
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrospun organic–inorganic nanohybrids as sustained release drug delivery systems†
Yanshan
Gao
ab,
Tian Wei
Teoh
b,
Qiang
Wang
 *a and
Gareth R.
Williams
*b
*a and
Gareth R.
Williams
*b
aCollege of Environmental Science and Engineering, Beijing Forestry University, 35 Qinghua East Road, Haidian District, Beijing 100083, P. R. China. E-mail: qiangwang@bjfu.edu.cn; qiang.wang.ox@gmail.com; Tel: +86 13699 130626
bUCL School of Pharmacy, University College London, 29-39 Brunswick Square, London WC1N 1AX, UK. E-mail: g.williams@ucl.ac.uk; Tel: +44 (0)20 77535868
First published on 9th November 2017
Abstract
In this work, the non-steroidal anti-inflammatory drugs ibuprofen (ibu) and ketoprofen (ket), both poorly soluble in water, were first intercalated into layered double hydroxide (LDH) nanoparticles. The drug–LDH composites were then mixed with poly(ε-caprolactone) (PCL) at 5% and 10% w/w ratios and processed into fibers via electrospinning, yielding organic–inorganic nanohybrids. PCL/drug fibers were additionally prepared as controls. The average diameter of the fibers ranged from 400 to 1000 nm. The fibers are found to be smooth and cylindrical, with the LDH-containing systems having more homogenous fibers than those without the inorganic filler. From in vitro drug release tests, it was determined that more than 90% of the intercalated ibu and ket were released from the drug–LDH nanohybrids within the first 4 hours. Similarly, more than 60% of the incorporated drug was freed from the PCL/drug fibers in this time period. However, the release rates of both ibu and ket from the drug–LDH loaded PCL fibers were significantly slower. Only 44–48% of ibu was released from the PCL/ibu–LDH system after 5 days, while the amount released in the case of ket was 20–25%. In addition, drug release was still ongoing after 5 days for all the PCL/drug–LDH samples. These systems are thus proposed to have potential as implantable drug delivery systems.
1. Introduction
Biodegradable and biocompatible polymers have been widely explored in drug delivery.1 Amongst these, poly(ε-caprolactone) (PCL) has received significant attention because of its slow degradation rate (≈3 years in vivo), which lends it to the preparation of long-lasting implants. PCL has been processed using a wide variety of pharmaceutical technologies, including with the electrospinning approach. The latter comprises a facile and scalable method to generate fibers from polymer solutions or melts.2,3In solution electrospinning, a polymer and functional component (such as a drug) are first dissolved in a volatile solvent. The resultant solution is ejected from a syringe fitted with a metal tip towards a metal collector plate, with a high potential difference applied between the two. The electrical energy causes the solvent to evaporate rapidly, resulting in one-dimensional fibers in which the drug is typically dispersed as a solid solution in the polymer. Usually the fiber diameter ranges from a few nanometers to a few micrometers.4–6 Electrospun drug loaded fibers have been widely explored for targeted release, for improving the dissolution rate of poorly water-soluble drugs, and for encapsulating multiple functional components.7–10
However, when fabricating nanofibers a burst release of drug is often observed even if the polymer filament is insoluble in water (as is the case for PCL). This arises because of the very high surface area-to-volume ratio of the fibers, which results in a large proportion of the incorporated drug molecules being present at or near the fiber surfaces. These molecules can rapidly diffuse into the release medium, leading to an initial burst release. A number of authors have explored this phenomenon,4,11,12 and the principal strategy used to ameliorate burst release has been to prepare core/shell fibers with drug present in the core component only. In such systems, the drug molecules must diffuse through the blank polymer shell before exiting the fibers, and thus the burst effect can often be prevented.13–16 However, the production of core/shell fibers is not a panacea, and several studies have shown that simply having a blank polymer shell surrounding a drug-loaded core does not always prevent the initial burst of release.17,18 A potentially more powerful route to preclude burst release is using triaxial electrospinning to generate three-layer fibers with a gradient distribution of drug.19
An alternative approach is to prepare fibers loaded with drug-containing nanoparticles. Some studies have been performed exploring this option, and it has been shown to have potential. For instance, when fibers of poly(lactic-co-glycolic acid) containing rhodamine B-loaded mesoporous silica nanoparticles were prepared, rhodamine release was slowed compared to the silica nanoparticles alone.20 Wang et al.21 studied composite PCL fibers containing chitosan nanoparticles loaded with naproxen or rhodamine B, and found the properties of the drug to have a major influence on the release rate. In other work, Polacco and co-workers prepared fibers of poly(lactic acid)/ε-caprolactone copolymers containing poly(DL-lactide-co-glycolide) and gelatin nanoparticles loaded with various drugs.22 It was postulated that these fibers could have applications in targeted delivery. Despite its evident promise, however, the production of nanoparticle-loaded fibers has been less explored than the core/shell fiber technique. In this work we sought to explore the use of layered double hydroxide (LDH) nanoparticles as drug carriers.
LDHs, also commonly called anionic clays, are a class of lamellar compounds made up of positively charged mixed-metal hydroxide layers with an interlayer region containing charge compensating anions and water molecules (Fig. 1(a)). They can be described by the general chemical formula [M1−xz+Mx3+(OH)2]q+(Xn−)q/n·yH2O, where Mz+ is typically a divalent cation (such as Mg2+, Zn2+, or Ca2+), M3+ is Fe3+ or Al3+, and X is a charge balancing interlayer anion (which can range from simple ions such as Cl−, NO3− or CO32− to complex biomolecules). Electrostatic interactions and hydrogen bonds between the layers and the contents of the interlayer space hold adjacent layers together, forming a three-dimensional structure.23,24 The earliest application of LDHs in relation to human health was their use as antacids and antipeptic reagents in 1996,25 and in the last decade nanometer-sized LDH materials (50–300 nm) have been increasingly explored as drug and gene carriers and delivery systems.26 Many drugs (e.g. the non-steroidal anti-inflammatory drugs ibuprofen (ibu) and ketoprofen (ket); Fig. 1(b)) are weak acids, and thus can be deprotonated to form anions and then incorporated between the layers of an LDH system. This can lead to drug release profiles different to traditional commercial formulations.12,27
 | ||
| Fig. 1 A schematic diagram showing (a) the general structure of an LDH, (b) the chemical structures of ibuprofen and ketoprofen, and (c) the strategy adopted in this work. | ||
Advantages often cited for LDHs in drug delivery are that they provide protection (e.g. to UV light or heat-mediated degradation) of the incorporated anions, can improve the solubility of poorly water-soluble drugs, and can reduce side effects.28 However, like electrospun fibers LDH–drug intercalates commonly suffer from an initial burst release which cannot be controlled. In this work, we sought to incorporate drug-loaded LDH particles into PCL fibers to determine whether the production of such organic–inorganic nanohybrids can resolve this issue. A schematic diagram illustrating our approach is depicted in Fig. 1(c). Ibuprofen and ketoprofen were used as model drugs with poor solubility, and because sustained release formulations of these active ingredients could be of major benefit for the treatment of diseases such as arthritis.
To date, there are a few reports of polymer/LDH composites prepared by electrospinning in the literature, but in general such studies are focused on the preparation of advanced electronics materials or absorbents, or use the LDH to improve the mechanical properties of the fibers. For instance, Birajdar et al.29 have used poly(vinylidene fluoride)/LDH fibers to remove Cu(II) ions from wastewater, and Qin and co-workers30 report that adding LDHs to poly(vinyl alcohol) fibers causes the tensile strength and elongation at break to increase. Romeo and co-workers31 generated PCL/LDH fibers using the Mg/Al LDH and found that when the interlayer anion was carbonate the LDH particles were deposited at the outside of the fibers, but when the guest was 12-hydroxydodecanoate the LDH particles were largely exfoliated and evenly dispersed throughout the polymer matrix. This is expected to be because the surfactant molecules surround the hydrophilic LDH layers and reduce the interfacial tension between these and the hydrophobic PCL. The size of the LDH particles has also been shown to affect the properties of LDH-loaded electrospun fibers.32
In the biomedical setting, Shafiei et al.33 fabricated PCL composites with different LDH loadings by electrospinning and determined that the blend fibers had improved mechanical properties over pure PCL fibers, with higher tensile strength and elongation at break values. In addition, the adhesion, proliferation, and differentiation of multipotent mesenchymal stem cells were significantly improved by the addition of the LDH. There are also a few studies employing polymer/LDH fibers for drug delivery. The first report came from Tammaro and co-workers,2 who encapsulated LDH particles intercalated with diclofenac into PCL by electrospinning. PCL/diclofenac fibers exhibited an extreme burst of release, while the composite PCL/LDH-diclofenac fibers could effectively retard release and mitigate the burst.
However, this is not universally observed. Miao et al.11 produced ibuprofen drug-delivery systems from PCL and poly(lactic acid) using both the neat drug and Mg/Al LDH intercalated with ibuprofen. The use of the LDH here did not have any significant effect on the amount of burst release observed: in most cases, the release profiles leveled off very quickly (within ca. 1 h of the start of the release experiment), and although the presence of LDH does affect the maximum percentage release reached it does not change the overall shape of the release profiles. In other work, amoxicillin-intercalated LDH has been loaded into PCL fibers; a burst of release approaching 50% of the loading occurred during the first day of the release experiment.34 The most recent report in this area comes from Yang et al. and is concerned with fibers based on poly(lactic-co-glycolic acid) and loaded with an Mg/Al LDH intercalated with flurbiprofen axetil.35 The presence of the LDH in the fibers could reduce the initial burst of release, and slowed down the rate at which the drug was freed into solution.
Where the polymer used for fiber formation is hydrophilic, the polar nature of the LDH particle surface should result in good compatibility between the two phases and the formation of a homogeneously-mixed composite with long term stability. In contrast, with a hydrophobic polymer such as PCL the two phases will have a tendency to separate, as noted by Romeo.31 Organic modification of the LDH surface using amphiphilic molecules such as surfactants can be used to ameliorate this problem. One route to achieve this is the aqueous miscible organic solvent treatment (AMOST), which results in stable, transparent dispersions of hydrophilic LDHs in nonpolar solvents.36,37 This simple technique uses organic solvents to wash LDH particles immediately after synthesis and can tune the surface of LDHs to be hydrophobic, enabling them to be effectively dispersed in apolar environments.38–40 AMOST modified LDHs have also been found to disperse better in hydrophobic polymers than unmodified LDH particles.
In this study, aqueous miscible organic layered double hydroxides (AMO-LDHs) were used to prepare PCL/LDH systems, with the aim of producing a homogeneous LDH dispersion in the polymer. We employed an LDH based on Mg and Al ([Mg2Al(OH)6]X·yH2O, where X represents the interlayer anion) because of the biocompatibility of these metals. First, the model drugs ibuprofen and ketoprofen were intercalated into the MgAl–LDH interlayer, and the products fully characterized. Next, the LDH nanoparticles were encapsulated into a PCL matrix via electrospinning. Following an in-depth characterization of the fibers, a systematic study exploring drug release from the PCL/drug, LDH–drug and PCL/LDH–drug systems was performed.
2. Experimental
2.1 Materials
Materials were procured as follows: poly(ε-caprolactone) (PCL; Mn = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–90000, Sigma-Aldrich), ibuprofen sodium salt (Santa Cruz Biotechnology, ≥98%), ibuprofen and ketoprofen (Sigma, ≥98%), MgCl2·6H2O (VWR, 99%), AlCl3·6H2O (Acros Organics, 99%), dichloromethane (DCM; VWR, 99%), acetone (Fisher Scientific, 99.98%), and sodium hydroxide (Fisher Scientific).
000–90000, Sigma-Aldrich), ibuprofen sodium salt (Santa Cruz Biotechnology, ≥98%), ibuprofen and ketoprofen (Sigma, ≥98%), MgCl2·6H2O (VWR, 99%), AlCl3·6H2O (Acros Organics, 99%), dichloromethane (DCM; VWR, 99%), acetone (Fisher Scientific, 99.98%), and sodium hydroxide (Fisher Scientific).
2.2 Synthesis of drug intercalated LDH
Ibuprofen intercalated MgAl–LDH was synthesized using a hydrothermal method.40,41 A metal precursor solution containing 2.032 g MgCl2·6H2O and 1.207 g AlCl3·9H2O (Mg:Al = 2:1) in 20 mL deionized water was added drop-wise into a second solution comprising 2.1 g of sodium ibuprofen dissolved in 20 mL of H2O. The pH was kept constant at ca. 9.5 using a NaOH (1 M) solution, and the subsequent slurry was transferred into a 100 mL Teflon-lined stainless steel autoclave. The autoclave was sealed and a hydrothermal reaction performed for 24 h at 100 °C, followed by product recovery through centrifugation and washing with deionized water until the supernatant pH was close to 7. A final wash was performed with acetone in order to produce AMO-LDH particles. Ketoprofen intercalated LDH was prepared in the same way, except that the Mg2+/Al3+ solution was added drop-wise into an alkaline solution containing 2.58 g of ketoprofen in 20 mL H2O.
2.3 Electrospinning
PCL solutions (10% w/v) were prepared by dissolving the polymer in a mixture of dichloromethane (DCM) and acetone (weight ratio 3:1) under stirring at room temperature. Ibuprofen and ketoprofen were added at concentrations of 2% and 5% (w/w, based on the dry weight of the polymer). The solutions were then stirred again until complete dissolution of the drug had occurred. Working fluids for producing drug–LDH loaded fibers were prepared by adding the relevant nanocomposite to a 10% w/v PCL solution in 3:1 DCM:acetone, at 5 and 10% w/w with respect to the dry weight of the polymer.
For electrospinning, solutions were loaded into a plastic syringe fitted with a flat-tipped needle with an inner diameter of 0.61 mm (Nordson EFD). The solution was ejected at a speed of 0.8 mL h−1, controlled by a syringe pump (KDS100, Cole-Parmer), with a distance of 15 cm between the tip of the needle and the collector. A voltage of 16 kV was applied between the needle and collector using a high-voltage power supply (HCP35-35000, FuG Elektronik). Fibers were deposited on a flat piece of steel covered with aluminum foil, forming mats which could be peeled off for subsequent experiments.
2.4 Characterization
The morphologies of the LDH particles and fibers were examined using a scanning electron microscope (SEM; FEI Quanta 200 instrument) with an accelerating voltage of 5.0 kV. The samples were sputter coated with a thin gold layer before assessment to prevent charging and to improve image quality. To calculate their mean size, the diameters of the fibers were measured at more than 100 points in the SEM images. Energy dispersive X-ray spectroscopy (EDX) was carried out using a Hitachi S3400N microscope fitted with an Oxford Instruments EDX detector, and data analysed using the Inca software.X-ray diffraction (XRD) patterns were recorded on a Rigaku MiniFlex 600 instrument in reflection mode with Cu Kα radiation (λ = 1.5418 Å). The accelerating voltage was set at 40 kV with 15 mA current. Diffraction patterns were obtained over the range of 2–70° at a scanning rate of 5° min−1. Fourier transform IR analyses were carried out on a Spectrum 100 instrument (PerkinElmer) over the range 650–4000 cm−1 at a resolution of 4 cm−1. Differential scanning calorimetry (DSC) analyses were performed using a Q2000 instrument (TA Instruments). Approximately 5 mg of each sample was loaded into an aluminum pan and heated from 0 to 160 °C at a rate of 10 °C min−1 under a nitrogen flow rate of ca. 50 mL min−1.
In the case of the LDH particles, the average hydrodynamic particle size and the size distribution were measured by dynamic light scattering (DLS) on a Zetasizer instrument (Malvern Instruments), after the particles were dispersed in ethanol via ultrasonication for 30 min. C, H, and N contents were determined using the combustion method on a Flash 2000 Elemental Analyser (Thermo Scientific).
2.5 In vitro drug release studies
Drug release from the drug–LDH intercalates was performed by dispersing 10 mg of the particles into 150 mL of phosphate buffered saline (PBS) at pH = 7.4 and stirring at 37 °C. At predetermined time points, 3 mL aliquots were taken out and replaced with fresh pre-heated phosphate buffer to maintain a constant volume. The samples were filtered through a 0.22 μm filter, after which the drug concentrations were measured using a UV-vis spectrophotometer (7315 spectrometer, Jenway) at 222 nm for ibuprofen and 204 nm for ketoprofen.For the fibers, 200 mg of each fiber mat was placed into 150 mL of PBS at 37 °C under magnetic stirring. At selected time intervals, 3 mL of the solution was withdrawn for analysis, and 3 mL of fresh pre-heated phosphate buffer added to maintain a constant volume. Drug concentrations were quantified using UV spectroscopy following identical procedures to those for the LDH systems.
3. Results and discussion
3.1 Synthesis of LDH–drug nanoparticles
XRD analysis was first used to verify the intercalation of drug ions into the LDH particles. The XRD patterns are shown in Fig. 2(a). Both drug–LDH samples have well-developed lamellar structures, characterized by a series of basal diffractions at low 2θ angles and weaker non-basal diffractions at higher angles.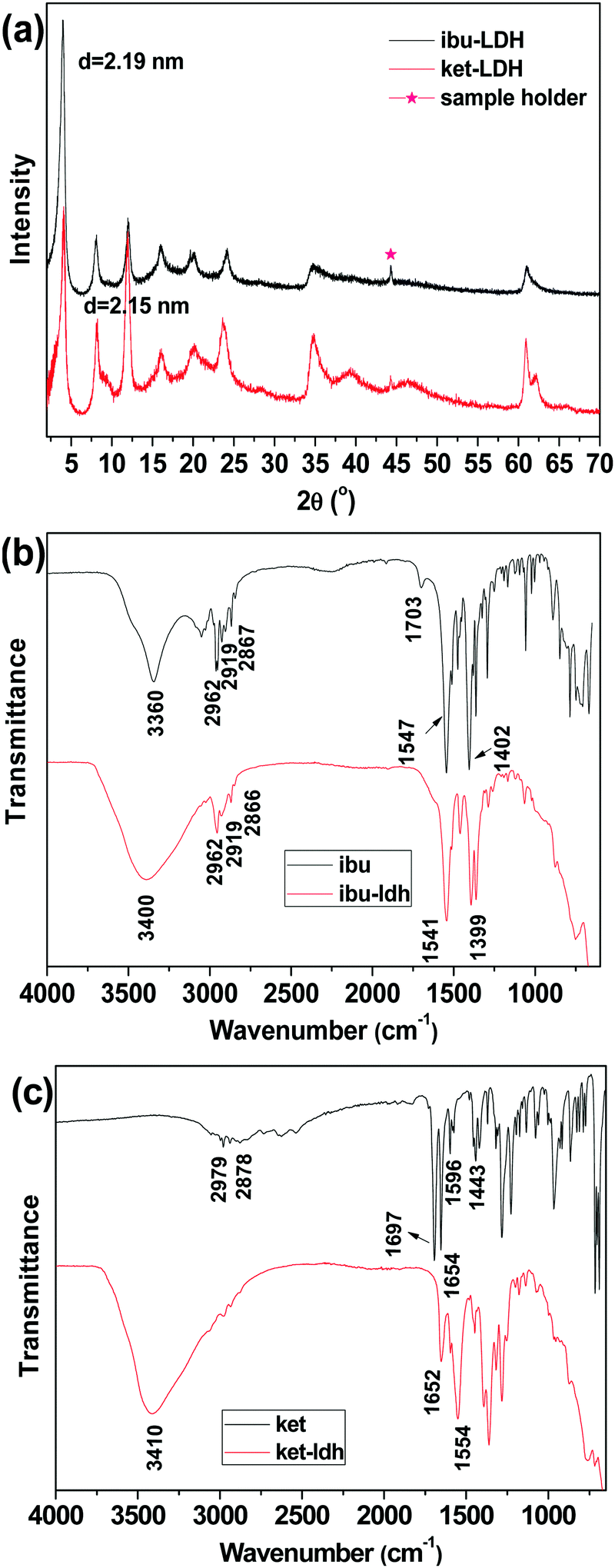 | ||
| Fig. 2 (a) XRD patterns of ibuprofen and ketoprofen intercalated LDH, with FT-IR spectra of (b) pure ibuprofen and ibu–LDH, and (c) pure ketoprofen and ket–LDH. | ||
The interlayer spacing (d003) values of Mg/Al LDH intercalated with inorganic anions (e.g. nitrate) is reported to be ∼0.88 nm.42 The distance between adjacent layers is larger with ibuprofen or ketoprofen anions in the interlayer space, at 2.19 and 2.15 nm respectively. This demonstrates that the drug ions were intercalated into the LDH particles, and these values are in good agreement with previous reports.27,43 The expanded interlayer separation is due to the molecular size and geometrical arrangement of the anions in the interlayer space.44 As the layer thickness of an LDH is 0.48 nm,45 the gallery heights (defined as the interlayer spacing – the layer thickness) of the ibu–LDH and ket–LDH hybrids are ca. 1.71 and 1.67 nm, respectively. This suggests that the ibu and ket ions are arranged as a slightly tilted bilayer with their long axes perpendicular to the metal hydroxide layer.46 Such an arrangement permits the carboxylate groups to interact with the layer surfaces.
IR spectra are given in Fig. 2(b and c). Fig. 2(b) depicts the FT-IR spectra of ibuprofen and the ibu–LDH nanohybrid. The characteristic bands at 2962, 2919, and 2867 cm−1 for pure ibuprofen are attributed to the alkyl stretching vibrations. The peaks at 1547 and 1402 cm−1 are due to asymmetric and symmetric RCOOH stretches, respectively. These have been observed at around 1550 and 1400 cm−1 in previous studies,27,47,48 and shift to 1541 and 1399 cm−1 in the ibu–LDH nanohybrids. This is expected to arise as a result of hydrogen bonding and electrostatic interactions between the RCOO− groups and the LDH layers in the intercalate.46 The weak bands at 1511 and 1466 cm−1 in the ibuprofen and ibu–LDH spectra are assigned to aromatic ring vibrations.
Pure ketoprofen (Fig. 2(c)) shows characteristic absorption peaks at 3054 and 2979–2878 cm−1, denoting stretching vibrations of aromatic and methyl C–H groups. The bands at 1697 and 1654 cm−1 are due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the carboxylic acid and ketone groups respectively.49 Peaks at 1596 and 1443 cm−1 can be attributed to CC stretching of the aromatic ring. As was observed for ibuprofen, the carboxylate stretches shift in position after intercalation, indicating the formation of interactions between the LDH layer and the guest anions.
O stretching of the carboxylic acid and ketone groups respectively.49 Peaks at 1596 and 1443 cm−1 can be attributed to CC stretching of the aromatic ring. As was observed for ibuprofen, the carboxylate stretches shift in position after intercalation, indicating the formation of interactions between the LDH layer and the guest anions.
The IR spectra of both ibu–LDH and ket–LDH also show a very broad band centered at approximately 3400 cm−1, which corresponds to v(OH) stretching vibrations of the hydroxyl groups of the host layers. The observations from IR spectroscopy thus concur with the XRD data, demonstrating successful intercalation of ibu and ket into the LDH gallery. Elemental analysis measurements (Table S1, ESI†) reveal that the average loading of ibuprofen and ketoprofen in the intercalates was 42.01 ± 2.55% and 45.89 ± 0.76% w/w, respectively.
The morphologies of the LDH particles were characterized using SEM analysis (Fig. 3). Both ibu–LDH and ket–LDH exist as aggregated plate-like nanoparticles. The lateral diameters of the particles are 147 ± 37 nm and 179 ± 35 nm, respectively. The hydrodynamic diameters measured by dynamic light scattering (DLS) are rather larger than those determined from the SEM images, with DLS giving sizes of 296 ± 3 nm and 315 ± 13 nm for the ibu– and ket–LDH systems respectively. This is consistent with previous work, for instance that reported by Xu et al.50 The larger particle size from DLS can partially be attributed to the particles being hydrated, but the large difference between the SEM and DLS results also suggests that the LDH–drug nanoparticles were not completely dispersed in the DLS experiment, and some aggregates were present.
 | ||
| Fig. 3 SEM images of (a) ibu–LDH and (b) ket–LDH, with particle size distributions. | ||
3.2 Electrospun fibers
XRD patterns, given in Fig. 4, provide information on the physical form of the drug and drug–LDH nanohybrids in the polymer matrix. Pure PCL has a semi-crystalline structure with two main reflections at 21.3° and 23.6°. When fibers were prepared with 2 and 5% w/w ibu (Fig. 4(a)), only the typical peaks of PCL can be seen in the patterns. This shows that the drug is amorphously dispersed in the polymer matrix after electrospinning, most likely as a solid solution, and is consistent with numerous previous reports of drug loaded electrospun fibers.11 For the PCL/ibu–LDH fibers, a reflection at 2θ = 4° is visible in Fig. 4(a), together with a weak peak at 8°. These correspond to the (003) and (006) reflections of ibu–LDH, indicating that the dispersed LDH retains an ordered structure after electrospinning. They are also more intense with the 10% w/w loading than with 5%, as would be intuitively expected. The overall drug loading in the fibers is calculated to be 4.20% w/w for the PCL/10% ibu–LDH fibers, and 2.10% for the PCL/5% ibu–LDH analogue. | ||
| Fig. 4 XRD patterns of the various (a) ibuprofen and (b) ketoprofen containing nanocomposites. | ||
The results with the ketoprofen systems are analogous to those with ibuprofen. The characteristic reflections of ketoprofen were completely absent in the PCL/ket nanofibers (Fig. 4(b)), even at 5% w/w loading, again confirming an amorphous drug distribution. Typical reflections of the ket–LDH hybrid can be seen in the PCL/ket–LDH fibers (Fig. 4(b)), with these being more distinctive at higher loading. The final drug loadings are 4.59 and 2.29% w/w for the PCL/10% ket–LDH and PCL/5% ket–LDH materials respectively.
SEM micrographs of the electrospun PCL–drug and PCL/LDH–drug composites are shown in Fig. 5. All the drug-loaded and LDH/drug-loaded fibers are largely smooth and cylindrical in morphology, but the LDH-free fibers show evidence of solvent inclusion in the form of merged fibers. The average diameter of the fibers ranges from 400 nm to 1000 nm. There is some influence of the drug or LDH–drug loading on the fiber diameters: in most (but not all) cases, the diameter increases with increasing drug or LDH–drug loading. The ketoprofen systems have consistently larger and more homogeneous diameters than their ibuprofen analogues. In general, the LDH-containing fibers have more homogeneous fiber diameters than the analogous LDH-free materials. The reasons for these variations are not entirely clear: the mechanism of electrospinning is extremely complicated, and many factors influence the fiber diameters due to a complex interplay between fluid dynamics, electrodynamics, and rheology.51,52
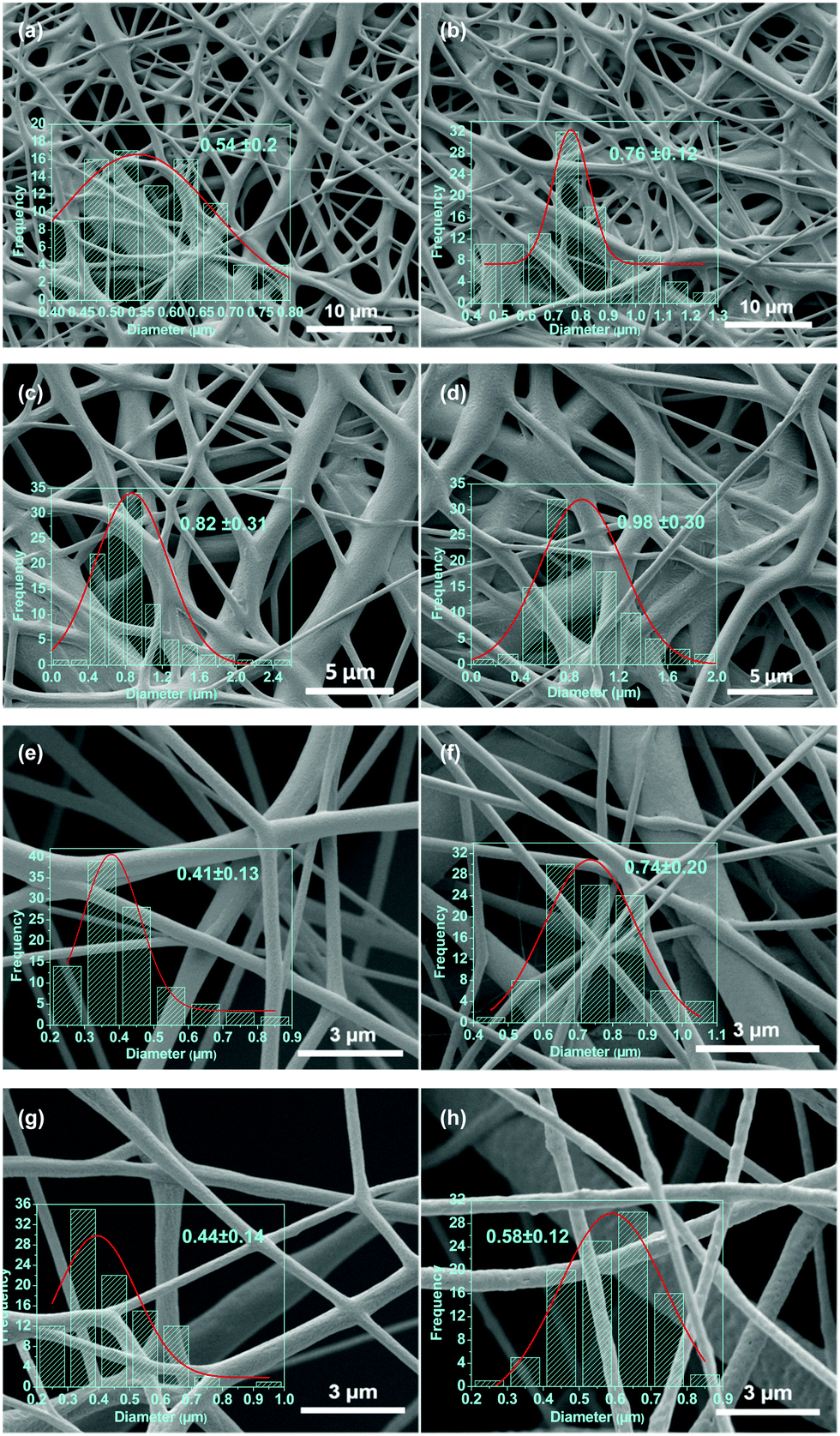 | ||
| Fig. 5 SEM images of (a) PCL/2% ibu, (b) PCL/2% ket, (c) PCL/5% ibu, (d) PCL/5% ket, (e) PCL/5% ibu–LDH, (f) PCL/5% ket–LDH, (g) PCL/10% ibu–LDH, and (h) PCL/10% ket–LDH fibers, with the corresponding diameter distributions. | ||
The drug–LDH distribution in the PCL composites was explored using SEM-EDX analyses. Elemental mapping results for Mg and Al are given in Fig. 6. Mg and Al are distributed evenly throughout the fibers, indicating that the drug loaded LDH nanoparticles were well dispersed as a result of the AMOST process. This renders the surfaces of the LDH particles somewhat hydrophobic, and thus they can efficiently mix with the PCL carrier matrix, rather than aggregating on the surface. In addition, the EDX also showed that the ratio of Mg/Al is around 2.
 | ||
| Fig. 6 SEM images, Mg, and Al elemental mapping of the PCL/drug–LDH fibers. (a) PCL/5% ibu–LDH, (b) PCL/5% ket–LDH, (c) PCL/10% ibu–LDH, and (d) PCL/10% ket–LDH. | ||
DSC analyses were performed to evaluate the crystallinity of the fiber systems, and the results are depicted in Fig. 7. The endothermic peak for pure PCL at 63.4 °C corresponds to its melting point, which is reported to lie the range from 59 to 64 °C.53,54 The DSC curves of ibuprofen and ketoprofen showed sharp endothermic peaks at 77.4 °C and 94.3 °C respectively, corresponding to their melting points and confirming the two raw materials to be crystalline. These values agree well with the literature.46,49,55,56 The DSC curves of physical mixtures of PCL and either drug show two endothermic peaks, corresponding to the melting points of the two components in the mixture. For the PCL + ibu physical mixture, the endothermic peaks are at 60.8 and 77.0 °C (Fig. 7(a)), whereas the PCL + ket mixture showed melts at 61.5 and 95.7 °C (Fig. 7(b)). Thus, as would be expected, simply making a physical mixture does not change the physical form.
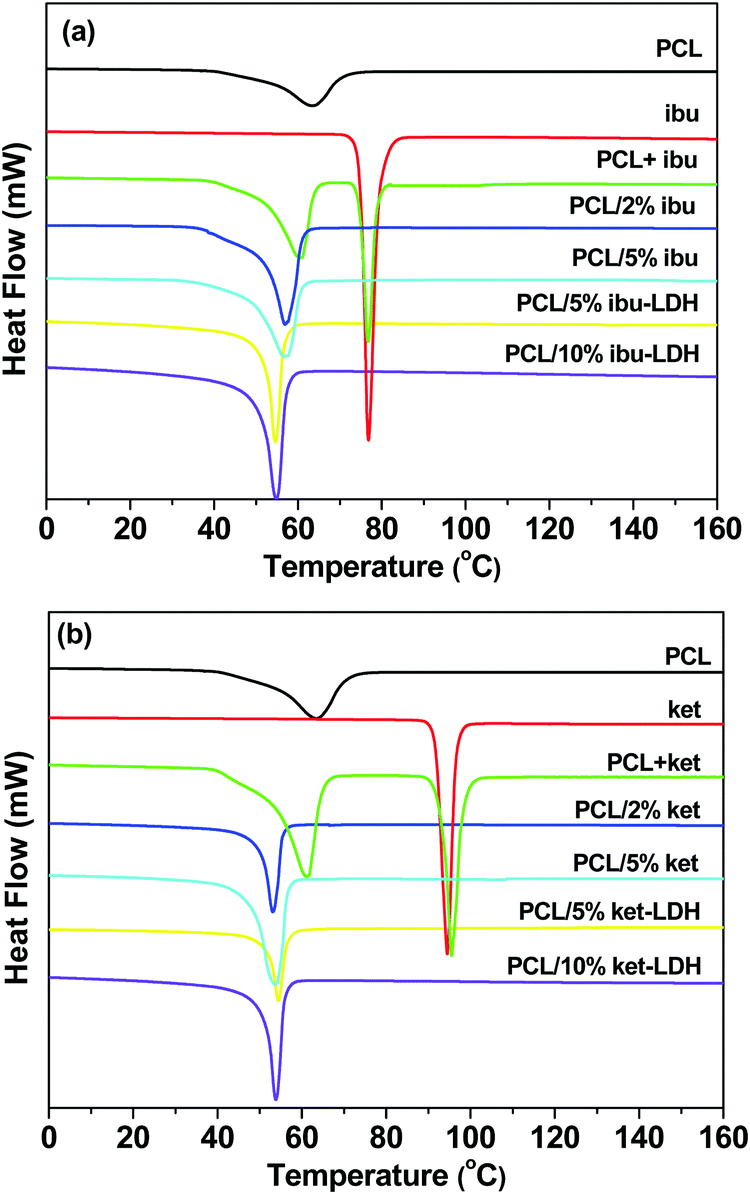 | ||
| Fig. 7 DSC curves (exo up) of the (a) ibuprofen and (b) ketoprofen based nanocomposites. PCL + ibu and PCL + ket denote physical mixtures of the drug and polymer with a mass ratio 5:1. | ||
In contrast, the DSC curves of the PCL based nanofibers showed only a single peak in the temperature range from 53 to 61 °C, which arises due to melting of the PCL. The peak is slightly shifted to lower temperatures compared to pure PCL, suggesting a reduction in the polymer's crystallinity and the size of its crystals. The degree of crystallinity (Xc) can be calculated from the melting peak areas using the following equation:57
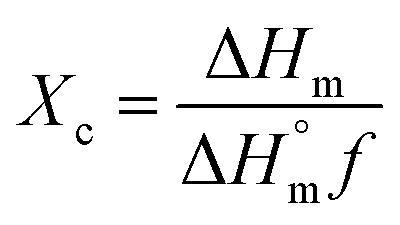
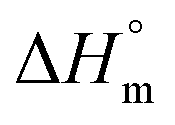 is the heat of fusion for a theoretically 100% crystalline PCL sample (taken to be 146 J g−1),2 ΔHm is the melting enthalpy of the sample of interest, and f is the weight fraction of polymer in the system. The results of these calculations are listed in Table 1. After incorporation of the drugs or LDH–drug nanohybrids in the PCL matrix, both ΔHm and Xc decreased, indicating that there are some interactions between PCL and the drug or drug–LDH particles.
is the heat of fusion for a theoretically 100% crystalline PCL sample (taken to be 146 J g−1),2 ΔHm is the melting enthalpy of the sample of interest, and f is the weight fraction of polymer in the system. The results of these calculations are listed in Table 1. After incorporation of the drugs or LDH–drug nanohybrids in the PCL matrix, both ΔHm and Xc decreased, indicating that there are some interactions between PCL and the drug or drug–LDH particles.
| Samples | T m (°C) | ΔHm (J g−1) | X c (%) |
|---|---|---|---|
| PCL | 63.43 | 79.35 | 54.35 |
| PCL/2% ibu | 56.91 | 68.17 | 47.64 |
| PCL/5% ibu | 56.84 | 56.37 | 40.64 |
| PCL/5% ibu–LDH | 55.19 | 47.99 | 34.60 |
| PCL/10% ibu–LDH | 54.82 | 47.61 | 36.23 |
| PCL/2% ket | 53.08 | 54.00 | 37.74 |
| PCL/5% ket | 53.71 | 52.86 | 38.11 |
| PCL/5% ket–LDH | 54.41 | 48.49 | 34.96 |
| PCL/10% ket–LDH | 53.82 | 46.92 | 35.71 |
The melting endotherms of ibuprofen and ketoprofen were both absent from the PCL/drug fibers. This concurs with the XRD data and confirms the PCL/ket and PLC/ibu fibers to be amorphous solid dispersions. Considering the PCL/LDH–drug fibers, again no endotherms are visible other than the melt of PCL. This is because the drug ions in the LDH interlayer spaces do not adopt the same arrangement as in the pure drug crystals, and the major degradation events of the LDH lie above the upper temperature of the DSC experiment.
3.3 In vitro drug release
Fig. 8 shows the cumulative drug release profiles from the drug loaded LDH nanoparticles and the PCL/drug fibers in PBS (pH = 7.4) at 37 °C. Significant bursts of release (>80% in the first 2 h) were seen with both the LDH–drug materials (Fig. 8(a)). Ibu–LDH initially releases more slowly but eventually reaches a higher release percentage than ket–LDH. This can perhaps be explained by the fact that ibuprofen is a smaller ion with greater charge density than ketoprofen, and thus should have stronger interactions with the LDH layers. This will lead to a higher energy barrier to release, causing ibuprofen to release more slowly. At the same time, however, ibuprofen is more water soluble than ketoprofen, and so eventually it is thermodynamically favorable for a larger percentage to release. No further release is observed after 4 h for both LDH–drug systems.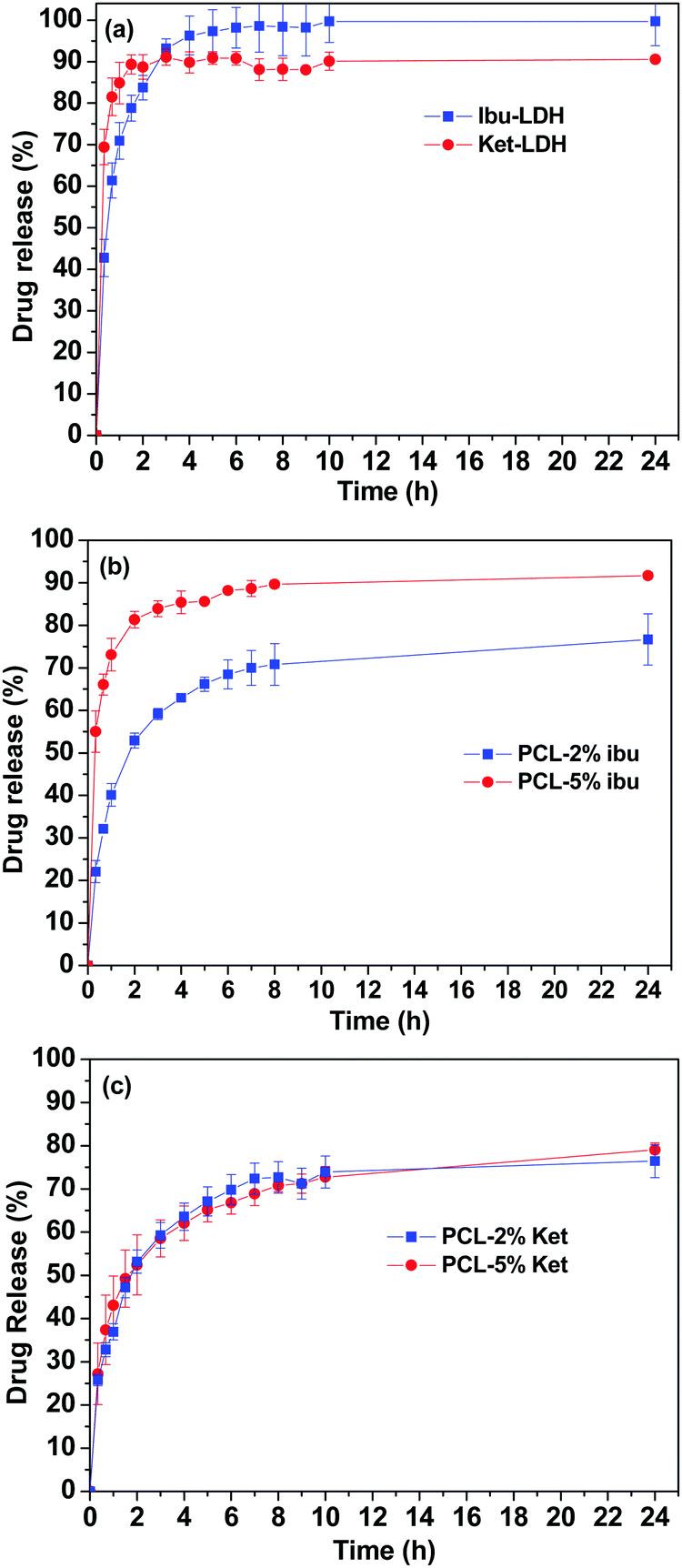 | ||
| Fig. 8 Drug release profiles of (a) the drug–LDH nanoparticles, (b) PCL/ibu fibers, and (c) PCL/ket fibers. The data points represent the mean ± S.D. of three independent experiments. | ||
Both drugs are released more slowly from the PCL/drug fibers than from the LDH interlayers (Fig. 8(b) and (c)). Over 60% of the incorporated drug is released within the first 4 h in all cases. With the PCL/ibu systems, an increase in the drug loading causes an acceleration in the release rate and extent: the PCL/5% ibu fibers free almost 85% of their drug content in 4 h. This can be explained because the higher loading will lead to more pores being formed in the fibers as drug molecules escape into solution, making it easier for ibuprofen molecules near the center of the fibers to diffuse to the release milieu. In contrast, the two ketoprofen loadings show essentially identical release profiles. This is likely to be a result of both stronger drug–polymer intermolecular interactions in the case of ketoprofen (PCL is hydrophobic, and ketoprofen more hydrophobic than ibuprofen), and its lower solubility.
The drug release profiles from the PCL/LDH–drug composites are included in Fig. 9. Fig. 9(a) and (b) give the drug release profile over 120 h, while Fig. 9(c) and (d) contain an enlargement of the first 24 h. The PCL/LDH–drug composites are able to reduce the initial burst of release profoundly (Table 2). The materials present a fast release in the initial period of the experiment, followed by a very slow release. The first stage is due to the presence of some LDH–drug particles at or close to the surface of fibers. Drug release from these depends on the ion exchange reaction time, which is very rapid and is completed in about 2 h.2 During this period, approximately 13 and 26% of the incorporated ibuprofen (or 13 and 10% of ketoprofen) are released from the systems with 5 and 10% LDH–drug loadings, respectively.
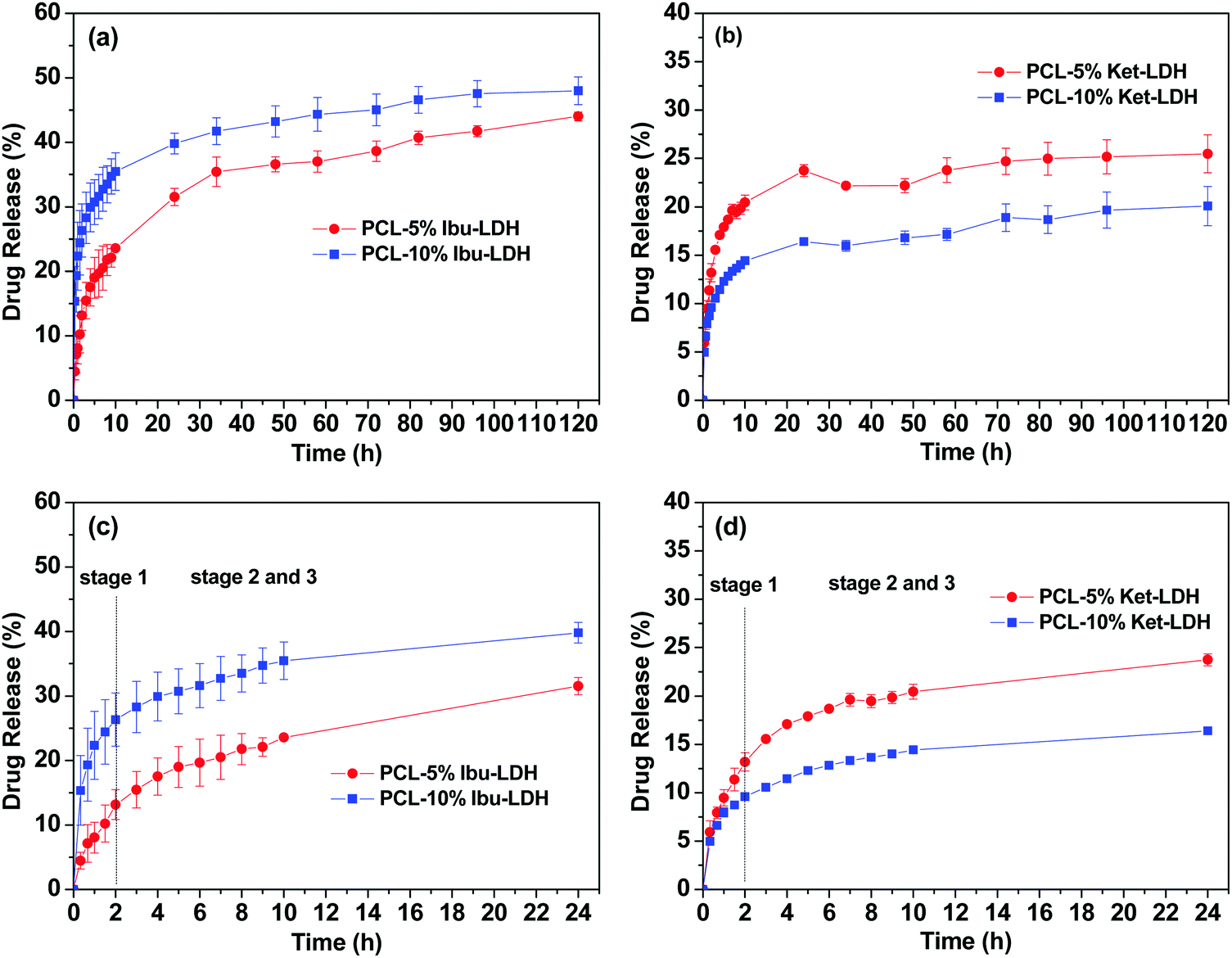 | ||
| Fig. 9 Drug release profiles for (a) PCL/ibu–LDH over 5 days, (b) PCL/ket–LDH over 5 days, with enlargements of the first 24 h for (c) PCL/ibu–LDH, and (d) PCL/ket–LDH. The data points represent the mean ± S.D. of three independent experiments. | ||
| Sample | % drug release after | |||
|---|---|---|---|---|
| 1 h | 2 h | 3 h | 4 h | |
| Ibu–LDH | 71 | 84 | 96 | 98 |
| Ket–LDH | 85 | 88 | 89 | 87 |
| PCL/2% ibu | 40 | 53 | 63 | 71 |
| PCL/5% ibu | 73 | 82 | 85 | 89 |
| PCL/2% ket | 36 | 53 | 64 | 73 |
| PCL/5% ket | 43 | 52 | 62 | 70 |
| PCL/5% ibu–LDH | 8 | 13 | 18 | 22 |
| PCL/10% ibu–LDH | 22 | 26 | 30 | 34 |
| PCL/5% ket–LDH | 9 | 13 | 17 | 19 |
| PCL/10% ket–LDH | 8 | 10 | 12 | 14 |
The second and third stages of release start after two hours, and are due to the drug escaping from the inside of the polymer matrix. Here, the release time depends on a number of factors: the diffusion behavior of the replacement phosphate ions entering the polymer/LDH matrix, the ion-exchange reaction time, and the diffusion of the drug ions escaping the fiber. This process is very slow and release extending for several days is observed.2 The fibers give an almost constant rate of release between 24–120 h, and fitting zero-order kinetics to the data over this period gives R2 values >0.9 (see Fig. S1, ESI†). After 5 days, approximately 44 and 48% of the ibuprofen loading is released for the 5 and 10% w/w fibers, while for ketoprofen the equivalent values are 25 and 20%.
The Ritger–Peppas and Bhaskar equations were also applied to the release data in the first 24 h. The former equation is expressed as:43
| Mt/M∞ = ktn |
The Bhaskar model assumes that diffusion through the particle is the rate limiting step to release, making it appropriate for ion-exchange processes. It takes the form:59
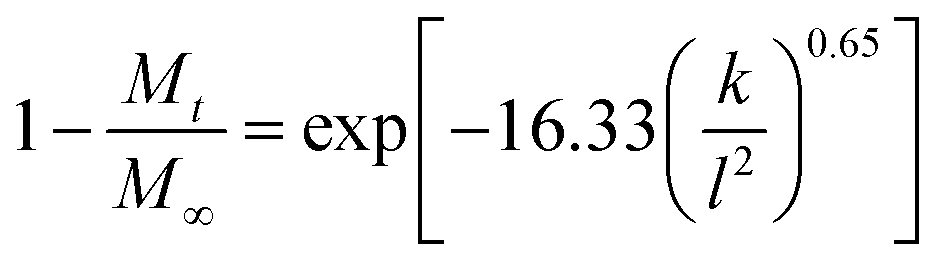
Fig. S2 (ESI†) depicts the fits obtained with both the Ritger–Peppas and Bhaskar equations: while the fits with the former are generally good with R2 > 0.96, the latter are clearly non-linear, with R2 typically below 0.9. The Peppas exponents are mostly below 0.45, which for a cylindrical dosage form such as a fiber is consistent with release governed by Fickian diffusion. The complexity of the system makes it difficult to be certain of the drug release mechanism, but the fact that the Peppas model gives good fits while the Bhaskar equation does not might indicate that the diffusion of drug through the PCL fibers is the rate limiting step to release (rather than ion-exchange in the LDH particles).
The use of the AMOST approach leads to significant improvements over previous work in this area: in the previous work by Miao et al.,11 it was found that the PCL/LDH nanofibers could slow the release of ibuprofen, but very rapid release was still observed in the first 1–2 h, and a plateau reached after 2 h. In contrast, our systems give a constant and sustained rate of release over at least 5 days, giving them great potential as implantable drug delivery systems.
4. Conclusions
Sustained drug release materials based on PCL fibers containing drug-intercalated LDH nanoparticles have been prepared and fully characterized. The fibers are found to be smooth and cylindrical, with the LDH-containing systems having more homogenous fibers than those without the inorganic filler. These systems are able to profoundly extend the drug release period over both PCL/drug fibers and LDH–drug particles alone. The use of ternary PCL/LDH–drug formulations also reduces the initial burst of release seen with the binary systems. More than 80% of the ibuprofen and ketoprofen loadings were released from the LDH–drug particles within 2 h, while release from the PCL/drug fibers reached at least 60% after 4 h and plateaus after 10 h. In contrast, the PCL/LDH–drug materials free only 10–26% of their drug loading after 2 h and extend release over more than 5 days. The PCL/ibu–LDH materials in particular give an almost constant rate of release from 10–120 h, and thus may have great potential in developing implantable drug delivery systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Mr David McCarthy and Mrs Kate Keen for the provision of SEM images, and Dr Eranka Illangakoon (UCL Mechanical Engineering) and Dr Tom Gregory (UCL Archaeology) for assistance with EDX analyses. We also thank the China Scholarship Council, British Council, and Royal Society of Chemistry for funding YG's work at UCL.References
- T. K. Dash and V. B. Konkimalla, Mol. Pharmaceutics, 2012, 9, 2365–2379 CrossRef CAS PubMed.
- L. Tammaro, G. Russo and V. Vittoria, J. Nanomater., 2009, 2009, 33–40 Search PubMed.
- S. R. Baker, S. Banerjee, K. Bonin and M. Guthold, Mater. Sci. Eng., C, 2016, 59, 203–212 CrossRef CAS PubMed.
- T. Potrč, S. Baumgartner, R. Roškar, O. Planinšek, Z. Lavrič, J. Kristl and P. Kocbek, Eur. J. Pharm. Sci., 2015, 75, 101–113 CrossRef PubMed.
- E. G. Bajsic, B. Mijovic, N. V. Penava, T. H. Grguric, M. Slouf and E. Zdraveva, J. Appl. Polym. Sci., 2016, 133, 43539 CrossRef.
- D. G. Yu, G. R. Williams, X. Wang, X. K. Liu, H. L. Li and S. W. A. Bligh, RSC Adv., 2013, 3, 4652–4658 RSC.
- M. Jin, D. G. Yu, C. F. G. C. Geraldes, G. R. Williams and S. W. A. Bligh, Mol. Pharmaceutics, 2016, 13, 2457–2465 CrossRef CAS PubMed.
- A. Baskakova, S. Awwad, J. Q. Jiménez, H. Gill, O. Novikov, P. Y. Khaw, S. Brocchini, E. Zhilyakova and G. R. Williams, Int. J. Pharm., 2016, 502, 208–218 CrossRef CAS PubMed.
- M. Mirjalili and S. Zohoori, J. Nanostruct. Chem., 2016, 6, 207–213 CrossRef.
- J. Cheng, Y. Yesl Jun, J. H. Qin and S.-H. Lee, Biomaterials, 2017, 114, 121–143 CrossRef CAS PubMed.
- Y. E. Miao, H. Zhu, D. Chen, R. Y. Wang, W. W. Tjiu and T. X. Liu, Mater. Chem. Phys., 2012, 134, 623–630 CrossRef CAS.
- A. I. Khan, L. X. Lei, A. J. Norquist and D. O'Hare, Chem. Commun., 2001, 2342–2343 RSC.
- C. Wang, K. W. Yan, Y. D. Lin and P. C. H. Hsieh, Macromolecules, 2010, 43, 6389–6397 CrossRef CAS.
- Y. Z. Zhang, X. Wang, Y. Feng, J. Li, C. T. Lim and S. Ramakrishna, Biomacromolecules, 2006, 7, 1049–1057 CrossRef CAS PubMed.
- W. J. Yang, N. Y. He and Z. Y. Li, J. Nanosci. Nanotechnol., 2016, 16, 9404–9412 CrossRef CAS.
- G. Z. Yang, J. J. Li, D. G. Yu, M. F. He, J. H. Yang and G. R. Williams, Acta Biomater., 2017, 53, 233–241 CrossRef CAS PubMed.
- D. Pedersbæk, M. T. Frantzen and P. Fojan, J. Self-Assem. Mol. Electron., 2017, 5, 17–30 CrossRef.
- M. Maleki, M. Latifi, M. Amani-Tehran and S. Mathur, Polym. Eng. Sci., 2013, 53, 1770–1779 CAS.
- D. G. Yu, X. Y. Li, X. Wang, J. H. Yang, S. W. Bligh and G. R. Williams, ACS Appl. Mater. Interfaces, 2015, 7, 18891–18897 CAS.
- B. T. Song, C. T. Wu and J. Chang, Regener. Biomater., 2015, 2, 229–237 CrossRef PubMed.
- Y. Z. Wang, B. C. Wang, W. L. Qiao and T. Y. Yin, J. Pharm. Sci., 2010, 99, 4805–4811 CrossRef CAS PubMed.
- G. Polacco, M. G. Cascone, L. Lazzeri, S. Ferrara and P. Giusti, Polym. Int., 2002, 51, 1464–1472 CrossRef CAS.
- A. I. Khan and D. O'Hare, J. Mater. Chem., 2002, 12, 3191–3198 RSC.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- G. Simoneau, Eur. J. Drug Metab. Pharmacokinet., 1996, 21, 351–357 CrossRef CAS PubMed.
- K. Ladewig, Z. P. Xu and G. Q. Lu, Expert Opin. Drug Delivery, 2009, 6, 907–922 CrossRef CAS PubMed.
- V. Ambrogi, G. Fardella, G. Grandolini and L. Perioli, Int. J. Pharm., 2001, 220, 23–32 CrossRef CAS PubMed.
- M. Wei, S. Shi, J. Wang, Y. Li and X. Duan, J. Solid State Chem., 2004, 177, 2534–2541 CrossRef CAS.
- M. S. Birajdar, S. D. Wanjale and S. P. Lonkar, J. Appl. Polym. Sci., 2013, 130, 4508–4515 CAS.
- Q. Qin, Y. Liu, S. C. Chen, F. Y. Zhai, X. K. Jing and Y. Z. Wang, J. Appl. Polym. Sci., 2012, 126, 1556–1563 CrossRef CAS.
- V. Romeo, G. Gorrasi and V. Vittoria, Biomacromolecules, 2007, 8, 3147–3152 CrossRef CAS PubMed.
- W. Y. Lv, Q. Q. Mei, M. Du, J. L. Xiao, W. J. Ye and Q. Zheng, J. Phys. Chem. C, 2016, 120, 14435–14443 CAS.
- S. S. Shafieia, M. Shavandia, G. Ahangarib and F. Shokrolahic, Appl. Clay Sci., 2016, 127-128, 52–63 CrossRef.
- E. Valarezo, L. Tammaro, S. Silvia González, O. Malagón and V. Vittoria, Appl. Clay Sci., 2013, 72, 104–109 CrossRef CAS.
- C. Y. Yang, T. H. Zhu, J. H. Wang, S. H. Chen and W. Y. Li, RSC Adv., 2015, 5, 69423–69429 RSC.
- Q. Wang and D. O'Hare, Chem. Commun., 2013, 49, 6301–6303 RSC.
- Q. Wang, X. Zhang, J. H. Zhu, Z. H. Guo and D. O'Hare, Chem. Commun., 2012, 48, 7450–7452 RSC.
- Y. S. Gao, Q. Wang, J. Y. Wang, L. Huang, X. R. Yan, X. Zhang, Q. L. He, Z. P. Xing and Z. H. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 5094–5104 CAS.
- Y. S. Gao, Q. Wang, L. Qiu, J. W. Wu, X. R. Yan, A. Umar, J. Guo, X. Zhang, J. Y. Wang and Z. H. Guo, Polym. Compos., 2016, 37, 3449–3459 CrossRef CAS.
- Y. S. Gao, Y. Zhang, G. R. Williams, D. O'Hare and Q. Wang, Sci. Rep., 2016, 6, 35502 CrossRef PubMed.
- Z. P. Xu, G. Stevenson, C. Q. Lu and G. Q. (Max) Lu, J. Phys. Chem. B, 2006, 110, 16923–16929 CrossRef CAS PubMed.
- Y. S. Gao, J. W. Wu, Z. Zhang, R. Jin, X. Zhang, X. R. Yan, A. Umar, Z. H. Guo and Q. Wang, J. Mater. Chem. A, 2013, 1, 9928–9934 CAS.
- M. Silion, D. Hritcu, I. M. Jaba, B. Tamba, D. Ionescu, O. C. Mungiu and I. M. Popa, J. Mater. Sci.: Mater. Med., 2010, 21, 3009–3018 CrossRef CAS PubMed.
- Q. Wang, Z. Wu, H. H. Tay, L. Chen, Y. Liu, J. Chang, Z. Zhong, J. Luo and A. Borgna, Catal. Today, 2011, 164, 198–203 CrossRef CAS.
- S. Bégu, A. A. Pouëssel, R. Polexe, E. Leitmanova, D. A. Lerner, J.-M. Devoisselle and D. Tichit, Chem. Mater., 2009, 21, 2679–2687 CrossRef.
- X. M. Lu, L. M. Meng, H. P. Li, N. Du, R. J. Zhang and W. G. Hou, Mater. Res. Bull., 2013, 48, 1512–1517 CrossRef CAS.
- C. R. Gordijo, C. A. S. Barbosa, A. M. D. C. Ferreira, V. R. L. Constantino and D. d. O. Silva, J. Pharm. Sci., 2005, 94, 1135–1148 CrossRef CAS PubMed.
- V. H. DeLeona, T. D. Nguyena, M. Narb, N. A. D'Souzab and T. D. Golden, Mater. Chem. Phys., 2012, 132, 409–415 CrossRef.
- G. Vijayaranga Vittal, R. Deveswaran, S. Bharath, B. V. Basavaraj and V. Madhavan, Int. J. Pharm. Invest., 2012, 2, 150–156 CrossRef CAS PubMed.
- Z. Zi, Gu, A. H. Wu, L. Li and Z. P. Xu, Pharmaceutics, 2014, 6, 235–248 CrossRef PubMed.
- S. Ramakrishna, K. Fujihara, W. E. Teo, T. C. Lim and Z. Ma, An introduction to electrospinning and nanofibers, World Scientific Publishing Co. Pte. Ltd, Singapore, 2005 Search PubMed.
- M. Jin, D. G. Yu, X. Wang, C. F. G. C. Geraldes, G. R. Williams and S. W. A. Bligh, Adv. Healthcare Mater., 2016, 5, 977–985 CrossRef CAS PubMed.
- V. R. Sinha, K. Bansal, R. Kaushik, R. Kumria and A. Trehan, Int. J. Pharm., 2004, 278, 1–23 CrossRef CAS PubMed.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- M. Subhabrota, R. Souvik and C. Subhadeep, Daru, J. Pharm. Sci., 2011, 19, 47–56 CAS.
- M. L. Vueba, L. A. E. Batista de Carvalho, F. Veiga, J. J. Sousa and M. E. Pina, Eur. J. Pharm. Biopharm., 2004, 58, 51–59 CrossRef CAS PubMed.
- S. K. Nayak, S. Mohanty and S. K. Samal, Mater. Sci. Eng., A, 2009, 523, 32–38 CrossRef.
- X. Y. Yang, H. M. Trinh, V. Agrahari, Y. Sheng, D. Pal and A. K. Mitra, AAPS PharmSciTech, 2016, 17, 294–306 CrossRef CAS PubMed.
- R. Bhaskar, S. R. S. Murthy, B. D. Miglani and K. Viswanathan, Int. J. Pharm., 1986, 28, 59–66 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb01825h |
| This journal is © The Royal Society of Chemistry 2017 |