and T. Kaneko, Data in Brief, 2016, 7, 123–128 CrossRef PubMed.
- M. Dinari and P. Asadi, RSC Adv., 2015, 5, 60745–60753 RSC.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 7, 671–675 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis route of polyimides, scanning electron microscopy images of S-IPNs, energy dispersive X-ray spectroscopy mapping, atomic force microscopy images, dielectric spectra, dynamic mechanical analysis curves, FTIR and 1H-NMR spectra, electrical breakdown and equiaxial stretching setups. See DOI: 10.1039/c6tc05193f |
| This journal is © The Royal Society of Chemistry 2017 |