
Immobilization of an antimicrobial peptide on silicon surface with stable activity by click chemistry†
Jingcai
He
a,
Junjian
Chen
a,
Guansong
Hu
b,
Lin
Wang
 *a,
Jian
Zheng
a,
Jiezhao
Zhan
b,
Yuchen
Zhu
b,
Chunting
Zhong
a,
Xuetao
Shi
a,
Sa
Liu
a,
Yingjun
Wang
ab and
Li
Ren
*b
*a,
Jian
Zheng
a,
Jiezhao
Zhan
b,
Yuchen
Zhu
b,
Chunting
Zhong
a,
Xuetao
Shi
a,
Sa
Liu
a,
Yingjun
Wang
ab and
Li
Ren
*b
aSchool of Materials Science and Engineering, South China University of Technology, Guangzhou 510641, China. E-mail: wanglin3@scut.edu.cn
bNational Engineering Research Center for Tissue Restoration & Reconstruction, South China University of Technology, Guangzhou 510006, China. E-mail: psliren@scut.edu.cn
First published on 15th November 2017
Abstract
Infections associated with biomedical implants and devices pose a serious clinical challenge in hospitals worldwide. Antimicrobial peptides (AMPs) have become a great prospect to inhibit this type of infection due to their broad-spectrum antimicrobial activity and low cytotoxicity. However, it is still a challenge to apply AMPs on the biomaterial surface as the activity of AMPs is sensitive to salt or enzyme. In the present study, we prepared a spacer molecule, poly[2-(methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (polySBMA), on a model silicon surface via surface-initiated atom transfer radical polymerization (SI-ATRP). We then modified the antimicrobial peptide HHC36 (KRWWKWWRR) with L-propargylglycine (PraAMP) to improve its salt-tolerant activity and integrated PraAMP onto the spacer molecule using click chemistry. We employed X-ray photoelectron spectroscopy (XPS), contact angle goniometry, and atomic force microscopy (AFM) to confirm the success of the immobilization process. We also characterized the antimicrobial activity and stability of the surface with an antimicrobial assay. The results reveal that the modified surface exhibits good antimicrobial activity to inhibit 98.26% of E. coli, 83.72% of S. aureus, and 81.59% of P. aeruginosa. Furthermore, as compared to the control group without the polySBMA spacer, the modified surface improved its resistance to enzymolysis. An in vitro CCK-8 assay also illustrated that this surface showed negligible cytotoxicity to mouse bone mesenchymal stem cells.
Introduction
Biomaterial-associated infection has become a serious problem in clinics.1–4 In the case of infection, the bacteria forms a biofilm on the biomaterial to inhibit the effects of antibiotics.5–7 The infection may lead to surgery failure, patient disability, and even death. Surface modification of the biomaterial with an antimicrobial agent, such as silver nanoparticles or an antibiotic, can markedly reduce the infection. These antimicrobial surfaces can kill the bacteria on the surface and inhibit the formation of the biofilm.8–10 However, the cytotoxicity of silver nanoparticles and bacterial drug-resistance caused by the antibiotic often limit their applications.11,12As a new antimicrobial agent, cationic antimicrobial peptides (AMPs) have been widely studied by researchers. AMPs are formed by amino acids and often have a positive charge and hydrophobicity.13,14 They exhibit broad-spectrum antimicrobial activity and low cytotoxicity.15 Particularly, AMPs do not result in bacterial drug-resistance due to their particular antimicrobial mechanism, which work by destroying the membrane of the bacteria.16,17 Recently, researchers have focused on improving the antimicrobial activity of biomaterials with AMPs.18–21 Among these AMPs, HHC36 (Seq: Lys-Arg-Trp-Trp-Lys-Trp-Trp-Arg-Arg, KRWWKWWRR) has the abovementioned advantages despite containing only 9 amino acids.22,23 Moreover, it exhibits a low immunological response and low cost due to its short sequence and has great potential for clinical applications. There have been some reports stating that the HHC36 peptide can improve the antimicrobial activity of a biomaterial after its integration.24
When applied to biomaterial surfaces, the bottleneck of AMPs often lies on their stability. Unlike other antimicrobial agents, the AMPs only exhibit activity with an appropriate orientation on the surface.25,26 However, the energy of the surface can disorder the orientation of the peptide, and the enzymes in the environment may degrade the peptide, destroying its orientation and markedly reducing its activity.27
Adding a spacer molecule between the substrate and the peptide is a promising method for improving the stability of the peptide. For example, as a widely used spacer molecule, PEG (polyethylene glycol) can optimize the orientation of the peptide on a surface. However, PEGylation can have an impact on the affinity of the peptide, and PEG is susceptible to oxidation and disintegration in biological environments containing aerobic and transition metal ions.28–31 Recently, another spacer molecule called zwitterion has been the focus of research efforts.32–34 Compared to PEG, zwitterion showed excellent thermal and chemical stabilities and salt-tolerant properties.35 In particular, poly(zwitterion)s have excellent resistance to the non-specific adsorption of protein due to their unique molecular structure containing both cationic and anionic groups,35–37 and they can increase the stability of the peptides without sacrificing affinity or bioactivity.30,31
In the present study, to prepare a novel antimicrobial surface by AMPs with improved stability, particularly an improved enzymolysis tolerance, we employed the poly(zwitterion) poly[2-(methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)-ammonium hydroxide (polySBMA) as the spacer molecule to integrate the HHC36 peptide. We first modified the N-terminus of the HHC36 peptide (AMP) with L-propargylglycine to obtain an alkyne-terminated peptide (PraAMP). Then, we chose silicon (Si) as the substrate due to its non-specific adsorption38 and immobilized polySBMA onto the Si substrate via surface-initiated atom transfer radical polymerization (SI-ATRP). Subsequently, we used a Cu(I)-catalyzed azide–alkyne cycloaddition reaction (CuAAC), a typical click chemistry reaction, to integrate PraAMP onto the silicon surface. This reaction is specific to the azide and alkyne groups and does not consume any of the active groups, such as the amino groups, in the peptide.22 Moreover, this reaction occurs at room temperature in water and optimizes the peptide's orientation as well as the activity on the surface.21,39,40 The whole surface preparation process is shown in Scheme 1. We characterized the modified surface using XPS, contact angle goniometry, and AFM and investigated its antimicrobial activity against E. coli, S. aureus, and P. aeruginosa. Moreover, we tested its biocompatibility towards mouse bone mesenchymal stem cells (mBMSCs) in vitro.
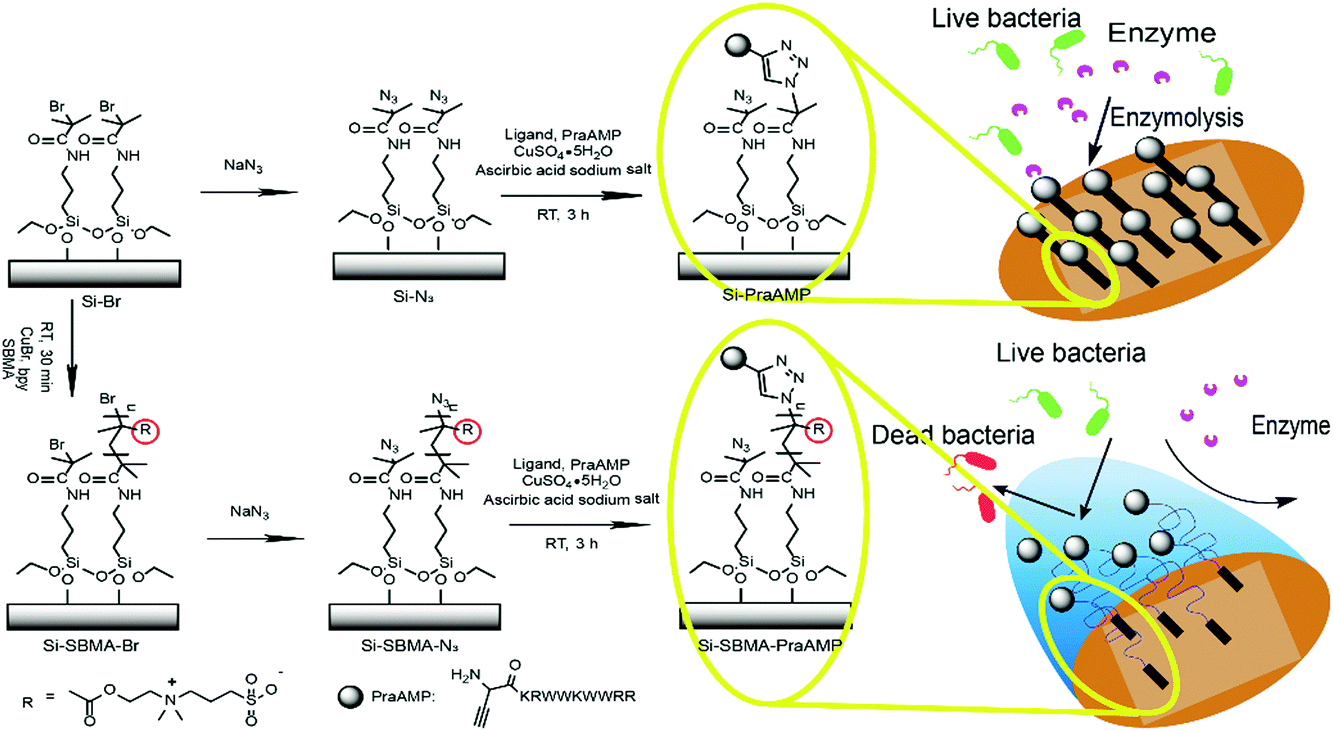 | ||
| Scheme 1 A schematic of the chemical immobilization of PraAMP on the Si surface. | ||
Experimental
Materials
[2-(Methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (SBMA, 97%), 2,2′-bipyridine (>99%), copper(I) bromide (Cu(I)Br, 98%), α-bromoisobutyryl bromide (BiBB, 98%), (3-aminopropyl)triethoxysilane (APTES, ≥98%), sodium azide (NaN3, ≥99.0% (T)) and tris(3-hydroxypropyltriazolylmethyl)amine (THPTA, 95%) were purchased from Sigma-Aldrich (Milwaukee, WI, USA). Triethylamine (TEA, ≥99.5%), hydrogen peroxide solution (H2O2, 30 wt% in H2O), L-ascorbic acid sodium (99%), N,N-dimethylformamide (DMF, ≥99.8%), ethanol (≥99.5%), dichloromethane (DCM, ≥99.9%), and methanol (≥99.9%) were purchased from Aladdin (Shanghai, China). Phosphate buffered saline (PBS) was purchased from Shanghai Life Biological technology Co., Ltd. The nutrient broth and LB agar used for the bacterial cultures were obtained from Guangdong Huankai Microbial Sci & Tech Co., Ltd. Dulbecco's modified eagle medium (DMEM, high glucose), fetal bovine serum (FBS) and Trypsin–EDTA were all purchased from Gibco® by Life Technologies (California, USA).Circular dichroism (CD)
The antimicrobial peptides, AMP, and PraAMP (their structures are shown in Scheme 1) were synthesized by GL Biochem Ltd (Shanghai, China). All CD spectra were obtained using a CHIRASCAN CD SPECTROMETER (Applied Photophysics, UK) in water using a 20 μM peptide. The ellipticities were reported as the mean residue ellipticity, [θ].Preparation of the initiator-functionalized silicon wafers
The wafers (5 mm × 5 mm × 1 mm) were sonicated in acetone, ethanol, and distilled water for 10 min and then dried under nitrogen. Then, the wafers were immersed in a piranha solution composed of 30% hydrogen peroxide and sulfuric acid (3/7, v/v) for 30 min, rinsed with distilled water, and then dried under nitrogen.After cleaning, the wafers were immersed in 30 mL of toluene containing 0.6 mL of 3-aminopropyltriethoxysilane (APTES) at 80 °C for 18 h, rinsed with toluene and ethanol, dried under nitrogen, and annealed in a vacuum oven at 120 °C for 1 h. Then, the wafers were treated with 40 mL dichloromethane (DCM) containing 0.5 mL of 2-bromoisobutyryl bromide (BiBB) and 1 mL of trimethylamine (TEA) at room temperature for 1 h, rinsed with DCM and ethanol, and dried under nitrogen. This sample was abbreviated as Si–Br.
Polymerization of SBMA via surface-initiated atom transfer radical polymerization (SI-ATRP)
SI-ATRP was synthesized following the methods previously reported in the literature.41,42 Briefly, 0.3 g of SBMA monomer, 18 mg of CuBr, and 39 mg of 2,2′-bipyridine were dissolved in a deoxygenated mixed solution comprising distilled water and methanol at a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Then, the mixture was injected into the degassed glass tube containing the wafers. The polymerization was performed with 90 rpm shaking under a nitrogen atmosphere at 37 °C for 30 min. After this, the wafers were rinsed with distilled water and dried under nitrogen. This sample was abbreviated as Si-SBMA-Br.
:1. Then, the mixture was injected into the degassed glass tube containing the wafers. The polymerization was performed with 90 rpm shaking under a nitrogen atmosphere at 37 °C for 30 min. After this, the wafers were rinsed with distilled water and dried under nitrogen. This sample was abbreviated as Si-SBMA-Br.
PraAMP functionalization of the wafers
To introduce the azido groups, the terminal bromide was reacted with NaN3 according to a literature procedure.43–45 Briefly, both the Si–Br and Si-SBMA-Br substrates were treated with 10 mL of DMF containing 0.26 g of sodium azide overnight at 60 °C, cleaned with distilled water, and dried under nitrogen. After this, the wafers were incubated in 225 μL of the click solution containing 120 μM of CuSO4, 240 μM of tris(3-hydroxypropyltriazolylmethyl)amine (THPTA), 6 mM of L-ascorbic acid sodium, and 100 μM alkyne-terminated antimicrobial peptide (PraAMP) for 2 h at 37 °C in the dark under a nitrogen environment. After the reaction, the wafers were washed three times with 5 mM ethylenediaminetetraacetic acid disodium salt (EDTA2Na) solution and PBS. These samples were abbreviated as Si-PraAMP (for Si–Br) and Si-SBMA-PraAMP (for Si-SBMA-Br).The whole surface preparation process is shown in Scheme 1.
Characterization of the surface
X-ray photoelectron spectroscopy (XPS) was conducted via a Thermo ESCALAB 250Xi operated using an Al Kα (1486.6 eV) monochromatic X-ray source with a power of 150 W and a 500 μm beam spot. The contact angle of the surface was determined using the liquid drop method via a contact angle goniometer (OCA15, Dataphysics, Germany). Atomic force microscopy (AFM) images were acquired using a MutiMode Nanoscope IIIa AFM (Digital Instruments Inc., Santa Barbara, CA).Antimicrobial assay
Single colonies of E. coli (ATCC 15224), S. aureus (ATCC 6538) or P. aeruginosa (ATCC 15442) were cultured in the medium at 37 °C with 150 rpm shaking for 12 h. Then, fresh medium was inoculated with the initial cultures and incubated to achieve a mid-log phase.The bacteria were separately diluted with PBS or medium to a concentration of 1.0 × 107 CFU mL−1 for different experiments. For the salt-tolerant property assay, the soluble AMP and PraAMP were employed at a concentration of 10 μM both in PBS and in the medium. For the antimicrobial assay of the surfaces, 10 μL of the bacterial suspension in PBS or the medium was added onto each sample to fully cover the substrate in a 48-well culture plate. After culturing for 2 h at 37 °C, the sample and the bacterial suspension were transferred to a new tube and 1 mL of PBS was added. The system was ultra-sonicated for 5 min to detach the bacteria from the surface. Then, 10 μL of the diluted bacterial suspension was placed onto an LB agar plate to evaluate the viability of the bacteria.
The live/dead assay was carried out as follows: after incubation with 10 μL of the E. coli suspension (107 CFU per mL in medium) on the surfaces for 2 h at 37 °C, the samples were washed three times with PBS and incubated in 100 μL of a mixed solution containing 0.05% fluorescein diacetate and 0.5% propidium iodide solution for 5 min at 25 °C in the dark. After this, the samples were washed three times with distilled water and examined using a Leica TCS SP8 Confocal System (Leica, Germany).
A field emission electron scanning microscopy (FESEM, ZEISS, MERLIN Compact, Germany) assay was carried out as follows: 10 μl of the E. coli suspension (107 CFU per mL in medium) was added to the indicated samples and incubated for 2 h. Then, the samples were washed with PBS, immersed with 100 μL of the fixative (4% methanal in PBS) at room temperature for 4 h, and kept at 4 °C overnight. Subsequently, the samples were dehydrated with 50%, 70%, 80%, and 90% of ethanol for 10 min consecutively, dehydrated twice with 100% ethanol for 15 min, and dried under nitrogen. The samples were then coated with platinum for 80 s at 20 mA and transferred to the FESEM for imaging.
For the enzymolysis test, the samples were treated with trypsin before the antimicrobial assay. Briefly, the surfaces were fully covered with 10 μL of a trypsin solution (1 mg mL−1) and incubated in an oven at 37 °C for 1, 3, and 5 min. Subsequently, the enzymolysis was completed with 10% FBS. The antimicrobial activity on the surfaces after the treatment was determined as described above.
The biocompatibility assay used for the sample
The biocompatibility of the sample to mouse bone mesenchymal stem cells (mBMSCs) was evaluated using a cell counting kit-8 (CCK-8) assay. The mBMSCs were cultured in high glucose Dulbecco's modified Eagle's medium (H-DMEM) with 10% fetal bovine serum (FBS) at 37 °C and 5% CO2 atmosphere. The medium was refreshed every third day. The cells were passaged when the confluence reached approximately 90%, and 5–10 (P5–P10) passaged cells were used for the subsequent CCK-8 assay. All the samples were sterilized using 75% ethanol for 2 h and washed three times with PBS. Then, 1 mL of the H-DMEM medium containing 30000 cells was added to each sample, and the system was cultured for 24 h. Subsequently, the samples were transferred to a new 48-well plate and washed three times with PBS. Then, 300 μL of the complete medium containing 30 μL of the CCK-8 solution was added. After culturing in the dark at 37 °C for 2 h, 100 μL of the incubated solution was transferred to a new 96-well plate, and the optical density (OD) of the solution was measured using the ELISA plate reader (Varioskan Flash 3001, Thermo, Finland) at 450 nm.
Statistics
The antimicrobial assay and biocompatibility assay were repeated at least three times and the results were expressed as the mean ± standard deviation. Statistical significance was calculated using GraphPad Prism 7.0 software. Statistical significance was defined as p < 0.01.Results and discussion
The MS results in Fig. S1 (ESI†) showed that the PraAMP was prepared successfully with a molecular weight of 1583.14 Da. The CD results in Fig. S2, ESI,† and Table 1 showed that both AMP and PraAMP had primarily α-helical and β structure (β-sheet and β-turn) features, with the α-helices in AMP and PraAMP making up 38.20% and 40.80% of the peptides, respectively, and the β structure making up 52.20% and 55.00% of the peptides, respectively. This result demonstrates that modification of the N-terminus of the AMP peptide with L-propargylglycine did not significantly change its secondary structure.| Samples abbreviation | AMP (%) | PraAMP (%) |
|---|---|---|
| α-Helix | 38.20 | 40.80 |
| β-Sheet | 31.20 | 35.10 |
| β-Turn | 21.00 | 19.90 |
| Rndm. coil | 6.80 | 6.70 |
L-Propargylglycine at the N-terminus of the AMP peptide (PraAMP) significantly improved the salt-tolerant properties of the peptide. The antimicrobial results in Fig. 1 showed that at the same concentration (10 μM), both AMP and PraAMP killed over 98% of E. coli in the medium. However, in PBS, PraAMP inhibited 93.36% of E. coli, whereas the AMP only inhibited 18.75% of E. coli. This result was similar to those of previously reported strategies used to improve the salt-tolerant properties of peptides by modifying their N-terminus.46,47
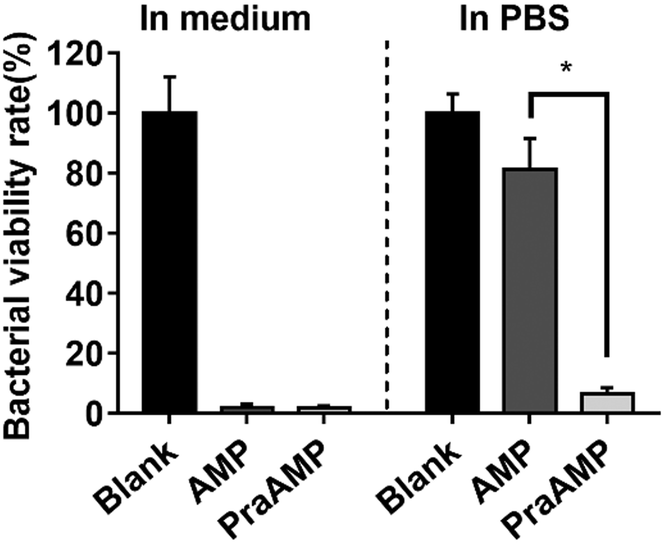 | ||
| Fig. 1 The antimicrobial activity of AMP and PraAMP against E. coli in medium and in PBS at a concentration of 10 μM (n = 3) (* denotes p < 0.001). | ||
As shown in Scheme 1, PraAMP was able to integrate onto the silicon surface via click chemistry. The XPS results in Fig. 2(a) show that the naked Si sample only had a simple C1s peak with no evident N1s peak. The XPS high-resolution C1s spectra in Fig. 2(b) and (c) show that after the reaction, the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N–H peaks (at 287.2 eV) became the major component of the high-resolution C1s spectra; this indicated the peptide contributions. In addition, the OC–O bond peak at 288.8 eV and C–N+ bond peak at 402.08 eV in Fig. 2(c) indicated the presence of an SBMA molecule in Si-SBMA-PraAMP. The N1s high-resolution spectra in Fig. 2(b) and (c) showed the OC–N peaks, which were also due to the peptide on Si-PraAMP and Si-SBMA-PraAMP.
C–N–H peaks (at 287.2 eV) became the major component of the high-resolution C1s spectra; this indicated the peptide contributions. In addition, the OC–O bond peak at 288.8 eV and C–N+ bond peak at 402.08 eV in Fig. 2(c) indicated the presence of an SBMA molecule in Si-SBMA-PraAMP. The N1s high-resolution spectra in Fig. 2(b) and (c) showed the OC–N peaks, which were also due to the peptide on Si-PraAMP and Si-SBMA-PraAMP.
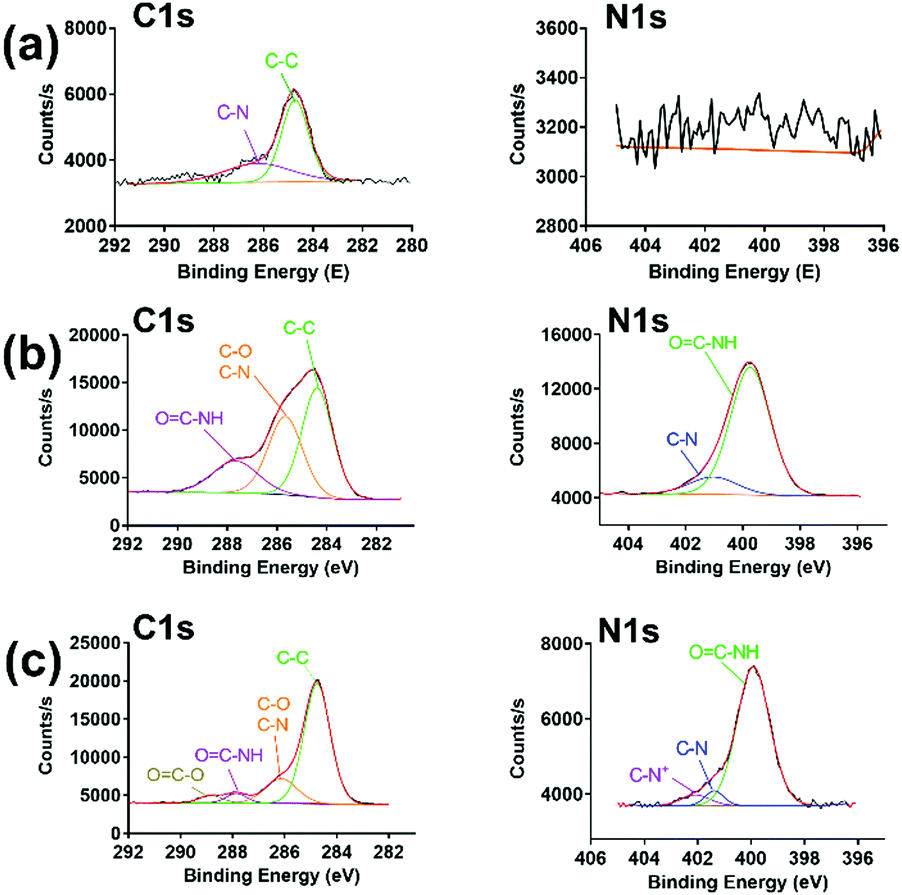 | ||
| Fig. 2 The high-resolution C1s and N1s spectra observed in the XPS obtained for the samples: (a) Si, (b) Si-PraAMP, and (c) Si-SBMA-PraAMP. | ||
After the integration of the peptide, the hydrophobicity and the morphology of the indicated surface changed. The water contact angle in Fig. 3(a) indicates that Si has a contact angle of 48.2 ± 0.9°. The integration of AMP increased the hydrophobicity of the surface. Compared to that of Si, the contact angle of Si-PraAMP increased to 67.5 ± 1.8°. For the sample containing SBMA, the contact angle of Si-SBMA decreased to 17.7 ± 4.3°; this was caused by the hydrophilicity of polySBMA. Compared to that of Si-SBMA, the contact angle of Si-SBMA-PraAMP increased to 42.7 ± 1.0°. In addition, before integration of the peptide, the AFM results in Fig. 3(b) showed that the original Si had a smooth and bulk morphology. After the integration of AMP, both Si-PraAMP and Si-SBMA-PraAMP exhibited a rough morphology (as shown in Fig. 3(c) and (d)).
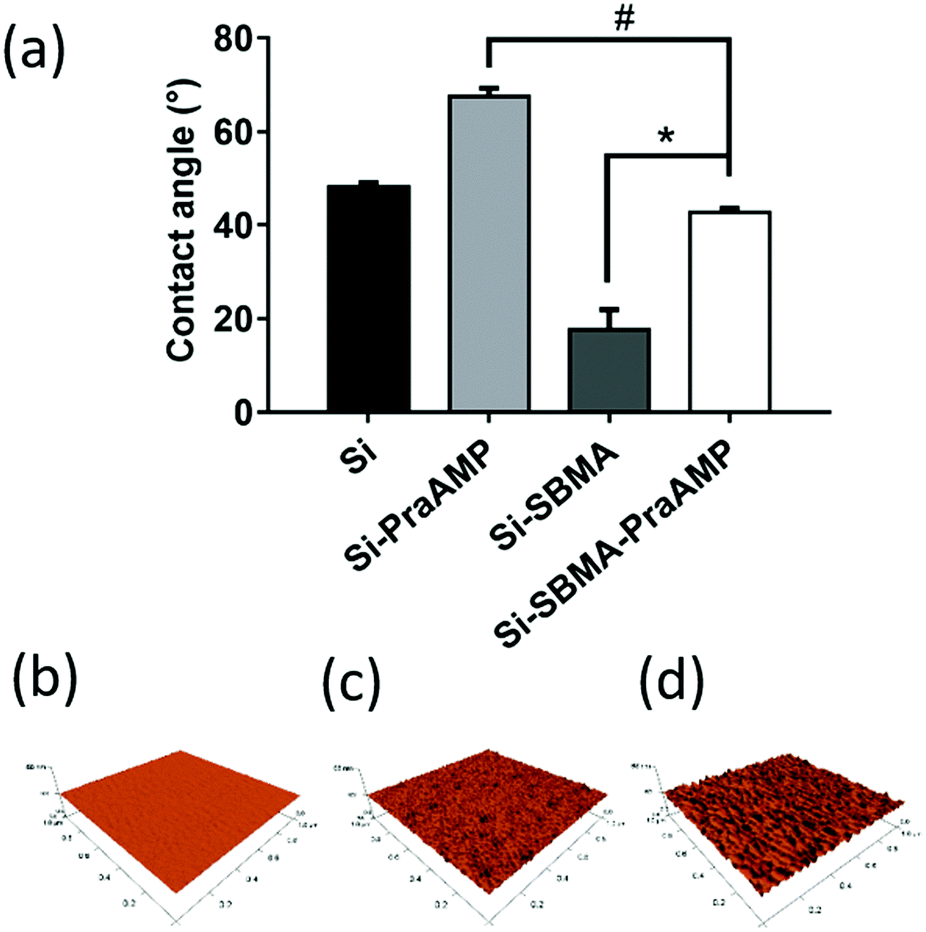 | ||
| Fig. 3 The water contact angle and AFM images of the samples: (a) water contact angle and the AFM images of (b) Si, (c) Si-PraAMP, and (d) Si-SBMA-PraAMP (# denotes p < 0.01, * denotes p < 0.001). | ||
The integration of PraAMP significantly increased the antimicrobial activity of the surface. The results in Fig. 4(a) show that after 2 h of culture, compared to Si, the Si-PraAMP and Si-SBMA-PraAMP killed 98.01% and 98.26% of E. coli in the medium and 91.86% and 93.80% of E. coli in PBS, respectively. Moreover, Fig. S3 and S4 show that relative to the blank Si group, Si-PraAMP inhibited 75.14% of S. aureus and 88.64% of P. aeruginosa, and Si-SBMA-PraAMP inhibited 83.72% of S. aureus and 81.59% of P. aeruginosa in the medium. These results demonstrate that PraAMP can retain its bioactivity after the immobilization process. The live/dead assay in Fig. 4(b) also showed that PraAMP on the surface killed the bacteria after 2 h, with E. coli on Si-PraAMP and Si-SBMA-PraAMP having red fluorescence and those on Si having green fluorescence. The further FESEM results in Fig. 4 (c) showed that PraAMP killed bacteria by destroying their membranes. The bacteria on Si had a smooth membrane. However, the membrane of E. coli was wrinkled, and holes appeared in the case of Si-PraAMP and Si-SBMA-PraAMP. These results were in accordance with the literature; this demonstrated that immobilized AMP destroyed the bacterial membrane and released their contents.48
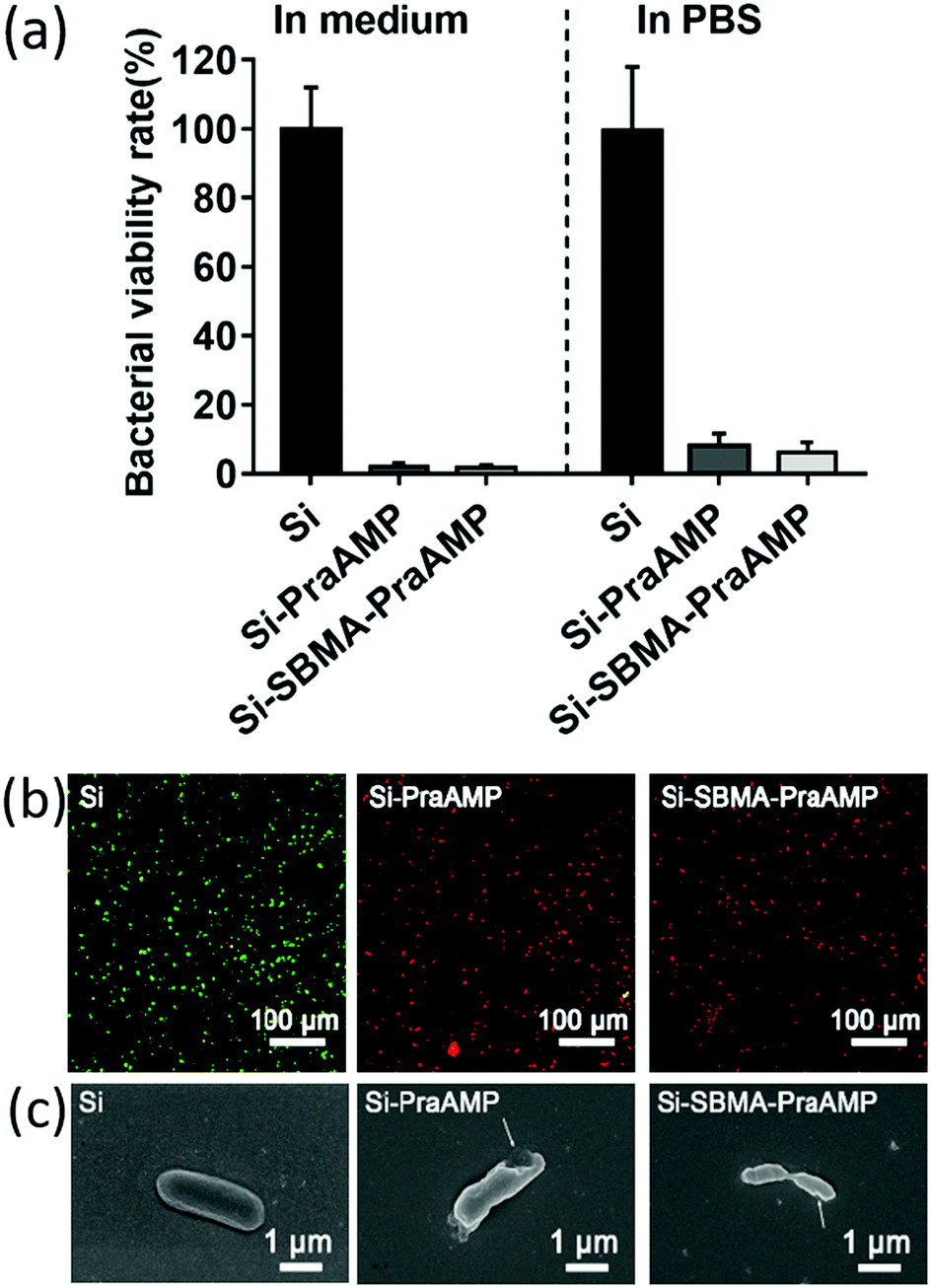 | ||
| Fig. 4 (a) The antimicrobial activity of the surface against E. coli in the medium or in PBS after 2 h (n = 3). (b) The live/dead assay of E. coli on the indicated sample in the medium after 2 h of culture the images were obtained by merging the images observed under the FITC and TRITC channels. The green bacteria were live, while the red bacteria were dead. (c) The FESEM images of E. coli on the indicated sample in the medium after 2 h of culture. | ||
Recently, Gomes et al.21 have mentioned that ascorbic acid sodium will have an impact on the arginine residues in the peptide during the CuAAC. In addition, guanidinium hydrochloride would be helpful for researchers to improve the CuAAC-containing peptide with arginine. We have also found that a Maillard-type reaction occurs during the integration of peptide. The details of the 9,10-phenanthrenequinone assay used for the Maillard-type reaction are described in the literature18,19 and shown in the supporting information. The results in Fig. S5 and Table S1 (ESI†) show that after 2 h of the reaction between PraAMP and ascorbic acid sodium, less than 40% of the arginine in PraAMP would be lost due to the Maillard-type reaction. However, the antimicrobial results demonstrated that it would not impact the activity of the peptide on the surface evidently, which was consistent with the literature.49,50
We have further demonstrated that Si-SBMA-PraAMP has a much better enzymolysis-tolerance than Si-PraAMP. The results in Fig. 5 indicate that after being treated with trypsin for 1 min, both Si-PraAMP and Si-SBMA-PraAMP maintained their antimicrobial activity as they killed 99.41% and 98.83% of E. coli, respectively. However, as the treatment time increased, Si-PraAMP lost its antimicrobial activity more readily. After 3 min of treatment, the peptide on Si-PraAMP began to decompose, and Si-PraAMP only killed 68.42% of the E. coli, whereas Si-SBMA-PraAMP killed 99.41% of the E. coli under the same conditions. After 5 min of the treatment, Si-PraAMP inhibited only 31.58% of E. coli on its surface, whereas Si-SBMA-PraAMP inhibited 84.80% of E. coli. These antimicrobial results indicate that the incorporation of polySBMA can markedly improve the peptide's enzymolysis-tolerance on the surface. This improved tolerance should result from the potential of the polySBMA layer, which can prevent the non-specific adsorption of the enzyme on the surface.51
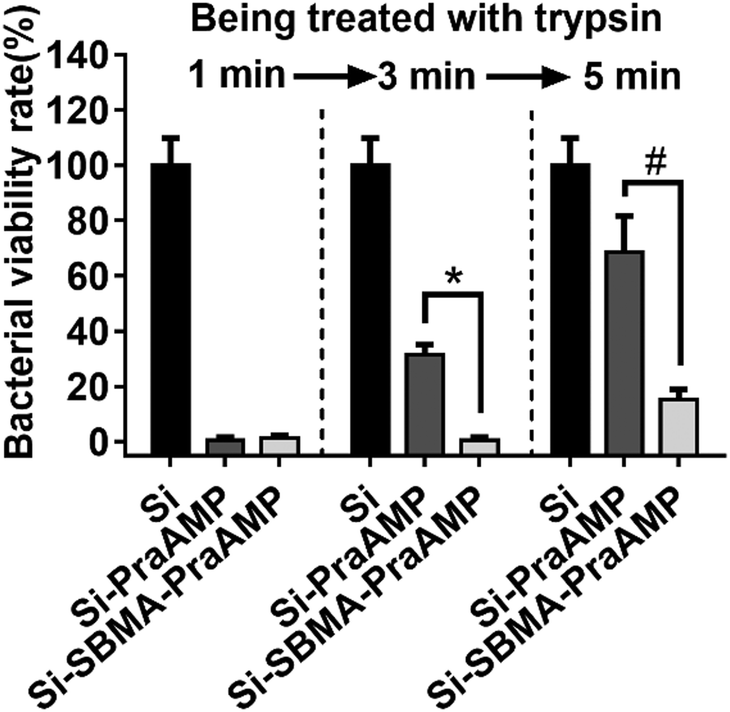 | ||
| Fig. 5 The antimicrobial activity of the surface against E. coli (in medium) after being treated with 1 mg mL−1 trypsin for 1, 3 or 5 min (n = 3) (# denotes p < 0.01, * denotes p < 0.001). | ||
The biocompatibility results shown in Fig. 6 illustrated that both Si-PraAMP and Si-SBMA-PraAMP had negligible cytotoxicity towards the mBMSCs. After 24 h of the culture, the CCK-8 results shown in Fig. 6(a) indicated that 0.96- and 1.04-folds of cells were on Si-PraAMP and Si-SBMA-PraAMP, respectively, as compared to the case of Si. Moreover, the fluorescence images in Fig. 6(b)–(d) show that the cells spread well on Si, Si-PraAMP, and Si-SBMA-PraAMP; this is consistent with the CCK-8 results.
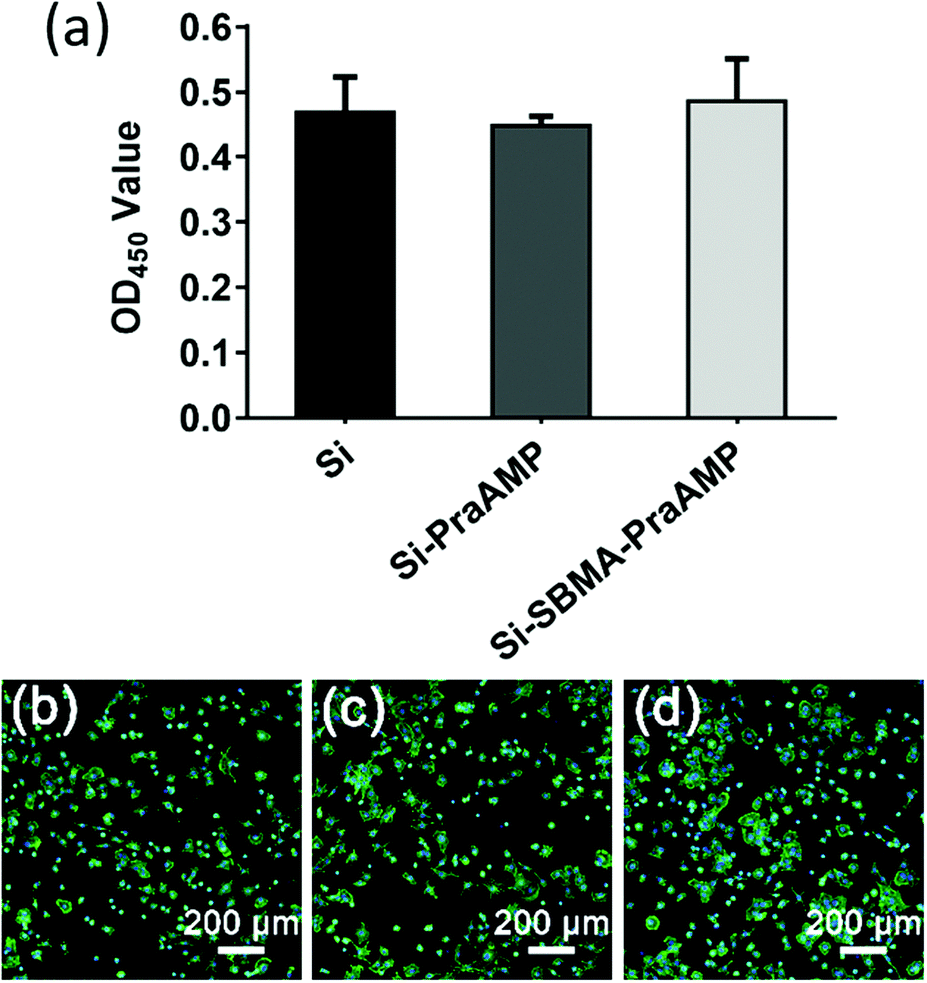 | ||
| Fig. 6 The biocompatibility of the samples towards mBMSCs. (a) The CCK-8 assay of the mBMSCs on the indicated sample after 24 h of culture (n = 3) and the morphology of the mBMSCs on (b) Si, (c) Si-PraAMP, and (d) Si-SBMA-PraAMP after 24 h of culture. | ||
Conclusions
Using a combination of SI-ATRP and click chemistry, we developed a method to modify a model silicon surface using a antimicrobial peptide with improved stability. L-Propargylglycine at the N-terminus of AMP was able to enhance its salt-tolerant properties, and the poly(zwitterion) spacer could increase the enzymolysis stability of the modified surface. The surface exhibited antimicrobial activity against E. coli, S. aureus, and P. aeruginosa and showed negligible cytotoxicity towards mBMSCs. This method may contribute to the development of enzymolysis-stable antimicrobial peptides for application in biomaterials.Author contributions
J. H., L. W. and L. R. designed the experiments and contributed to writing the manuscript. J. C. participated in CCK-8 assay. G. H participated in the Maillard-type reaction assay. The other authors performed the preparation of the surface and its characterization. All authors participated in the discussion and review of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51232002, 51302088, and 51273072), the GuangZhou Important Scientific and Technological Special Project (201508020123), the Guangdong Scientific and Technological Project (2014B090907004), and the Guangdong Natural Science Funds for Distinguished Young Scholar (2016A030306018).Notes and references
- D. Campoccia, L. Montanaro and C. R. Arciola, Biomaterials, 2006, 27, 2331–2339 CrossRef CAS PubMed.
- R. O. Darouiche, Clin. Infect. Dis., 2003, 36, 1284–1289 CrossRef PubMed.
- K. A. Poelstra, N. A. Barekzi, A. M. Rediske, A. G. Felts, J. B. Slunt and D. W. Grainger, J. Biomed. Mater. Res., 2002, 60, 206–215 CrossRef CAS PubMed.
- R. O. Darouiche, N. Engl. J. Med., 2004, 350, 1422–1429 CrossRef CAS PubMed.
- C. R. Arciola, D. Campoccia, P. Speziale, L. Montanaro and J. W. Costerton, Biomaterials, 2012, 33, 5967–5982 CrossRef CAS PubMed.
- K. J. Grobe, J. Zahller and P. S. Stewart, J. Ind. Microbiol. Biotechnol., 2002, 29, 10–15 CrossRef CAS PubMed.
- E. Drenkard and F. M. Ausubel, Nature, 2002, 416, 740–743 CrossRef CAS PubMed.
- F. Paladini, M. Pollini, D. Deponti, A. Di Giancamillo, G. Peretti and A. Sannino, J. Mater. Sci.: Mater. Med., 2013, 24, 1105–1112 CrossRef CAS PubMed.
- N. J. Hickok and I. M. Shapiro, Adv. Drug Delivery Rev., 2012, 64, 1165–1176 CrossRef CAS PubMed.
- A. L. P. F. Caroni, C. R. M. de Lima, M. R. Pereira and J. L. C. Fonseca, Colloids Surf., B, 2012, 100, 222–228 CrossRef CAS PubMed.
- X. Chen and H. J. Schluesener, Toxicol. Lett., 2008, 176, 1–12 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS PubMed.
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 CrossRef CAS PubMed.
- N. K. Brogden and K. A. Brogden, Int. J. Antimicrob. Agents, 2011, 38, 217–225 CAS.
- M. Kazemzadeh-Narbat, J. Kindrachuk, K. Duan, H. Jenssen, R. E. W. Hancock and R. Z. Wang, Biomaterials, 2010, 31, 9519–9526 CrossRef CAS PubMed.
- F. Costa, I. F. Carvalho, R. C. Montelaro, P. Gomes and M. C. Martins, Acta Biomater., 2011, 7, 1431–1440 CrossRef CAS PubMed.
- K. Matsuzaki, Biochim. Biophys. Acta, 2009, 1788, 1687–1692 CrossRef CAS PubMed.
- F. Costa, S. Maia, J. Gomes, P. Gomes and M. C. L. Martins, Acta Biomater., 2014, 10, 3513–3521 CrossRef CAS PubMed.
- F. Costa, S. Maia, P. Gomes and M. C. L. Martins, Biomaterials, 2015, 52, 531–538 CrossRef CAS PubMed.
- C. D. Fjell, H. Jenssen, K. Hilpert, W. A. Cheung, N. Panté, R. E. W. Hancock and A. Cherkasov, J. Med. Chem., 2009, 52, 2006 CrossRef CAS PubMed.
- M. Barbosa, N. Vale, F. Costa, M. C. L. Martins and P. Gomes, Carbohydr. Polym., 2017, 165, 384–393 CrossRef CAS PubMed.
- L. Wang, J. J. Chen, C. Z. Cai, L. Shi, S. Liu, L. Ren and Y. J. Wang, J. Mater. Chem. B, 2015, 3, 30–33 RSC.
- K. Hilpert, M. Elliott, H. Jenssen, J. Kindrachuk, C. D. Fjell, J. Korner, D. F. H. Winkler, L. L. Weaver, P. Henklein, A. S. Ulrich, S. H. Y. Chiang, S. W. Farmer, N. Pante, R. Volkmer and R. E. W. Hancock, Chem. Biol., 2009, 16, 58–69 CrossRef CAS PubMed.
- J. J. Chen, Y. C. Zhu, Y. C. Song, L. Wang, J. Z. Zhan, J. C. He, J. Zheng, C. T. Zhong, X. T. Shi, S. Liu, L. Ren and Y. J. Wang, J. Mater. Chem. B, 2017, 5, 2407–2415 RSC.
- M. Gabriel, K. Nazmi, E. C. Veerman, A. V. N. Amerongen and A. Zentner, Bioconjugate Chem., 2006, 17, 548–550 CrossRef CAS PubMed.
- M. D. Steven and J. H. Hotchkiss, J. Appl. Polym. Sci., 2008, 110, 2665–2670 CrossRef CAS.
- Z. Y. Ong, J. C. Cheng, Y. Huang, K. J. Xu, Z. K. Ji, W. M. Fan and Y. Y. Yang, Biomaterials, 2014, 35, 1315–1325 CrossRef CAS PubMed.
- S. F. Chen, J. Zheng, L. Y. Li and S. Y. Jiang, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS PubMed.
- C. J. Morris, K. Beck, M. A. Fox, D. Ulaeto, G. C. Clark and M. Gumbleton, Antimicrob. Agents Chemother., 2012, 56, 3298–3308 CrossRef CAS PubMed.
- A. J. Keefe and S. Y. Jiang, Nat. Chem., 2012, 4, 59–63 CrossRef CAS PubMed.
- M. C. Parrott and J. M. DeSimone, Nat. Chem., 2012, 4, 13–14 CrossRef CAS PubMed.
- Z. Zhang, S. F. Chen and S. Y. Jiang, Biomacromolecules, 2006, 7, 3311–3315 CrossRef CAS PubMed.
- H. Wang, G. F. Yue, C. Q. Dong, F. L. Wu, J. Wei, Y. Yang, Z. Y. Zou, L. F. Wang, X. P. Qian, T. Zhang and B. R. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 4550–4559 CAS.
- Q. Shao and S. Y. Jiang, Adv. Mater., 2015, 27, 15–26 CrossRef CAS PubMed.
- J. B. Schlenoff, Langmuir, 2014, 30, 9625–9636 CrossRef CAS PubMed.
- Z. Zhang, H. Vaisocherova, G. Cheng, W. Yang, H. Xue and S. Y. Jiang, Biomacromolecules, 2008, 9, 2686–2692 CrossRef CAS PubMed.
- M. C. Sin, Y. M. Sun and Y. Chang, ACS Appl. Mater. Interfaces, 2014, 6, 861–873 CAS.
- L. Wang, J. Chen, L. Shi, Z. Shi, L. Ren and Y. Wang, Chem. Commun., 2014, 50, 975–977 RSC.
- Y. Zhu, B. Gupta, B. Guan, S. Ciampi, P. J. Reece and J. J. Gooding, ACS Appl. Mater. Interfaces, 2013, 5, 6514–6521 CAS.
- K. A. Kilian, T. Boecking and J. J. Gooding, Chem. Commun., 2009, 630–640, 10.1039/b815449j.
- L. Pingsheng, C. Qiang, L. Li, L. Sicong and S. Jian, J. Mater. Chem. B, 2014, 2, 7222–7231 RSC.
- K. Takasu, K. Kushiro, K. Hayashi, Y. Iwasaki, S. Inoue, E. Tamechika and M. Takai, Sens. Actuators, B, 2015, 216, 428–433 CrossRef CAS.
- P. Chapman, R. E. Ducker, C. R. Hurley, J. K. Hobbs and G. J. Leggett, Langmuir, 2015, 31, 5935–5944 CrossRef CAS PubMed.
- Y. H. Ma, X. X. Bian, L. He, M. T. Cai, X. X. Xie and X. L. Luo, Appl. Surf. Sci., 2015, 329, 223–233 CrossRef CAS.
- F. He, B. W. Luo, S. J. Yuan, B. Liang, C. Choong and S. O. Pehkonen, RSC Adv., 2014, 4, 105–117 RSC.
- H. L. Chu, H. Y. Yu, B. S. Yip, Y. H. Chih, C. W. Liang, H. T. Cheng and J. W. Cheng, Antimicrob. Agents Chemother., 2013, 57, 4050–4052 CrossRef CAS PubMed.
- I. Y. Park, J. H. Cho, K. S. Kim, Y. B. Kim, M. S. Kim and S. C. Kim, J. Biol. Chem., 2004, 279, 13896–13901 CrossRef CAS PubMed.
- X. Li, P. Li, R. Saravanan, A. Basu, B. Mishra, S. H. Lim, X. D. Su, P. A. Tambyah and S. S. J. Leong, Acta Biomater., 2014, 10, 258–266 CrossRef CAS PubMed.
- C. M. Santos, A. Kumar, S. S. Kolar, R. Contreras-Caceres, A. McDermott and C. Z. Cai, ACS Appl. Mater. Interfaces, 2013, 5, 12789–12793 CAS.
- E. J. Bodner, N. S. Kandiyote, M. Y. Lutskiy, H. B. Albada, N. Metzler-Nolte, W. Uhl, R. Kasher and C. J. Arnusch, RSC Adv., 2016, 6, 91815–91823 RSC.
- A. T. Nguyen, J. Baggerman, J. M. J. Paulusse, H. Zuilhof and C. J. M. van Rijn, Langmuir, 2012, 28, 604–610 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The 9,10-phenantroquinone assay, MASS spectrometry, circular dichroism, antimicrobial activity against S.aureus and P. aeruginosa. See DOI: 10.1039/c7tb02557b |
| This journal is © The Royal Society of Chemistry 2018 |