
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Prevention of encrustation and blockage of urinary catheters by Proteus mirabilis via pH-triggered release of bacteriophage†
Scarlet
Milo
a,
Hollie
Hathaway
a,
Jonathan
Nzakizwanayo
b,
Diana R.
Alves
bc,
Patricia Pérez
Esteban
d,
Brian V.
Jones
be and
A. Toby A.
Jenkins
 *a
*a
aDepartment of Chemistry, University of Bath, BA2 7AY, UK. E-mail: a.t.a.jenkins@bath.ac.uk; Tel: +44 (0) 1225 386118
bSchool of Pharmacy and Biomolecular Sciences, University of Brighton, Brighton, East Sussex BN2 4GJ, UK
cThe Blond McIndoe Research Foundation Ltd, Queen Victoria Hospital, East Grinstead, West Sussex RH19 3DZ, UK
dDepartment of Biochemical Engineering, UCL, Bernard Katz Building, Gordon Street, London, WC1H 0AH, UK
eQueen Victoria Hospital NHS Foundation Trust, East Grinstead, RH19 3DZ, UK
First published on 22nd June 2017
Abstract
The crystalline biofilms of Proteus mirabilis can seriously complicate the care of patients undergoing long-term indwelling urinary catheterisation. Expression of bacterial urease causes a significant increase in urinary pH, leading to the supersaturation and precipitation of struvite and apatite crystals. These crystals become lodged within the biofilm, resulting in the blockage of urine flow through the catheter. Here, we describe an infection-responsive surface coating for urinary catheters, which releases a therapeutic dose of bacteriophage in response to elevated urinary pH, in order to delay catheter blockage. The coating employs a dual-layered system comprising of a lower hydrogel ‘reservoir’ layer impregnated with bacteriophage, capped by a ‘trigger’ layer of the pH-responsive polymer poly(methyl methacrylate-co-methacrylic acid) (EUDRAGIT®S 100). Evaluation of prototype coatings using a clinically reflective in vitro bladder model system showed that catheter blockage time was doubled (13 h to 26 h (P < 0.05)) under conditions of established infection (108 CFU ml−1) in response to a ‘burst-release’ of bacteriophage (108 PFU ml−1). Coatings were stable both in the absence of infection, and in the presence of urease-negative bacteria. Quantitative and visual analysis of crystalline biofilm reduction show that bacteriophage constitute a promising strategy for the prevention of catheter blockage, a clinical problem for which there is currently no effective control method.
Introduction
Catheter-associated urinary tract infection (CAUTI) is the most common healthcare-associated infection worldwide, accounting for approximately 150–250 million cases globally per year,1 manifesting as an estimated cost of £125 million per year in the UK alone.2 The most severe CAUTI sequelae occur as a result of infection by the urease-producing motile bacteria Proteus mirabilis (P. mirabilis), which colonise the catheter surface, forming extensive biofilm communities embedded within an exopolymeric matrix. P. mirabilis expresses a potent bacterial urease enzyme, which generates ammonia as a by-product of urea hydrolysis, thus elevating the pH of the urine and surrounding biofilm. Under these conditions, local supersaturation and precipitation of struvite [MgNH4PO4·6H2O] and apatite [Ca10(PO4)6CO3] causes accumulation of crystalline aggregates, which become embedded within the organic matrix surrounding the cells on the luminal surfaces. The continued development of such biofilms and accretion of crystalline material leads to the eventual obstruction of the flow of urine through the catheter. Incontinence can develop owing to urine leakage around the catheter, and reflux of infected urine to the kidneys may result in serious symptomatic episodes such as pyelonephritis, endotoxic shock and septicaemia.3–5A range of approaches have been evaluated to prevent bacterial attachment and subsequent catheter blockage, including antiseptic-coated silver alloy6 and antimicrobial impregnated minocycline and rifampin7 approaches. However, all preventative measures thus far have proved disappointing in clinical use,8–10 and all available catheter types remain susceptible to infection by P. mirabilis.11,12 Other attempted approaches include impregnation with nitrofurazone,13 electrical currents,14 release of nitric oxide,15–17 and modulation of urinary pH to prevent precipitation of inorganic salts.4 A recent randomised controlled clinical trial evaluating nitrofurazone-coated catheters alongside silver-coated and plain catheters concluded that neither of these commercially available catheters showed a clinically significant reduction of infection.18,19 Indeed, international guidelines now state that the evidence is insufficient to support their use in both short term (<30 days) and long term (>30 days) indwelling catheterisation, and nitrofurazone-coated catheters are no longer available on the market.8
CAUTI currently presents a worldwide therapeutic problem as, owing to their prevalence, uropathogenic infection has become a major contributor to global antibiotic use and resistance. Infections caused by P. mirabilis are notoriously difficult to eliminate once established in the catheterised urinary tract, and often respond poorly to conventional antibiotic therapy.1 Furthermore, infections as a result of P. mirabilis have been found to persist despite multiple catheter changes, and periods without catheterisation. A striking study by Sabbuba et al. showed that the same strain of P. mirabilis colonised one patient for 121 days despite 8 catheter changes, an 8 day course of antibiotics, and a 20 day period during which the urinary tract remained uncatheterised.20
As medical procedures become ever more reliant upon implanted devices for diagnosis and treatment of infectious disease, further emphasis is placed on safeguarding antibiotic therapy as the panacea of modern medicine. However, recent research has begun to focus on alternative forms of antimicrobials in the battle against bacterial pathogens. One such treatment utilises bacteriophage (phage), the natural predatory viruses of bacteria that selectively infect, and in the case of lytic phage, hijack the host's biosynthetic machinery in favour of viral replication, causing the bacterium to lyse rapidly.21 Despite pre-dating the discovery of chemical antibiotics by several decades,22,23 phage therapy was largely supplanted by its chemical successor. Today, interest in phage therapy is undergoing a revival as the antibiotic paradigm shift occurs throughout western medicine and several advantages over conventional therapeutics have been recognised, including high strain specificity (therefore unlikely to disturb normal flora),24,25 self-replication (facilitating low-dosage treatment),26 and easy manipulation of the phage genome (providing the possibility of ‘designer’ engineered phage to treat problematic infections27). Although phage resistance has been reported, largely owing to changes in phage-receptor molecules,28,29 this may be offset via the use of phage ‘cocktails’, where multiple strains of phage are directed against the target species.21,30–32
One of the major challenges in determining the effect of a therapeutic agent comes in achieving adequate local delivery to the site of infection. Often, controlled drug delivery systems rely on sustained passive delivery, although this often results in exposure of bacterial pathogens to sub lethal doses of chemical antibiotics, thus potentially contributing to the continued development of multidrug resistant species.33,34 Recently, emphasis has been placed on developing methods of active, or triggered release of antimicrobial agents in response to external stimuli (e.g. pH,35–37 temperature,38,39 or biomarker signals40), in order to achieve a ‘burst response’ of the active cargo. Triggered release formulations can be used to reduce the amount of drug necessary to cause the same therapeutic effect in patients, as well as ensuring that the cargo is released only in the physiological location required, thus reducing systemic dosage and its associated complications.41
Previous studies investigating the ability of phage to reduce P. mirabilis biofilm biomass, either by entropic confinement of phage within a hydrogel, or simple administration of phage within an aqueous suspension has resulted in significant reduction P. mirabilis populations.21,30,31,42 However, despite the successful outcomes of these investigations, the issue of formulating a working triggered release system has not been addressed.
We have previously reported a novel infection-responsive surface coating that utilises urinary pH elevation to provide a visual warning of catheter blockage by P. mirabilis biofilms. The dual-layered polymeric coating consisted of a lower hydrogel ‘reservoir’ layer (poly(vinyl alcohol)), employed to encapsulate the self-quenching dye 5(6)-carboxyfluorescein. This was capped and sealed by an upper layer of the pH-sensitive polymer EUDRAGIT®S 100 (an anionic co-polymer of methacrylic acid and methyl methacrylate). Elevation of urinary pH facilitated the swelling of the upper EUDRAGIT®S 100 layer, resulting in the release and consequent fluorescence switch on of the carboxyfluorescein contained in the lower hydrogel matrix. The subsequent visual colour-change of the urine provided a clear visual signal throughout the closed drainage system of imminent catheter blockage.43
The aim of this study was to evaluate the potential for triggered phage delivery from an infection-responsive coating, in response to an increase in urinary pH by P. mirabilis. Though there is potential for this technology to be translated to deliver a variety of antimicrobial agents, the use of lytic bacteriophage was employed in this work in order to evaluate the efficacy of a biological antimicrobial in inhibiting catheter blockage. The potential for bacteriophage to constitute an effective countermeasure for such a severe consequence of long-term indwelling catheterisation has a wide-reaching impact, not only on improvement of patient health and welfare, but also in extending catheter life, thus reducing associated healthcare costs. Prototype coatings were evaluated using a clinically relevant in vitro bladder model system, in which the sterile closed drainage system was replicated and the catheterised urinary tract accurately represented.
Materials and methods
Materials
Triethyl citrate, talc, Luria-Bertani (LB) broth, Tryptic Soy Broth (TSB), Tryptic Soy Agar, (TSA), yeast extract, vegetable peptone number 1, tryptone, sodium chloride, magnesium sulphate, Tris–hydrochloride (pH 7.5), gelatine, acetone, isopropanol, sodium hydroxide, anhydrous sodium sulphate, magnesium chloride hexahydrate, tri-sodium citrate, sodium oxalate, potassium di-hydrogen orthophosphate (potassium phosphate monobasic), potassium chloride, ammonium chloride, calcium chloride, urea, 5(6)-carboxyfluorescein, poly(vinyl alcohol) (MW 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600–18600 g mol−1), and agarose were all purchased from Sigma-Aldrich (Poole, Dorset, UK). Bacteriological agar was obtained from Oxoid, (Basingstoke, Hampshire, UK).
600–18600 g mol−1), and agarose were all purchased from Sigma-Aldrich (Poole, Dorset, UK). Bacteriological agar was obtained from Oxoid, (Basingstoke, Hampshire, UK).
Eudragit S100 was kindly donated by Evonik Industries, Darmstadt, Germany. Uncoated all-silicone Foley catheters were obtained from Bard (Crawley, West Sussex, UK).
Material preparation
:1 mixture of ammonia (33% v/v) and hydrogen peroxide (30% v/v) for 10 minutes with constant shaking, then rinsed with sterile deionised water and dried under nitrogen. Catheters were then placed in (3-aminopropyl triethoxysilane) (APTES) (1% v/v) in N,N-dimethylformamide (DMF) for 16 h. Surface modified catheters were rinsed with DMF, followed by sterile deionised water and dried under nitrogen. Water contact angle measurements were made to ensure the hydrophilicity of the catheter surface.
600–18600 gmol−1, 20% w/v) was dissolved in deionised water and heated to 97 °C with constant stirring to facilitate dissolution.
:2, and an average molecular weight of 150000 gmol−1 was used (Evonik Industries, Germany). The organic dip-coating solution was prepared according to the technical information44 as described previously.43 The solution was stored at room temperature until required.
Coating of Foley catheters
To the cooled PVA solution (20 °C) was added high titre phage lysate (1010 plaque-forming units per ml (PFU ml−1)) at a 1:1 ratio, to form a final PVA concentration of 10% w/v. Catheters were coated with the gel/phage solution (100 μl) between the retention balloon and the tip, and stored overnight at −20 °C to promote cryogenic gelation. Catheters were thawed at room temperature (2 hours) before coating with the pH-sensitive trigger layer. Catheters were manually dip-coated 20 times in the EUDRAGITsS 100 solution, with a 5 minute solvent evaporation period between each coating, to achieve a final coating thickness of approximately 500 μm. Coated catheters were stored at 4 °C until required.
Microbiology
Analysis
Results and discussion
In this study, the potential for bacteriophage as an effective countermeasure for encrustation and blockage of urinary catheters was investigated. Previous work developed an infection-responsive coating for diagnosis of CAUTI, showing that molecules of the self-quenching dye 5(6)-carboxyfluorescein can be successfully incorporated into a hydrogel coating and released in response to elevated urinary pH caused by P. mirabilis infection.43In vitro infection models for evaluation of prototype coatings
To evaluate the ability of the dual-layered polymeric coating to delay catheter blockage, performance was assessed using the in vitro bladder model system (Fig. S1, ESI†), originally described by Stickler et al.48 This model provides an accurate physiological representation of the catheterised urinary tract, mimicking the full closed drainage system currently used in clinical practice. In comparison, previous studies have utilised simple static models of biofilm formation on catheter sections,21 flow models that use only the central catheter lumen,31,42 or those that do not accurately represent the full closed drainage system. Whilst these studies have provided fundamental information about the general properties of biofilm formation and the success of various treatment options, the in vitro bladder models used in this study assess biofilm formation and catheter blockage under conditions in which the design features of the catheter and the hydrodynamics of the catheterised bladder are taken into consideration.Experiments assessing the stability and efficacy of prototype coatings were carried out using out an initial P. mirabilis inoculum of 108 CFU ml−1. This was a simulation of an established infection, therefore testing the coatings under conservative ‘worst case scenario’ conditions. Furthermore, bladder model inoculums were left to establish for 1 hour before urinary flow was restored, meaning that coatings were tested under established biofilm conditions rapidly after model start. Although this resulted in a delay of catheter blockage, rather than a complete prevention (which has been observed previously via use of a lower starting inoculum),30 it provides a far more accurate representation of the bladder during long-term indwelling catheterisation, particularly after a catheter change, where colonisation by P. mirabilis may already be established within the bladder.
Activation of catheter coating: effect on catheter blockage
In order to evaluate the capacity of the triggered-release bacteriophage coating to delay catheter blockage and reduce biofilm biomass on the luminal surfaces, pH of artificial urine media, bacteriophage release and consequent reduction in viable bacterial population were monitored quantitatively throughout the course of the bladder model experiments. Regular measurements at 0, 2, 4 and 6 hours after model start served to observe changes in the biological and chemical conditions within the bladder models during crystalline biofilm formation, whilst the final measurement was made at the point of blockage of the uncoated control (13 hours after model start), and served to compare the conditions within the bladders containing coated and uncoated catheters at this point.The blockage of the catheters and subsequent stemming of artificial urine flow was defined as the experimental end-point. Parallel experiments evaluating the response of the coating to infection by E. coli were also undertaken, in order to determine the ability of the system to discriminate between urease positive (P. mirabilis), and urease negative (E. coli) species. Since E. coli species are the most commonly encountered pathogens within the urinary tract (including the early stage colonisation of the catheterised urinary tract), differential switch-on of the phage release coating is essential, in order for the burst release of phage to be of sufficient concentration during the later-stage colonisation of P. mirabilis.49 During the experiments, no significant phage release was observed in models devoid of P. mirabilis. Indeed, coatings remained intact to the naked eye in uninoculated models, as well as those infected with the urease negative pathogen. Further controls evaluating the effect of the polymeric coating (not containing phage) were also performed in parallel. No significant effect on urinary pH, bacterial population or time to blockage was observed in the presence of the dual-layered coating in comparison to uncoated catheters. Graphical representation of the change in urinary pH, CFU ml−1 and PFU ml−1 including full experimental controls can be observed in the ESI† (Fig. S2–S4).
Inoculation with P. mirabilis and subsequent urease production caused a substantial increase in pH within two hours of the model start, owing to the expression of urease from P. mirabilis and production of ammonia within the urine. Consequent formation of the carboxylate anion results in swelling of the EUDRAGIT® network, thereby exposing the PVA reservoir layer to the artificial urine media, resulting in release of bacteriophage from the coating via diffusion (Fig. 1).
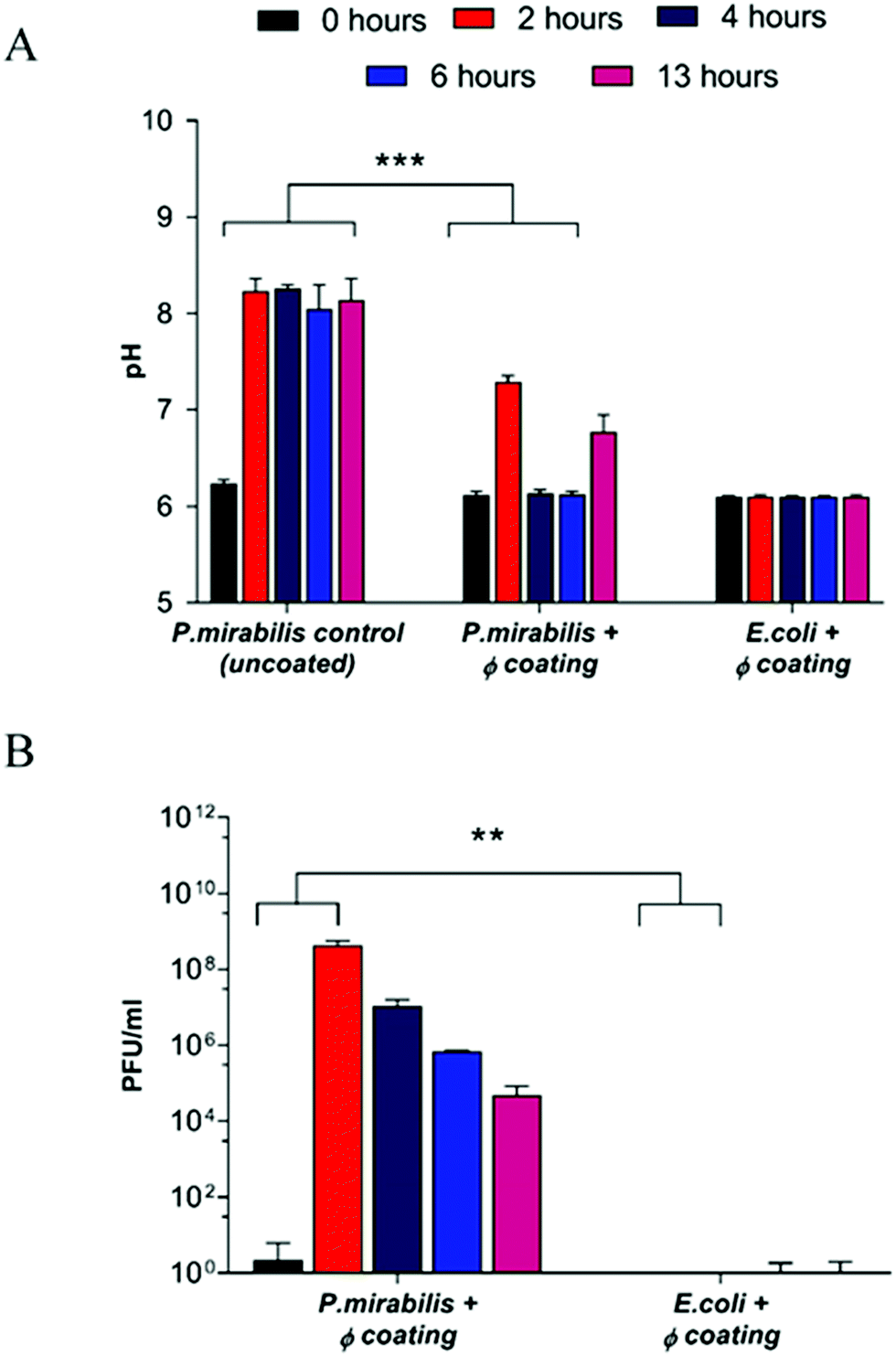 | ||
| Fig. 1 Analysis of in vitro bladder model conditions at regular intervals after model start (0, 2, 4, 6 hours), and at the blockage time of the uncoated control (13 hours). (A) Measured pH of residual bladder model urine. ***P < 0.001. (B) Release of bacteriophage (φ) from the catheter coating. **P < 0.005. Data shown is the mean of triplicate repeats. Error bars represent standard error of the mean (SEM). | ||
Since the threshold pH for EUDRAGIT® swelling occurs at pH 7,44 elevation of urinary pH above this value results in a burst response of phage from the coating (P = 0.0008). The concentration of phage released after 2 hours (4.3 × 108 PFU ml−1) (P = 0.0048) is sufficient to cause bacterial cell death according to previous experiments investigating efficacy of infection and replication of phage against P. mirabilis (data not shown), and corresponded to a urinary pH change of 2 pH units, from the ‘healthy’ 6.24 to the infected 8.24 after 2 hours. The elevated pH was maintained within the uncoated control bladder, eventually resulting in blockage after 13 hours, indicating the speed at which biofilm formation occurs once an infection is established. In contrast, a reduction of urinary pH back into the healthy range was observed post-release in the phage-coated catheter model. Gradual decrease of the phage population within the bladder model, and hence the failure of the coating to prevent catheter blockage altogether is due primarily to the elution of particles from the bladder post-release (as confirmed by titre measurements of residual bag urine upon completion of the experiment). Controls exhibited negligible phage release throughout all measured time points, indicating the stability of the catheter coating for the duration despite exposure to a continuously flowing culture.
Notable also is the stability of the phage within the coating, which remained viable throughout the entire experimental procedure, including encapsulation within the hydrogel reservoir, coating with an organic polymeric mixture, storage at 4 °C, and exposure to the challenging conditions of the bladder (involving both neutral and alkaline pHs, as well as the high ionic strength of the urine media). Phage stability with respect to clinical application is considered to be a barrier to their eventual use within the clinical setting,50 hence the proof of stability within a hydrogel matrix, as well as the maintenance of infectivity in the face of more representative physiological conditions presented in this work signify the successfully maintained viability of the viruses despite the removal of the phage from its natural conditions and exposure to experimental interferents.
As a result of triggered phage release from the catheter coatings, a significant decline in the bladder-dwelling P. mirabilis concentration was observed (Fig. 2) (P = 0.0016). Decline in viable cell numbers can be seen to be proportional with the release profile of phage from coated catheters. In contrast, bladders containing uncoated catheters sustained cell populations in the region of 108 CFU ml−1, leading to eventual blockage of the catheter lumens via the crystalline biofilms of P. mirabilis. Fig. S2 (ESI†) shows that no significant changes in bacterial cell numbers were observed in the presence of the dual-layered coating not containing phage, indicating that any reduction in cell numbers is as a direct result of lysis via phage infection.
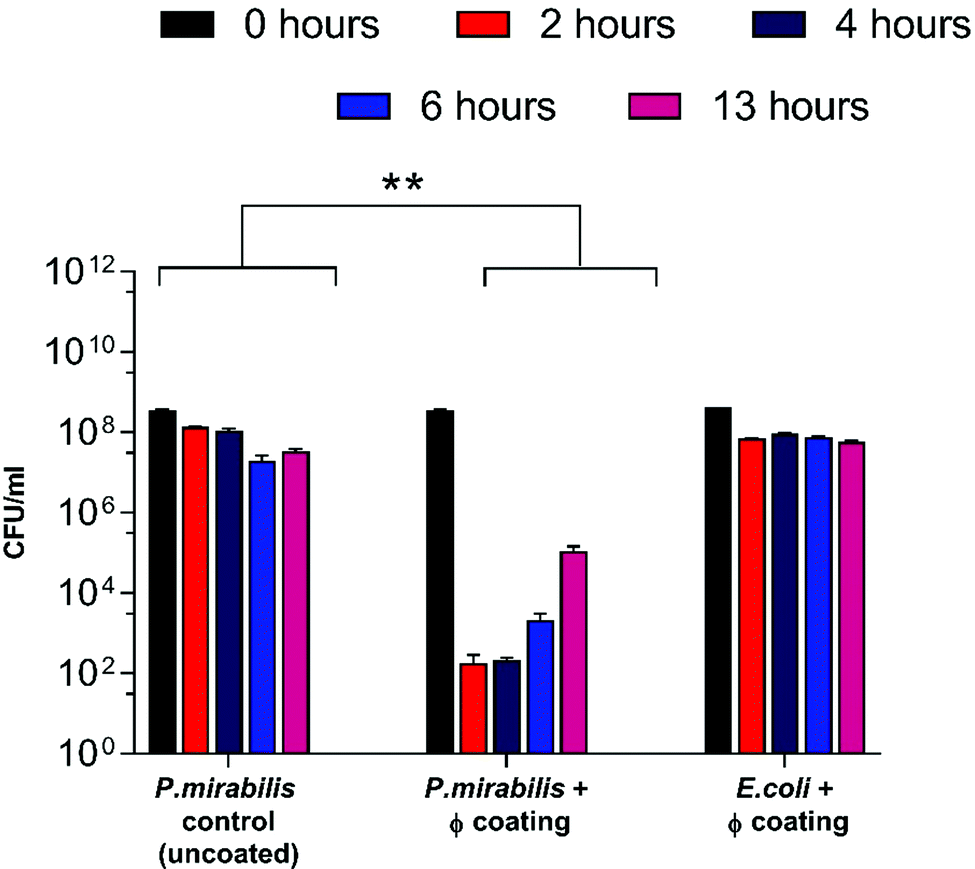 | ||
| Fig. 2 Analysis of P. mirabilis population within in vitro bladder models (containing uncoated and phage (φ) coated catheters) at regular intervals after model start (0, 2, 4, 6 hours), and at the blockage time of the uncoated control (13 hours). **P < 0.005. Data shown is the mean of triplicate repeats. Error bars represent standard error of the mean (SEM). | ||
Nevertheless, despite an approximately 6-log reduction in P. mirabilis concentration within 2 hours of coating activation, triggered phage release was successful only in delaying catheter blockage, not preventing it completely. The gradual increase in CFU ml−1 between 4–13 hours after model start is likely due to the proportional elution of phage form the bladders observed previously. However, it is also possible that the failure to prevent blockage was multifactorial, and that the development of resistance to the phage used also contributed to the eventual crystalline biofilm formation. The emergence of resistance may be associated with a number of different factors which affect the rate of phage adsorption to the bacterial host, including alteration of the structure or exposure of the receptor site, restriction modification, or other mechanisms of abortive infection such as the presence of clustered regularly interspaced short palindromic repeats (CRISPRs) within the bacterial genome.51 Although comparable resistance has been observed in other studies of phage therapy over a similar time frame,52 the fact that this proof-of-concept study utilised a single phage for ease of measurement is worth noting. The issue of resistance to phage may be eased by the use of phage ‘cocktails’, where two or more species of phage are used to target and significantly reduce biofilm formation in a number of applications. Indeed, treatment of uropathogenic isolates with phage cocktails have shown recent success in vitro,21,30,52 although ensuring an effective cocktail of strains requires detailed study into host range profiles, and in-depth understanding of phage-host interaction and binding under conditions encountered within the catheterised urinary tract. Additionally, recent advances in therapy using bacteriophage products, namely bacteriophage-encoded endolysins, which are utilised in end stages of phage infection, and care capable of enzymatically degrading the bacterial cell wall through digestion of the peptidoglycan polymeric material, resulting in cell death by osmolysis.38 Endolysins have been use synergistically with antibiotics to give increased antibiotic activity against otherwise resistant Gram-negative strains.53 Other factors potentially hindering phage efficacy include the inhibition of phage adhesion to their bacterial hosts owing to the accumulation of struvite and apatite crystalline precipitates, or the physiological state of the cells themselves, since log-phase cells are lysed more efficiently than those in a state of partial or full metabolic quiescence (such as those within a biofilm).54 As a consequence of the phage burst response and subsequent reduction in viable cell count of P. mirabilis, time to blockage of the phage coated catheter was doubled in comparison to the uncoated control (Fig. 3) (P = 0.0199).
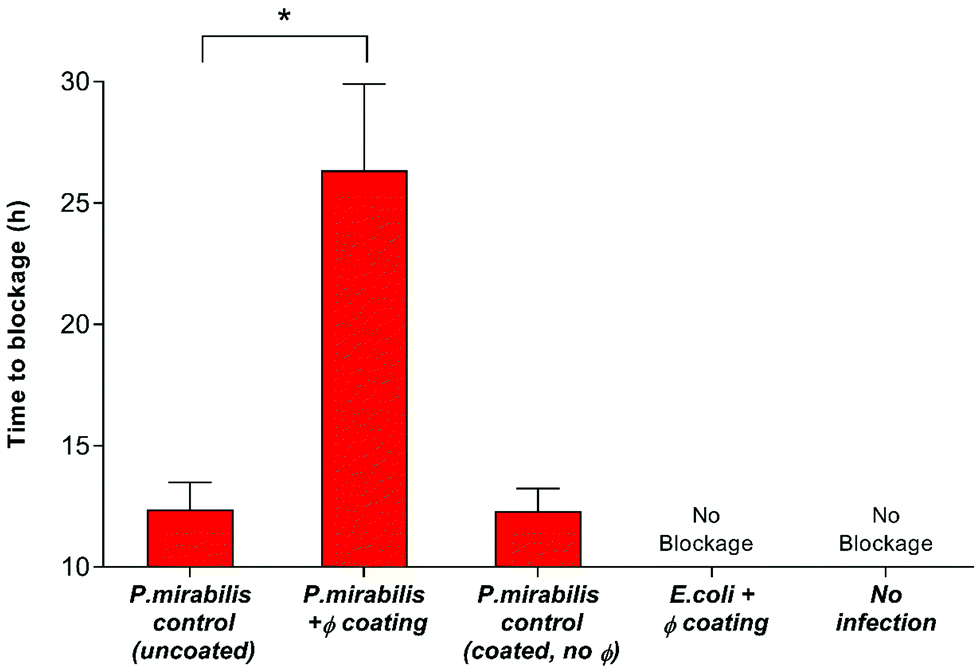 | ||
| Fig. 3 Impact of bacteriophage treatment on catheter blockage. In vitro models of the catheterised urinary tract replicating established P. mirabilis infection were used to evaluate the impact of triggered phage (φ) release on blockage and encrustation. The time at which the catheters became blocked and urine ceased to accumulate in the drainage bags was used as the experimental end point. Data represents the mean of 3 independent replicates. *P < 0.05. Error bars represent standard error of the mean (SEM). | ||
Our findings were in agreement with previous studies which assessed the ability of lytic bacteriophage to reduce catheter biofilms caused by common uropathogenic species including P. mirabilis, E. coli, Pseudomonas aeruginosa and Staphylococcus epidermis.21,30,31,42,52,55–57 Former investigation into the impact of bacteriophage therapy on an early infection model (starting inoculum of 103 CFU ml−1) have shown a delay in catheter blockage of more than 8 days.30 Hence, the blockage delay presented in this work may translate to days, rather than hours, when assessed using early-stage colonisation. Achieving such delay of catheter blockage has the potential to improve the health and welfare of many patients worldwide, as well as relieve some of the substantial demand that this issue places on the resources of the health service. Currently, there are no effective strategies for preventing catheter encrustation and blockage. Prophylactic treatment of bacteriuria with systemic antibiotics is strongly discouraged, owing to the development of multidrug resistance. Indeed, the most effective preventative strategy developed to date in the implementation of a closed drainage system, which was introduced almost a century ago.58 Hence, the successful application of targeted release phage in vitro, which is able to delay catheter blockage whilst both avoiding systemic treatment and the use of chemical antibiotics represents a major step forward in providing an effective solution for infections of this type.
Activation of catheter coating: effect of crystalline biofilm formation
Visual examination of biofilm formation of catheter surfaces showed a marked reduction of biofilm biomass on those coated with phage. Models containing uncoated and coated catheters were halted after 13 hours (blockage time of control) and catheter sections from varying luminal locations were viewed using SEM (Fig. 4).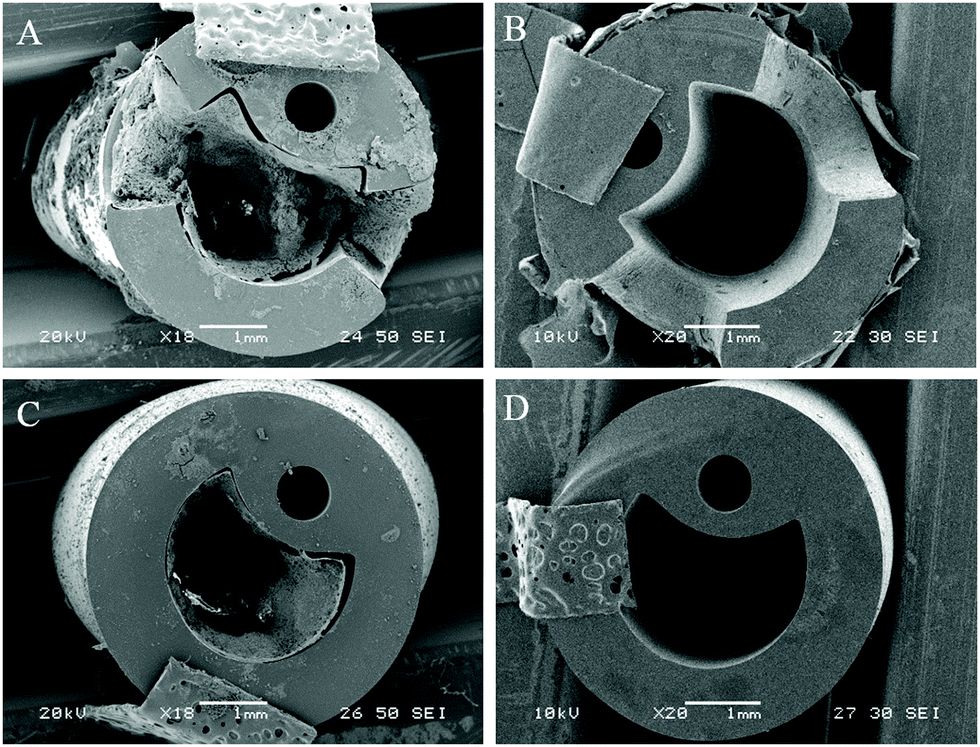 | ||
| Fig. 4 SEM visualisation of catheter cross-sections comparing levels of encrustation and blockage by P. mirabilis. (A and C) Uncoated control. (B and D) With dual-layered bacteriophage coating. All catheters were removed from in vitro bladder models 13 hours after model start. (A and B) Show cross sections of catheter eyeholes. (C and D) Show cross sections of catheter lumen 1 cm below retention balloon. | ||
At the time of blockage of the uncoated control catheters, which showed prominent encrustations, the phage-coated catheters can be seen to be devoid of visible biofilm deposits. The average pH of residual bladder urine at this time point was 6.77, which was evidently insufficient to cause precipitation of struvite and apatite salts within the urine.
Quantification of crystalline biofilm formation at this time point was performed using AAS (Fig. 5). The presence of low levels of calcium apatite in the biofilms of the phage-treated bladder was confirmed using AAS, despite it being invisible in the visual SEM results. This is likely due to the distinct stages of P. mirabilis forming its complex crystalline biofilms as recently described,59 which suggest that accumulation of diffuse crystalline material accumulates within the bladder before defined crystals become embedded within the biofilm.
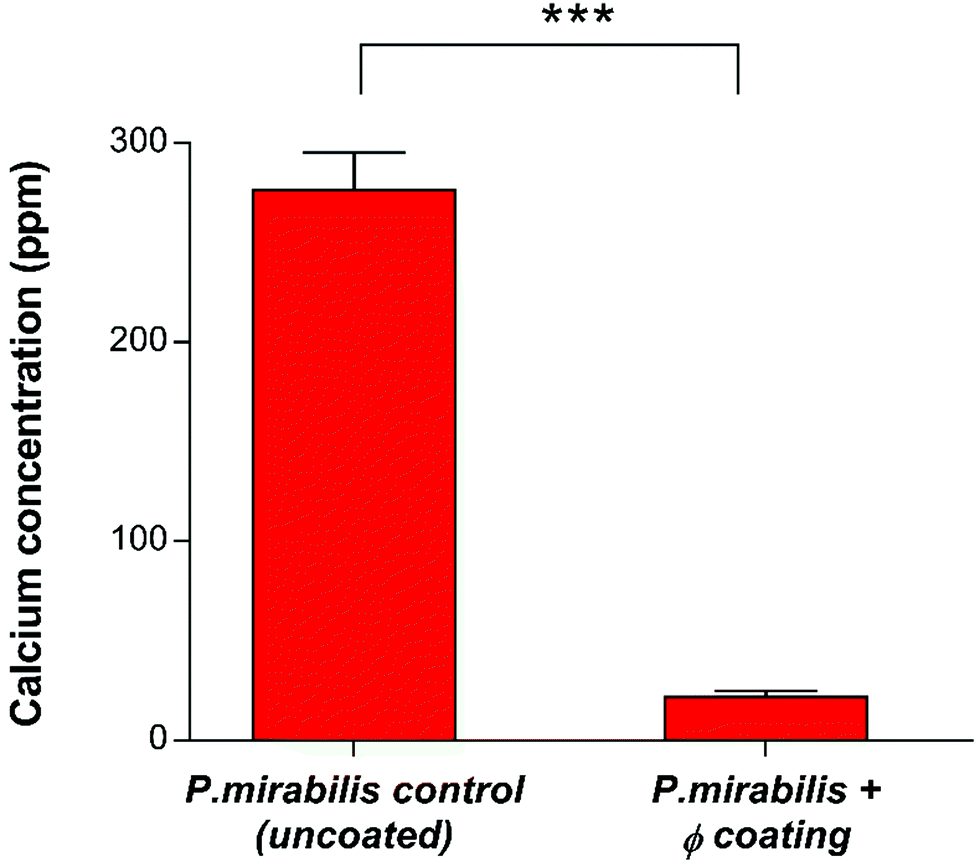 | ||
| Fig. 5 Quantitative analysis of crystalline biofilm biomass on catheter surfaces by Atomic Absorption Spectroscopy (AAS). Comparison of uncoated and phage (φ) coated catheters 13 hours post-inoculation with P. mirabilis. ***P < 0.001. Data shown is the mean of triplicate repeats. Error bars represent standard error of the mean (SEM). | ||
The ability of bacteriophage to mitigate the growth of P. mirabilis biofilms observed in this work is in agreement with previous research into phage efficacy in this setting,21 where the use of a phage cocktail to eradicate E. coli and P. mirabilis biofilms showed promise. Significant reductions in E. coli biofilm populations (3–4 log) were observed, however only a 1 log reduction was observed for P. mirabilis. It was suggested that the possible reason for this reduced efficacy may be related to phage-dependent factors such as the production of depolymerases and the penetration of the extracellular matrix. More recently, evaluation of phage cocktails on P. mirabilis biofilms,31 and pre-treated hydrogel silicone catheters to prevent adhesion of P. aeruginosa and P. mirabilis42 have showed a 1 log, and approximately 2 log reduction in P. mirabilis (single and mixed species) populations respectively. The significantly greater 6-log reduction observed here is likely owing to the initiation of a burst response from the polymeric film, resulting in exposure of viable phage particles to exponential growth-phase bacterial cells and subsequent rapid and substantial decline in bacterial population, owing the successful administration of phage after the ‘proliferation threshold’ necessary for active phage treatment.60 Additionally, limitations on cocktail utility have been described previously.61 Whilst it is logical to assume that phage cocktails are inherently more effective against bacterial populations in comparison to monophages, in reality the potential for mixed coinfections, and the limitations of the cocktails to inhibit bacterial resistance may in fact hinder phage productivity. Hence, it is possible that phage cocktails used in previous studies may not have used compatible phage types, resulting in reduced infection robustness or interference by one or more of the participating phage types. Indeed, treatment via phage cocktail may not effectively prevent the development of phage-resistant mutant populations. Such populations will not be present in sufficiently high densities to support phage amplification able to control population growth. Once the phage-resistant bacteria have replicated enough to support active phage treatment, then cocktail use against them will be equivalent to the primary treatment. Such secondary treatment is only possible if phage types are supplied either in sequential doses or continuously.61 Nevertheless, development of suitable, compatible phage cocktails is essential for the future of in situ phage therapy such as the work described here, although an in-depth understanding of phage kinetics, interference, competition and synergy is required first in order to produce an effective treatment with a broad host-range and lower likelihood of resistance evolution.
Conclusion
In summary, this study demonstrates the potential of phage therapy in the control of CAUTI, in particular the delay of blockage caused by P. mirabilis device colonisation. Delivery of therapeutic phage to the infected catheter was achieved locally via the formulation of a dual-layered polymeric architecture consisting of a lower ‘reservoir’ layer of PVA hydrogel (containing phage), capped by an upper ‘trigger’ layer of the copolymer EUDRAGIT®S 100, which has been deemed safe for human consumption and is already used in the manufacturing of drug-loaded enteric delivery vehicles.62 This has been demonstrated by an approximately 6-log reduction of cells within the in vitro bladder model system after coating initiation via increased urinary pH. Reduction in bacterial population and subsequent prevention of struvite and apatite crystal aggregation after phage treatment in situ was successful in doubling the time to catheter blockage, thus increasing catheter lifetime and potentially reducing the likelihood of serious symptomatic episodes within patients undergoing long-term indwelling catheterisation. Bacteriophage release was achieved in direct response to successful infection of the catheter luminal surfaces by P. mirabilis, resulting in an infection-responsive ‘burst’ release of phage into an actively growing bacterial population capable of sustaining and amplifying phage numbers in situ, without external interference or treatment administration. Moreover, production and degradation of the dual-layered polymeric coating was found to have no impact on bacterial or phage viability within the bladder models, meaning that any observed killing was resultant exclusively from phage infection. This research represents a novel delivery system for the delivery of a long-overlooked biological therapeutic, which shows significant promise in the prevention of encrustation and blockage of urinary catheters, although further insight into the development of effective phage cocktails, as well as phage-biofilm interactions is necessary in order to prevent blockage via controlled in situ release.Acknowledgements
The authors wish to acknowledge the Annette Charitable Trust for PhD student support of SM, and the BBSRC/Public Health England for support of HH. BVJ and JN were supported by the Dunhill Medical Trust (R394/1114) and the Medical Research Council (MR/N006496/1). BVJ is was also supported by the QVH Charitable Trust during this study. DA was supported directly by the Queen Victoria Hospital NHS Foundation Trust, the Blond McIndoe Research Foundation and the University of Brighton. Thanks also to Ms Diana Lednitzky and Dr Philip Fletcher their help with the SEM, and Mr Alan Carver for elemental analysis.References
- H. M. Zowawi, P. N. A. Harris, M. J. Roberts, P. A. Tambyah, M. A. Schembri, M. D. Pezzani, D. A. Williamson and D. L. Paterson, Nat. Rev. Urol., 2015, 12, 1–15 CrossRef PubMed.
- R. Plowman, N. Graves, M. Griffin, A. Swan, B. Cookson and L. Taylor, Eurosurveillance, 2000, 5, 4 Search PubMed.
- S. M. Jacobsen, D. J. Stickler, H. L. T. Mobley and M. E. Shirtliff, Clin. Microbiol. Rev., 2008, 21, 26–59 CrossRef CAS PubMed.
- D. J. Stickler and S. D. Morgan, J. Med. Microbiol., 2006, 55, 489–494 CrossRef CAS PubMed.
- C. Armbruster and H. Mobley, Nat. Rev. Microbiol., 2012, 10, 743–754 CrossRef CAS PubMed.
- K. Davenport and F. Keeley, J. Hosp. Infect., 2005, 60, 298–303 CrossRef CAS PubMed.
- R. O. Darouiche, J. A. Smith Jr., H. Hanna, C. B. Dhabuwala, M. S. Steiner, R. J. Babaian, T. B. Boone, P. T. Scardino, J. I. Thornby and I. I. Raad, Urology, 1999, 54, 976–981 CrossRef CAS PubMed.
- L. E. Fisher, A. L. Hook, W. Ashraf, A. Yousef, D. A. Barrett, D. J. Scurr, X. Chen, E. F. Smith, M. Fay, C. D. J. Parmenter, R. Parkinson and R. Bayston, J. Controlled Release, 2015, 202, 57–64 CrossRef CAS PubMed.
- J. H. Crabtree, R. J. Burchette, R. A. Siddiqi, I. T. Huen, L. L. Hadnott and A. Fishman, Peritoneal Dial. Int., 2003, 23, 368–374 CAS.
- T. Lam, O. Mi, E. Fisher, K. Gillies and S. Maclennan, Cochrane Database Syst. Rev., 2014, 9 Search PubMed.
- N. Morris, D. Stickler and C. Winters, Br. J. Urol., 1997, 80, 58–63 CAS.
- S. D. Morgan, D. Rigby and D. J. Stickler, Urol. Res., 2009, 37, 89–93 CrossRef CAS PubMed.
- D. G. Maki and P. A. Tambyah, Emerging Infect. Dis., 2001, 7, 342–347 CrossRef CAS PubMed.
- M. Gabi, L. Hefermehl, D. Lukic, R. Zahn and J. Vo, Urol. Res., 2011, 39, 81–88 CrossRef PubMed.
- D. O. Schairer, J. S. Chouake, J. D. Nosanchuk and A. J. Friedman, Virulence, 2012, 3, 271–279 CrossRef PubMed.
- H. Ren, J. Wu, A. Colletta, M. E. Meyerhoff and C. Xi, Front. Microbiol., 2016, 7, 1–8 Search PubMed.
- H. Ren, A. Colletta, D. Koley, J. Wu, C. Xi, T. C. Major, R. H. Bartlett and M. E. Meyerhoff, Bioelectrochemistry, 2015, 104, 10–16 CrossRef CAS PubMed.
- R. Pickard, T. Lam, G. Maclennan, K. Starr, M. Kilonzo, G. Mcpherson, K. Gillies, A. Mcdonald, K. Walton, B. Buckley, C. Glazener, C. Boachie, J. Burr, J. Norrie, L. Vale, A. Grant and J. N. Dow, J.-Lancet, 2012, 380, 1927–1935 CrossRef CAS.
- T. M. Hooton, S. F. Bradley, D. D. Cardenas, R. Colgan, S. E. Geerlings, J. C. Rice, S. Saint, A. J. Schaeffer, P. A. Tambayh, P. Tenke, L. E. Nicolle and Infectious Diseases Society of America, Clin. Infect. Dis., 2010, 50, 625–663 CrossRef PubMed.
- N. A. Sabbuba, E. Mahenthiralingam and D. J. Stickler, J. Clin. Microbiol., 2003, 41, 4961–4965 CrossRef CAS PubMed.
- L. Carson, S. P. Gorman and B. F. Gilmore, FEMS Immunol. Med. Microbiol., 2010, 59, 447–455 CrossRef CAS PubMed.
- F. D'Herelle, L'academie des Sciences Paris, 1917, 165, 373–375 Search PubMed.
- F. Twort, Lancet, 1915, 186, 1241–1243 CrossRef.
- M. Skurnik and E. Strauch, Int. J. Med. Microbiol., 2006, 296, 5–14 CrossRef CAS PubMed.
- S. Mccallin, C. Barretto, S. Sultana, B. Berger, S. Huq, L. Krause, R. Bibiloni, B. Schmitt, G. Reuteler and H. Brüssow, Virology, 2013, 443, 187–196 CrossRef CAS PubMed.
- C. R. Merril, D. Scholl and S. Adhya, Nat. Rev. Drug Discovery, 2003, 2, 489–497 CrossRef CAS PubMed.
- Z. Moradpour and A. Ghasemian, Biotechnol. Adv., 2011, 29, 732–738 CrossRef CAS PubMed.
- J. Uchiyama, S. Sakurai, T. Ujihara, M. Kuroda, M. Ikeuchi and T. Tani, J. Infect. Chemother., 2005, 11, 211–219 CrossRef PubMed.
- A. Sulakvelidze and Z. Alavidze, Antimicrob. Agents Chemother., 2001, 45, 649–659 CrossRef CAS PubMed.
- J. Nzakizwanayo, A. Hanin, D. R. Alves, B. McCutcheon, C. Dedi, J. Salvage, K. Knox, B. Stewart, A. Metcalfe, J. Clark, B. F. Gilmore, C. G. M. Gahan, A. T. A. Jenkins and B. V. Jones, Antimicrob. Agents Chemother., 2016, 60, 1530–1536 CrossRef PubMed.
- L. D. R. Melo, P. Veiga, N. Cerca, A. M. Kropinski, C. Almeida, J. Azeredo and S. Sillankorva, Front. Microbiol., 2016, 7, 1–12 Search PubMed.
- P. Chadha, O. Prakash and S. Chhibber, Microb. Pathog., 2016, 99, 68–77 CrossRef CAS PubMed.
- L. Rodrigues, in Bacterial Adhesion, ed. D. Linke and A. Goldman, Springer Netherlands, Netherlands, 1st edn, 2011, pp. 351–367 Search PubMed.
- R. I. Aminov, M. Otto and A. Sommer, Front. Microbiol., 2010, 1, 1–7 Search PubMed.
- L. Lu and L. D. Unsworth, Biomacromolecules, 2016, 17, 1425–1436 CrossRef CAS PubMed.
- Z. Lu, J. Zhang, Z. Yu, Q. Liu, K. Liu, M. Li and D. Wang, New J. Chem., 2017, 41, 432–436 RSC.
- N. J. Irwin, C. P. McCoy, D. S. Jones and S. P. Gorman, Pharm. Res., 2013, 30, 857–865 CrossRef CAS PubMed.
- H. Hathaway, J. Ajuebor, L. Stephens, A. Coffey, U. Potter, J. M. Sutton and A. T. A. Jenkins, J. Controlled Release, 2017, 245, 108–115 CrossRef CAS PubMed.
- H. Hathaway, D. R. Alves, J. Bean, P. P. Esteban, K. Ouadi, J. Mark Sutton and A. T. A. Jenkins, Eur. J. Pharm. Biopharm., 2015, 96, 437–441 CrossRef CAS PubMed.
- J. E. Bean, D. R. Alves, M. Laabei, P. P. Esteban, N. T. Thet, M. C. Enright and A. T. A. Jenkins, Chem. Mater., 2014, 26, 7201–7208 CrossRef CAS.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS PubMed.
- S. M. Lehman and R. M. Donlan, Antimicrob. Agents Chemother., 2015, 59, 1127–1137 CrossRef PubMed.
- S. Milo, N. T. Thet, D. Liu, J. Nzakizwanayo, B. V. Jones and A. T. A. Jenkins, Biosens. Bioelectron., 2016, 81, 166–172 CrossRef CAS PubMed.
- Evonik, Eudragit S100 Technical Information, http://eudragit.evonik.com/product/eudragit/en/products-services/eudragit-products/enteric-formulations/s-100/Pages/default.aspx, accessed 24 July 2016.
- B. V. Jones, R. Young, E. Mahenthiralingam and D. J. Stickler, Infect. Immun., 2004, 72, 3941–3950 CrossRef CAS PubMed.
- D. R. Alves, A. Gaudion, J. E. Bean, P. P. Esteban, T. C. Arnot, D. R. Harper, W. Kot, L. H. Hansen and M. C. Enright, Appl. Environ. Microbiol., 2014, 80, 6694–6703 CrossRef CAS PubMed.
- V. Levering, Q. Wang, P. Shivapooja, X. Zhao and G. Lopez, Adv. Healthcare Mater., 2014, 3, 1588–1596 CrossRef CAS PubMed.
- D. Stickler, N. Morris and C. Winters, Methods Enzymol., 1999, 310, 498–501 Search PubMed.
- C. E. Armbruster, S. Smith, A. Johnson, V. DeOrnellas, K. Eaton, A. Yep, L. Mody, W. Wu and H. L. T. Mobley, Infect. Immun., 2017, 85, 1–23 CrossRef PubMed.
- D. V. Dixon, Z. Hosseinidoust and N. Tufenkji, Langmuir, 2014, 30, 3184–3190 CrossRef CAS PubMed.
- R. M. Donlan, Trends Microbiol., 2009, 17, 66–72 CrossRef CAS PubMed.
- W. Fu, T. Forster, O. Mayer, J. J. Curtin, S. Lehman and R. M. Donlan, Antimicrob. Agents Chemother., 2010, 54, 397–404 CrossRef CAS PubMed.
- R. Thummeepak, T. Kitti, D. Kunthalert and S. Sitthisak, Front. Microbiol., 2016, 7, 1–8 Search PubMed.
- S. M. Soto and S. M. Soto, Adv. Biol., 2014, 2014, 1–13 CrossRef.
- K. S. Liao, S. M. Lehman, D. J. Tweardy, R. M. Donlan and B. W. Trautner, J. Appl. Microbiol., 2012, 1131530, 1530–1539 CrossRef PubMed.
- A. Chibeu, E. Lingohr, L. Masson, A. Manges, J. Harel, H. W. Ackermann, A. Kropinski and P. Boerlin, Viruses, 2012, 4, 471–487 CrossRef CAS PubMed.
- J. J. Curtin and R. M. Donlan, Antimicrob. Agents Chemother., 2006, 50, 1268–1275 CrossRef CAS PubMed.
- B. W. Trautner, R. H. Hull and R. O. Dariouche, Curr. Opin. Infect. Dis., 2005, 18, 37–41 CrossRef PubMed.
- S. a. Wilks, M. J. Fader and C. W. Keevil, PLoS One, 2015, 10, 1–13 Search PubMed.
- S. T. Abedon and C. Thomas-Abedon, Curr. Pharm. Biotechnol., 2010, 11, 28–47 CAS.
- B. K. Chan and S. T. Abedon, in Advances in Medical Microbiology, ed. A. I. Laskin, S. Sariaslani and G. M. Gadd, Elsevier, San Diego, CA, USA, 1st edn, 2012, vol. 78, pp. 17–19 Search PubMed.
- H. Saade, R. Diaz de León-Gómez, F. J. Enríquez-Medrano and R. G. López, J. Biomater. Sci., Polym. Ed., 2016, 27, 1126–1138 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb01302g |
| This journal is © The Royal Society of Chemistry 2017 |