
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface enhancement of a molecularly imprinted polymer film using sacrificial silica beads for increasing L-arabitol chemosensor sensitivity and detectability
Marcin
Dabrowski
a,
Maciej
Cieplak
a,
Krzysztof
Noworyta
 a,
Matthias
Heim
b,
Witold
Adamkiewicz
a,
Alexander
Kuhn
b,
Piyush Sindhu
Sharma
*a and
Wlodzimierz
Kutner
*ac
a,
Matthias
Heim
b,
Witold
Adamkiewicz
a,
Alexander
Kuhn
b,
Piyush Sindhu
Sharma
*a and
Wlodzimierz
Kutner
*ac
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: psharma@ichf.edu.pl; wkutner@ichf.edu.pl
bUniv. Bordeaux, CNRS, ISM, ENSCBP, 16 Avenue Pey Berland, 33607 Pessac, France
cFaculty of Mathematics and Natural Sciences, School of Sciences, Cardinal Stefan Wyszynski University in Warsaw, Woycickiego 1/3, 01-815 Warsaw, Poland
First published on 14th July 2017
Abstract
Molecular imprinting in polymers leads, among others, to synthetic receptors of high selectivity, comparable to that of their biological counterparts. Deposition of a thin non-porous molecularly imprinted polymer (MIP) film directly on a transducer surface enables fabrication of chemosensors for various health relevant biocompounds. However, the sensitivity of a chemosensor with such an MIP film as the recognition unit is limited, mostly because of slow analyte diffusion through this film. Herein, a simple procedure was developed to enhance, in a controlled way, the active surface area of an L-arabitol imprinted polymer film. For this, a macroporous MIP-(L-arabitol) film was synthesized and simultaneously deposited on a gold electrode of a quartz crystal resonator transducer by potentiodynamic electropolymerization. This large surface area film effectively enhanced analytical signals of mass changes at a quartz crystal microbalance. Hence, the L-arabitol limit of quantification was ∼16-fold better than that of the corresponding non-porous MIP film of the same mass.
Introduction
Development of robust and sensitive sensors for real-time analytical detection is important in clinical diagnosis. Generally, these sensors contain recognition units derived either from biological1,2 or chemically synthesized materials3,4 to provide the desired sensing selectivity. Depending on the recognition materials used, these sensors are categorized as bio- or chemosensors. Recognition units prepared from chemically synthesized materials are considered advantageous in comparison with biological ones. The major disadvantage associated with biosensors includes limited stability (because of poor tolerance of extreme solution acidity or basicity, temperature, some organic solvents, and exposure to external fields, e.g., an electromagnetic or ultrasonic field), short lifetime, low availability, inadequate reproducibility, and high cost because of the necessity of tedious purification. Moreover, different receptor orientations on the transducer surface can affect the performance of biosensors. In contrast, chemical sensors appear more robust and reproducible. In these chemosensors, selective analyte determination is attained by the deposition of synthetic receptors directly on the transducer surface. A lot of effort has been focused on designing and synthesizing new materials as alternatives to biological counterparts.5–7One of such research approaches involves imprinting of analytes in polymers.7–9 In this approach, synthetic materials are prepared by copolymerization of functional and cross-linking monomers in the presence of a template in a porogenic solvent. During this synthesis, first, the monomers bearing suitable functionalities and the template molecules are pre-organized in solution to form a complex via either noncovalent or covalent interactions between their complementary binding functionalities. The structures of these complexes are then “frozen” with a cross-linking monomer to result in a molecularly imprinted polymer (MIP) matrix. After subsequent template removal, vacant molecular cavities, complementary in their size and shape to the template molecule, are left in this matrix. These cavities are then capable of selective binding of analyte molecules, even in the presence of similarly structured molecules of interferenes.
MIP preparation as a thin non-porous film, directly deposited on the transducer surface, is the most common way to integrate receptors for devising a chemosensor. Towards that, different strategies have already been developed.10 However, mass transport of analytes in non-porous films is hindered because of slow diffusion to less accessible imprinted cavities that are buried deeply inside the MIP film. This mass transport hindrance severely limits the sensitivity of MIP chemosensors. Therefore, several attempts have been made to enhance this transport. One possible solution involves the enhancement of the MIP film active surface area.11 This can easily be achieved by preparing an MIP as the porous film. One approach, which was proposed recently to increase this area, is based on growing the MIP film in between self-assembled nanostructures, e.g., colloidal silica or spherical polymer beads,11–13 similarly to what has been reported for metal films with controlled porosity.14–17 In addition to a large (surface area)-to-volume ratio, well-defined and uniform macroporous structures were produced that way.11 The resulting films were capable of binding analytes more effectively than the non-porous MIP films.12,13,18
Clinical statistics shows that the Candida yeasts19,20 have been responsible for the growing number of invasive fungal infections in recent years.21 Despite the introduction of novel antifungal therapeutic treatments, the rate of mortality caused by these infections is still very high.21 Early diagnosis of these infections seems to be crucial for successful therapy.22 Now, there is no adequate method in terms of sensitivity and selectivity for the diagnosis of invasive candidiasis in humans. Results from blood and tissue cultures are still considered the ‘gold standard’, although a number of non-culture methods have been developed including antibody and antigen tests, DNA amplification tests, and detection of Candida metabolites.
A method that has gained considerable attention in recent years involves the determination of D-arabitol in body fluids. This polyol is produced in vitro by most pathogenic and medically important Candida sp.23,24 Moreover, it is produced by the human organism in an equimolar ratio with the L-enantiomeric form. Physiological concentrations of D- and L-arabitol in blood plasma and urine are in the micromolar range25 and their abnormal concentrations may indicate the Candida sp. infection. However, the concentration of D-arabitol in body fluids can be affected by many other factors of patients' variability, e.g., renal function. This effect lowers the accuracy of the diagnosis. To overcome this deficiency, two strategies were developed. Both of them rely on referencing the D-arabitol concentration to concentrations of some internal standards, which indicate the efficiency of renal function. These two standards are L-arabitol and creatinine. Therefore, more accurate methods of diagnosis of infections caused by Candida sp. involve the determination of the (D-arabitol)-to-creatinine ratio, or the (D-)-to-(L-arabitol) ratio in body fluids.26,27
In our previous effort to elaborate chemosensors for the above polyols, we have designed synthetic receptors for D-arabitol and integrated them with suitable transducers.28 However, the sensitivity and limit of detection (LOD) of these former chemosensors were insufficient for arabitol determination in blood plasma samples. Therefore, there is a clear need to develop a sugar-alcohol templated MIP film with superior sensitivity. Moreover, it is reasonable to target the less abundant L-enantiomer to obtain an enantioselective chemosensor.
For this reason, herein, we explored the use of colloidal crystals based on silica beads as sacrificial nanostencils for electrodeposition of macroporous MIP-(L-arabitol) films with an inverse opal structure. The obtained films were then used as recognition units in piezoelectric microgravimetry (PM) measurements at an electrochemical quartz crystal microbalance, EQCM. Undoubtedly, the major advantage of this synergistic combination of methods is the ability to determine L-arabitol at concentrations much lower than those attained with a non-porous MIP film. Hence, this decreased LOD should enable early and immediate initiation of treatment and protection of a patient, thus avoiding unnecessary complications.21,29 Therefore, monitoring the health status of patients under antifungal treatment should become relatively straightforward.30
Materials and methods
Chemicals
2,2′-Bithiophene-boronic acid and 3,3′-bithiophene were purchased from Combi-Blocks, while acetonitrile, 3-aminopropyltriethoxysilane, tetraethoxysilane, and tetrabutylammonium perchlorate were from Sigma-Aldrich. L-Arabitol, D-arabitol, xylitol, and ribitol were bought from Alfa-Aesar. Ethanol, sodium hydroxide, and hydrochloric acid were supplied by POCH. Potassium hexacyanoferrate(II) was from CHEMPUR. Milli-Q water with 18.2 MΩ cm resistivity was used for all of the Langmuir and Langmuir–Blodgett (LB) experiments.Apparatus
An AUTOLAB electrochemistry system (Metrohm Autolab), equipped with a PGSTAT12 potentiostat and a FRA2 frequency response analyzer, was used for the potentiodynamic and differential pulse voltammetry (DPV) experiments. A three-electrode one-compartment V-shaped glass minicell was used for the electrochemical experiments. A 1 mm diameter Pt disk electrode, a saturated Ag|AgCl electrode, and Pt foil were used as the working, reference, and auxiliary electrode, respectively.Model EQCM 5610 and EQCM 5710 quartz crystal microbalances, controlled by the EQCM 5710-S2 software, all of the Institute of Physical Chemistry (Warsaw, Poland), were used to perform PM experiments under flow-injection analysis (FIA) and batch analysis conditions, respectively. The resonance frequency change was measured with 1 Hz resolution using 14 mm diameter, AT-cut, plano–plano, unpolished gold-coated quartz crystal resonators, Au-QCRs31 (relative surface area, Rsa = 2.0), of 10 MHz resonance frequency with 5 mm diameter and 100 nm thick Au film electrodes evaporated over Ti underlayer films on both resonator sides. However, only one side was wetted by the working solution and the Au film electrode of this side was used both as the working electrode and the substrate for the MIP film. Then, the Au-QCRs were rinsed with methanol, next with acetone, and then dried in air. Pt coil and Ag wire were used as the counter and pseudo-reference electrode, respectively.
A Zepto model plasma cleaner of 100 W power from Diener Electronic was used for cleaning and hydrophilizing the surfaces of the Au-QCR. A rectangular (7 × 75) cm2 Langmuir trough type BAM 601 with an accessory from Nima Technology, controlled by NIMA TR620 software, was used to perform the Langmuir experiments and LB transfer of silica nanobeads onto the hydrophilized surfaces of Au-QCRs. A MPW Medical Instruments centrifuge model MPW-351R with the rotor no. 11457 was used for the centrifugation of nanobead suspensions for the Langmuir and LB experiments. The MIP films were imaged with scanning electron microscopy (SEM) using a Nova NanoSEM 450 microscope of FEI Nova.
Chemosensor fabrication
Purified and dehydrated nanobeads were suspended in a mixed solvent solution of anhydrous ethanol and chloroform (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, v:v) with the nanobead concentration of ∼10 mg mL−1. Then, the resulting suspension was spread over the air–water interface of the Langmuir trough filled with Milli-Q water. After organic solvent evaporation, the nanobeads present at the air–water interface were compressed with a barrier speed of 10 cm2 min−1 to the target LB transfer pressure of 8.0 mN m−1.36
:4, v:v) with the nanobead concentration of ∼10 mg mL−1. Then, the resulting suspension was spread over the air–water interface of the Langmuir trough filled with Milli-Q water. After organic solvent evaporation, the nanobeads present at the air–water interface were compressed with a barrier speed of 10 cm2 min−1 to the target LB transfer pressure of 8.0 mN m−1.36
The Au-QCRs were rinsed, consecutively, with water, methanol, and acetone. Subsequently, they were treated with 50 W oxygen plasma under the oxygen pressure of 0.25 kPa for 10 min. A freshly plasma cleaned Au-QCR was used as the substrate for the LB deposition of the colloidal nanobead crystals. This substrate was immersed in the trough liquid, and then withdrawn four times, at the speed of 38 mm min−1 (the maximum speed available) and 1 mm min−1, respectively. After the first withdrawal, the sample was dried and heated to promote adhesion of the nanobeads of the first layer to the Au electrode surface.
An EQCM stagnant solution system, comprising an EP-21 potentiostat of ELPAN, was used for the deposition with two potential cycles from 0.50 to 1.20 V vs. an Ag|AgCl pseudo reference electrode at 100 mV s−1 scan rate. An acetonitrile solution of 1 mM L-arabitol (template), 2 mM 2,2′-bithiophene boronic acid (functional monomer), 4 mM 3,3′-bithiophene (cross-linking monomer), and 0.1 M (TBA)ClO4 (supporting electrolyte) was used for the electrochemical syntheses of MIP films. After electropolymerization, the MIP films were rinsed with acetonitrile to remove unreacted substrates and the electrolyte, and then dried in air.
A non-imprinted polymer (NIP) control film was deposited from the template-free solution using the same electropolymerization procedure.
Results and discussion
The nature of interaction between a template and functional monomers determines the performance of the designed MIP. Moreover, the number of recognition sites in the MIP directly depends on the nature and extent of the monomer–template interactions in the pre-polymerization solution.39 For instance, if the pre-polymerization complex involves only non-covalent interactions, the pre-polymerization solution at equilibrium will be a mixture of functional monomers, and non-complexed, partially complexed, and fully complexed templates. This equilibrium can generate heterogeneous binding sites and, ultimately, decrease the efficiency of molecular imprinting. This deficiency can be avoided by promoting pre-polymerization complex formation through reversible covalent bonding.Reversible covalent bonding of boronic acids with cis-1,2- or 1,3-diols leads to the formation of five- or six-membered cyclic esters, respectively. These interactions were proven to be sufficiently strong. Hence, binding saccharides at millimolar or sub-millimolar concentration levels was possible, thus allowing boronic-acid-based saccharide sensing in biologically relevant environments.6,40 Therefore, we used boronic acid derivatized functional monomers to imprint the L-arabitol polyol. Vicinal diols of arabitol were covalently bound by the 2,2′-bithiophene-5-boronic acid functional monomer for this imprinting.
Chemosensor fabrication
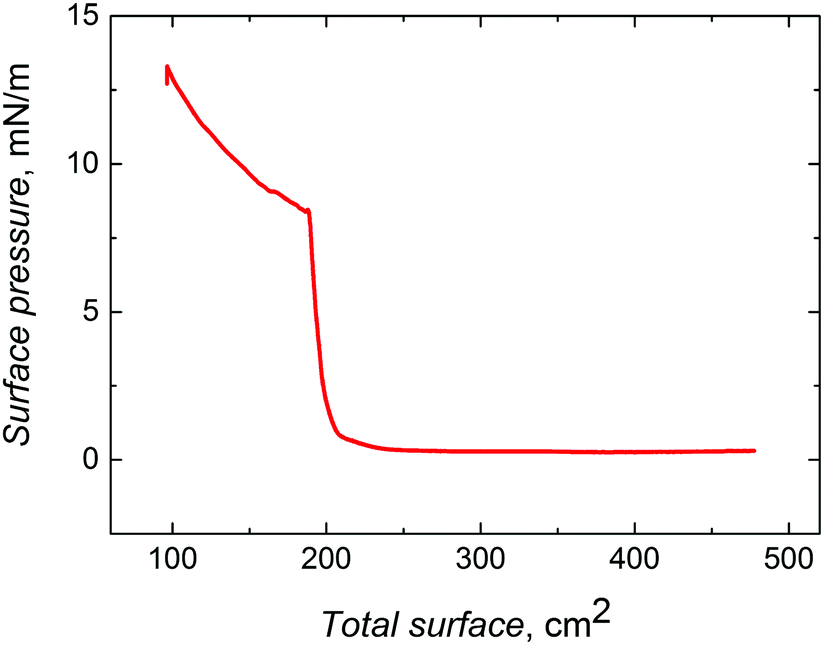 | ||
| Fig. 1 Compression isotherm for ∼330 nm silica nanobeads, which were surface functionalized with 3-aminopropyltriethoxysilane. The barrier speed was 10 cm2 min−1. | ||
The SEM images (Fig. 2) confirmed the presence of four layers of silica nanobeads and their hexagonal-like packing, characteristic of a colloidal crystal. Although the structure of this crystal was imperfect, importantly, each nanobead was in contact with several other nanobeads. Fabrication of highly ordered colloidal crystals under our experimental conditions was difficult because the surface of the chemically unpolished Au-QCRs is relatively rough.31
 | ||
| Fig. 2 The SEM images of side views of (a) a four-layer 330 nm diameter silica nanobead colloidal crystal, (b) a non-porous MIP film, and (c) a macroporous MIP film deposited on Au electrodes of quartz crystal resonators. | ||
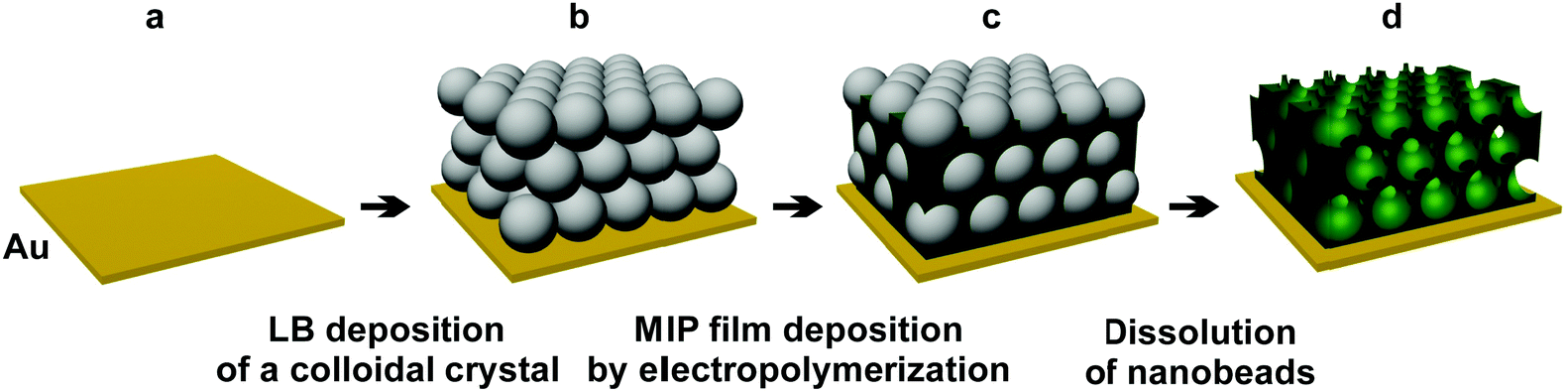 | ||
| Scheme 1 Preparation of (a) the Au electrode, (b) the silica nanobead colloidal crystals by the LB technique, and then (c) deposition of the MIP by electropolymerization, followed by (d) the formation of the macroporous MIP film by dissolution of the silica beads. | ||
During potentiodynamic potential cycling, electro-oxidation at ∼1.1 V resulted in the generation of a bithiophene cation radical, which then initiated the polymerization.42 3,3′-Bithiophene acts as an acceptor of the cation radical and, hence, passively participates in the electropolymerization. Therefore, the cross-linking monomer was added to the solution for electropolymerization at a much higher concentration than that of the functional monomer. The dynamic resistance change resulting from film deposition was relatively small, equaling ∼60 Ω, thus indicating a very low visco-elasticity change of the polymer film and that way showing that the films were rigid.
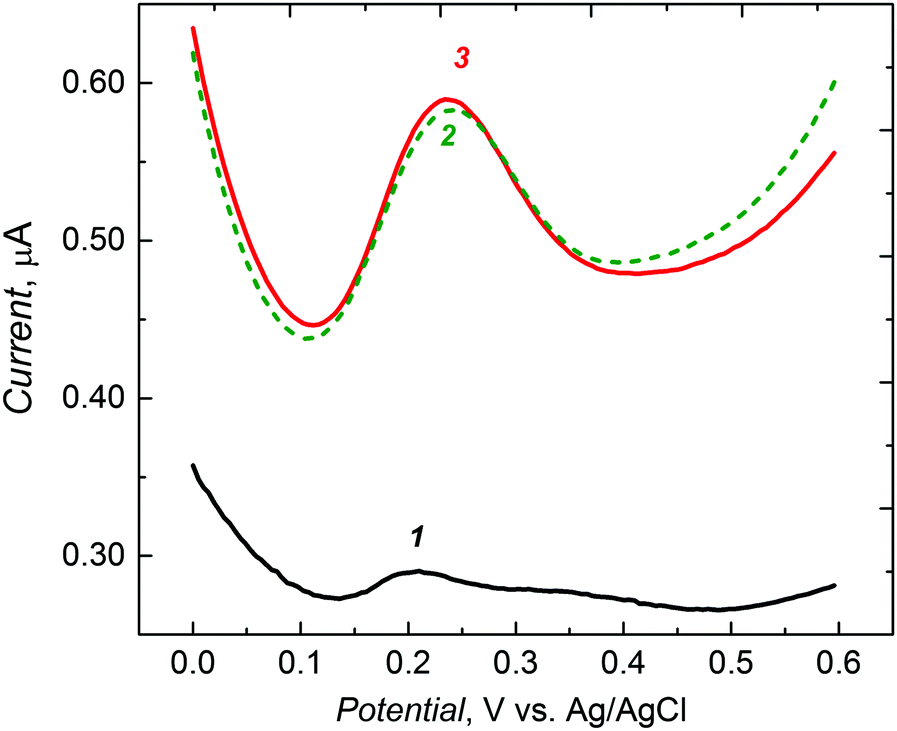 | ||
| Fig. 3 Differential pulse voltammograms for 10 mM K4Fe(CN)6 in 0.1 M KCl for a 1 mm diameter Pt disk electrode coated with the L-arabitol templated non-porous MIP film (1) before and after L-arabitol extraction with 0.1 M HCl for (2) 1 h and (3) 1.5 h. | ||
The SEM imaging of MIP films allowed deeper insights into film morphology of the macroporous film structure (Fig. 2). Polythiophene deposited by electropolymerization does not form a typical inverse opal-like crystal structure (Scheme 1) but, instead, a structure composed of hollow spheres43 (Fig. 2c). Nevertheless, the 3-D structure of the macroporous MIP film obtained in this way was mechanically stable. The diameter of each pore in this structure was the same and close to 330 nm. This corresponds to the diameter of the employed silica nanobeads. Moreover, most of the pores were interconnected, with small channels, with neighboring pores. Unfortunately, some pores of the outer polymer layer were closed (Fig. 2c). However, the pore closing film was relatively thin. Therefore, it only negligibly hindered permeation of the target analyte into the pores. Moreover, the specific surface area of the film composed of hollow spheres was higher than that of a typical inverse opal-like structure because of the presence of additional outer sides of the pore walls exposed to the solution. Therefore, introduction of macroporosity increased significantly the specific surface area of the polymer.
From the PM experiment, the density of the macroporous and non-porous MIP film was determined. The thickness of the non-porous MIP film was 200 nm, whereas that of the macroporous film was ∼700 nm. The mass of both MIPs (non-porous and macroporous) was the same (3.47 μg). The calculated density (mass/volume) of the non-porous film was ∼890 kg m−3, whereas the apparent density of the macroporous MIP film was ∼3.5 times lower (∼247 kg m−3).
When injected to the PM-FIA carrier liquid, the sample solution of L-arabitol flows over the (L-arabitol)-extracted MIP thin film, and then L-arabitol is temporarily captured by this film. This L-arabitol binding increases the mass of the MIP film and, hence, decreases the resonant frequency of the Au-QCR (curve 1 in Fig. 4a). When the concentration of the L-arabitol in the carrier liquid decreases, it is eluted from the film with excess of the liquid and the resonant frequency increases back to its original baseline level. This reversible change in resonant frequency during L-arabitol binding and releasing confirms that the MIP film does not suffer from any detectable memory effects. Moreover, a series of injections for each calibration curve were repeated several times and the first sample of the following series was injected just after the last injection of the preceding one. Under these conditions, we also did not observe any memory effect.
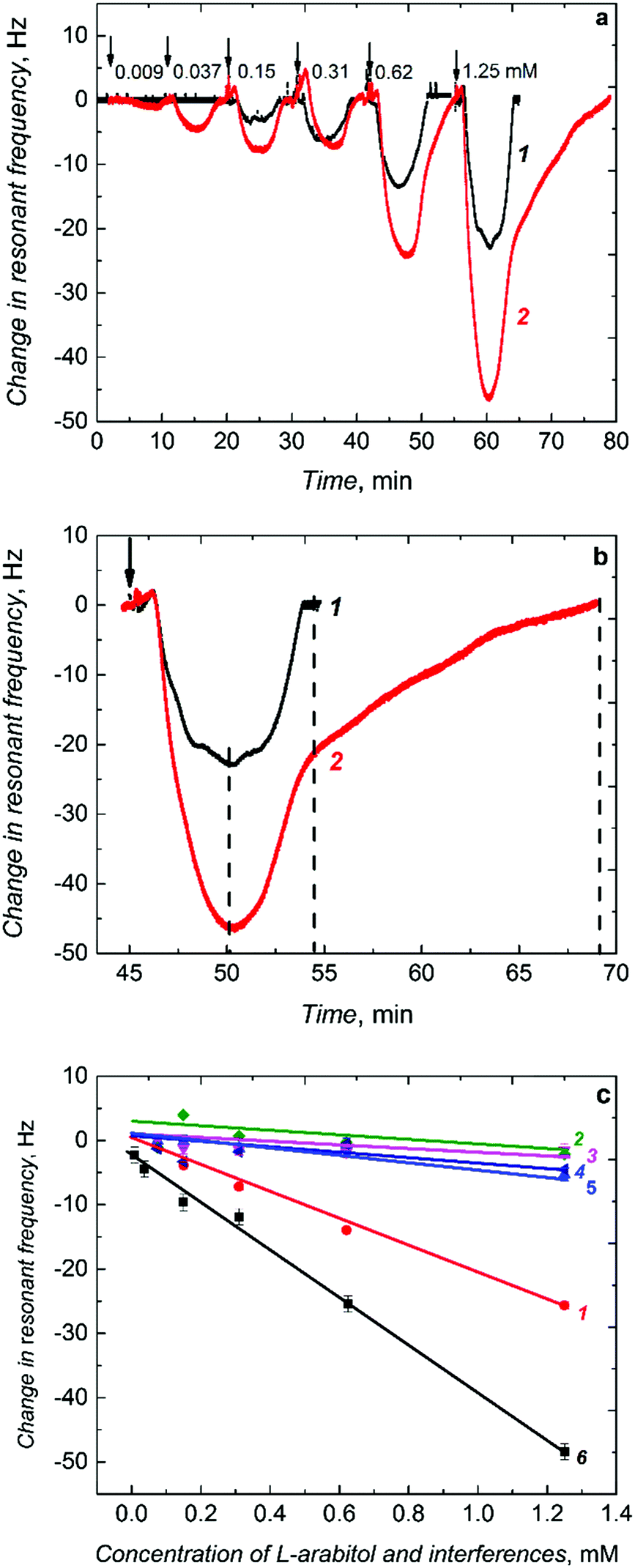 | ||
| Fig. 4 (a) Time dependence of the resonant frequency change due to L-arabitol sample injection under FIA conditions for a piezoelectric microgravimetry chemosensor with the (1) non-porous and (2) macroporous (L-arabitol)-templated MIP film after L-arabitol extraction. (b) An expanded view of the 1.25 mM L-arabitol injection over the (1) non-porous and (2) macroporous MIP film. (c) Calibration plots for the L-arabitol analyte (1 and 6) and interferences (2 – D-arabitol, 3 – xylitol, and 4 – ribitol) for piezoelectric microgravimetry chemosensors with the non-porous MIP film (1, 2, 3, 4), the macroporous MIP film (6), and the non-porous NIP film (5). | ||
The non-porous MIP film chemosensor determined L-arabitol in the concentration range of 0.15 to 1.25 mM (curve 1 in Fig. 4a). Interestingly, when the non-porous MIP film coated Au-QCR was replaced with a macroporous MIP film coated Au-QCR in the FIA system, the chemosensor performance was enhanced appreciably (curve 2 in Fig. 4a). A 100% sensitivity was gained for the macroporous MIP film compared to that of the non-porous MIP film (Table 1). This gain enabled the chemosensor to determine L-arabitol at a concentration as low as 9 μM, i.e., ∼16 times lower than that determined with the non-porous MIP film (0.15 mM). Apparently, this higher detectability was due to macroporosity of the MIP film with interconnecting channels, prepared using silica colloidal crystals, similarly to what has been observed for other sensors based on porous metals.47,48
| Chemosensor recognition unit | Analytical parameter | Value ± st. dev. |
|---|---|---|
| Non-porous (L-arabitol)-MIP | Sensitivity to L-arabitol analyte, Hz mM−1 | −19.78 ± 0.53 |
| Linear dynamic concentration range, mM | 0.150–1.250 | |
| Limit of quantitation, mM | 0.150 | |
| Sensitivity to interferences, Hz mM−1 | ||
| D-Arabitol | −0.88 ± 0.82 | |
| Xylitol | −1.42 ± 0.97 | |
| Ribitol | −1.76 ± 1.63 | |
| Macroporous (L-arabitol)-MIP film | Sensitivity to L-arabitol analyte, Hz mM−1 | −36.51 ± 1.27 |
| Linear dynamic concentration range, mM | 0.009–1.250 | |
| Limit of quatitation, μM | 9 | |
| Non-porous NIP film | Sensitivity to L-arabitol, Hz mM−1 | −4.21 ± 0.99 |
The film macroporosity facilitated binding of L-arabitol molecules to MIP molecular cavities by enhancing their diffusion deeper into the film. Interestingly, the non-porous and macroporous film reached binding saturation at a similar time (Fig. 4b). However, higher and stable binding of L-arabitol was not only evident by studying the time dependence of the association, but also by following the dissociation with time. Approximately twice as much time was necessary to extract the analyte completely (dissociation of the covalent bond formed between L-arabitol and boronic acid) in comparison to the non-porous MIP film (Fig. 4b). The LOD for the macroporous chemosensor was sufficient to determine L-arabitol at concentrations typical for blood plasma, i.e., in the micromolar range. Table 1 summarizes the comparative analytical parameters of these two MIP chemosensors.
The presence of selective molecular cavities imprinted in the polymer was confirmed by determining the imprinting factor. This factor is defined by the ratio of the sensitivity of the MIP chemosensor (−19.78 Hz mM−1) (Table 1) to that of the NIP chemosensor (−4.21 Hz mM−1) (Table 1). This ratio was as high as ∼4.5. Such a high value indicates a homogenous recognition site distribution in the MIP matrix and confirms suitability of the functional monomer. Moreover, selectivity of the chemosensor for L-arabitol was determined as the ratio of sensitivity to the L-arabitol analyte and to potentially interfering compounds including D-arabitol, xylitol, and ribitol (Fig. 4c and Table 1). Advantageously, the chemosensor did not respond to xylitol and ribitol, close structural analogs of L-arabitol. Moreover, it was completely insensitive to D-arabitol, an enantiomer of opposite chirality.
Conclusions
A relatively simple and inexpensive procedure was developed for the fabrication of an MIP chemosensor for the selective determination of L-arabitol. The chemosensor revealed very significant enantio- and stereoselectivity with respect to other sugar alcohols. Colloidal crystal templating for increasing the specific surface area of the MIP film substantially improved the detectability to reach concentrations comparable to those typical found for L-arabitol in blood plasma. This procedure of L-arabitol determination is an attractive alternative for the currently used clinical determination procedures. We anticipate that the herein developed protocol of molecular imprinting has the potential of use in practical clinical applications for the determination of the D- to L-arabitol ratio in body fluids and, therefore, opens the door to early diagnosis of fungal infections. Furthermore, this protocol can easily be adapted for other types of analytes and, therefore, might help to improve the performance of several other electrochemical and piezomicrogravimetric chemosensors.Acknowledgements
The present research was funded by the Polish National Science Centre (NCN) through Grant No. 2011/N/ST4/03491 to M. D. Moreover, the authors acknowledge NCN (Grant No. 2011/03/D/ST4/02596 to P. S. S.), the European Regional Development Fund (Grant No. POIG.01.01.02-00-008/08 to P. S. S., K. N. and W. K.), and the European 7th Framework Programme (Grant No. FP7-REGPOT-CT-2011-285949-NOBLESSE to W. K.) for financial support.References
- P. T. Kissinger, Biosens. Bioelectron., 2005, 20, 2512–2516 CrossRef CAS PubMed.
- P. D'Orazio, Clin. Chim. Acta, 2003, 334, 41–69 CrossRef.
- Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848–2866 RSC.
- L. E. Santos-Figueroa, M. E. Moragues, E. Climent, A. Agostini, R. Martınez-Manez and F. Sancenon, Chem. Soc. Rev., 2013, 42, 3489–3613 RSC.
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS PubMed.
- J. F. Teichert, D. Mazunin and J. W. Bode, J. Am. Chem. Soc., 2013, 135, 11314–11321 CrossRef CAS PubMed.
- F. L. Dickert, P. Lieberzeit, S. G. Miarecka, K. J. Mann, O. Hayden and C. Palfinger, Biosens. Bioelectron., 2004, 20, 1040–1044 CrossRef CAS PubMed.
- J. Wackerlig and R. Schirhagl, Anal. Chem., 2016, 88, 250–261 CrossRef CAS PubMed.
- A. Speltini, A. Scalabrini, F. Maraschi, M. Sturini and A. Profumo, Anal. Chim. Acta, 2017, 974, 1–26 CrossRef CAS PubMed.
- E. L. Holthoff and F. V. Bright, Anal. Chim. Acta, 2007, 594, 147–161 CrossRef CAS PubMed.
- P. S. Sharma, M. Dabrowski, F. D'Souza and W. Kutner, TrAC, Trends Anal. Chem., 2013, 51, 146–157 CrossRef CAS.
- L. Li, L. Yang, Z. Xing, X. Lu and X. Kan, Analyst, 2013, 138, 6962–6968 RSC.
- R. B. Pernites, S. K. Venkata, B. D. B. Tiu, A. C. C. Yago and a. R. C. Advincula, Small, 2012, 8, 1669–1674 CrossRef CAS PubMed.
- B. D. B. Tiu, S. B. Tiu, A. M. Wen, P. Lam, N. F. Steinmetz and R. C. Advincula, Langmuir, 2016, 32, 6185–6193 CrossRef CAS PubMed.
- B. D. B. Tiu, R. B. Pernites, S. B. Tiu and R. C. Advincula, Colloids Surf., A, 2016, 495, 149–158 CrossRef CAS.
- J. C. Yang and J. Y. Park, ACS Appl. Mater. Interfaces, 2016, 8, 7381–7389 CAS.
- S. Reculusa, M. Heim, F. Gao, N. Mano, S. Ravaine and A. Kuhn, Adv. Funct. Mater., 2011, 21, 691–698 CrossRef CAS.
- R. I. Boysen, L. J. Schwarz, D. V. Nicolau and M. T. W. Hearn, J. Sep. Sci., 2017, 40, 314–335 CrossRef CAS PubMed.
- B. P. Guery, M. C. Arendrup, G. Auzinger, E. Azoulay, M. B. Sa, E. M. Johnson, E. Muller, C. Putensen, C. Rotstein, G. Sganga, M. Venditti, R. Z. Crespo and B. J. Kullberg, Intensive Care Med., 2009, 35, 55–62 CrossRef PubMed.
- M. A. Johnson and D. R. Bundle, Chem. Soc. Rev., 2013, 42, 4327–4344 RSC.
- L. Alcazar-Fuoli and E. Mellado, Br. J. Haematol., 2014, 166, 471–484 CrossRef PubMed.
- B. P. Guery, M. C. Arendrup, G. Auzinger, E. Azoulay, M. B. Sa, E. M. Johnson, E. Muller, C. Putensen, C. Rotstein, G. Sganga, M. Venditti, R. Z. Crespo and B. J. Kullberg, Intensive Care Med., 2009, 35, 206–214 CrossRef CAS PubMed.
- L. Lehtonen, V.-J. Anttila, T. Ruutu, J. Salonen, J. Nikoskelainen, E. Eerola and P. Ruutu, J. Clin. Microbiol., 1996, 2175–2179 CAS.
- G. Sigmundsdottir, B. Christensson, L. J. Bjorklund, K. Hakansson, C. Pehrson and L. Larsson, J. Clin. Microbiol., 2000, 3039–3042 CAS.
- J. H. Huck, N. M. Verhoeven, E. A. Struys, G. S. Salomons, C. Jakobs and M. S. van der Knaap, Am. J. Hum. Genet., 2004, 74, 745–751 CrossRef CAS PubMed.
- M. C. Arendrup, O. J. Bergmann, L. Larsson, H. V. Nielsen, J. O. Jarløv and B. Christensson, Clin. Microbiol. Infect., 2010, 16, 855 CrossRef CAS PubMed.
- L. Larsson, C. Pehrson, T. Wiebe and B. Christensson, J. Clin. Microbiol., 1994, 32, 1855–1859 CAS.
- M. Dabrowski, P. S. Sharma, Z. Iskierko, K. Noworyta, M. Cieplak, W. Lisowski, S. Oborska, A. Kuhn and W. Kutner, Biosens. Bioelectron., 2016, 79, 627–635 CrossRef CAS PubMed.
- R.-J. Bensadoun, L. L. Patton, R. V. Lalla and J. B. Epstein, Support Care Cancer, 2011, 19, 737–744 CrossRef PubMed.
- T. Kiehn, E. Bernard, J. Gold and D. Armstrong, Science, 1979, 206, 577–580 CAS.
- L. Stobinski, L. Zommer, I. Flis-Kabulska, M. Mecina, B. Gniewinska, W. Szulc and W. Kutner, J. Electrochem. Soc., 2000, 147, 4203–4205 CrossRef CAS.
- S. Reculusa, C. Poncet-Legrand, S. Ravaine, C. Mingotaud, E. Duguet and E. Bourgeat-Lami, Chem. Mater., 2002, 14, 2354–2359 CrossRef CAS.
- S. Reculusa and S. Ravaine, Appl. Surf. Sci., 2005, 246, 409–414 CrossRef CAS.
- S. Reculusa and S. Ravaine, Chem. Mater., 2003, 15, 598–605 CrossRef CAS.
- S. Reculusa, P. Massé and S. Ravaine, J. Colloid Interface Sci., 2004, 279, 471–478 CrossRef CAS PubMed.
- B. v. Duffel, R. H. A. Ras, F. C. D. Schryver and R. A. Schoonheydt, J. Mater. Chem., 2001, 11, 3333–3336 RSC.
- M. Heim, S. Reculusa, S. Ravaine and A. Kuhn, Adv. Funct. Mater., 2012, 22, 538–545 CrossRef CAS.
- A. Kochman, A. Krupka, J. Grissbach, W. Kutner, B. Gniewinska and L. Nafalski, Electroanalysis, 2006, 18, 2168–2173 CrossRef CAS.
- K. Karim, F. Breton, R. Rouillon, E. V. Piletska, A. Guerreiro, I. Chianella and S. A. Piletsky, Adv. Drug Delivery Rev., 2005, 57, 1795–1808 CrossRef CAS PubMed.
- X. Wu, Z. Li, X.-X. Chen, J. S. Fossey, T. D. James and Y.-B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 RSC.
- P. N. Bartlett, P. R. Birkina and M. A. Ghanem, Chem. Commun., 2000, 1671–1672 RSC.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- P. N. Bartlett, P. R. Birkin, M. A. Ghanema and C.-S. Toh, J. Mater. Chem., 2001, 11, 849–853 RSC.
- A. Pietrzyk, W. Kutner, R. Chitta, M. E. Zandler, F. D'Souza, F. Sannicolo and P. R. Mussini, Anal. Chem., 2009, 81, 10061–10070 CrossRef CAS PubMed.
- A. Pietrzyk, S. Suriyanarayanan, W. Kutner, R. Chitta, M. E. Zandler and F. D'Souza, Biosens. Bioelectron., 2010, 25, 2522–2529 CrossRef CAS PubMed.
- D. A. Buttry and M. D. Ward, Chem. Rev., 1992, 92, 1355–1379 CrossRef CAS.
- T. Yutthalekha, C. Wattanakit, V. Lapeyre, S. Nokbin, C. Warakulwit, J. Limtrakul and A. Kuhn, Nat. Commun., 2016, 7, 12678 CrossRef CAS PubMed.
- C. Wattanakit, Y. B. S. Come, V. Lapeyre, P. A. Bopp, M. Heim, S. Yadnum, S. Nokbin, C. Warakulwit, J. Limtrakul and A. Kuhn, Nat. Commun., 2014, 5, 3325 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |