
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocompatible pH-responsive nanoparticles with a core-anchored multilayer shell of triblock copolymers for enhanced cancer therapy†
Elizabeth
Ellis
abc,
Kangyi
Zhang
c,
Qianyu
Lin
d,
Enyi
Ye
c,
Alessandro
Poma
a,
Giuseppe
Battaglia
ae,
Xian Jun
Loh
 cd and
Tung-Chun
Lee
*ab
cd and
Tung-Chun
Lee
*ab
aDepartment of Chemistry, University College London (UCL), UK
bInstitute for Materials Discovery, University College London (UCL), UK. E-mail: tungchun.lee@ucl.ac.uk
cInstitute for Materials Research and Engineering (IMRE), Agency for Science Technology and Research (A*STAR), Singapore
dDepartment of Materials Science and Engineering, National University of Singapore, Singapore
eDepartment of Chemical Engineering, University College London (UCL), UK
First published on 12th May 2017
Abstract
Drug nanocarriers are synthesised via a facile self-assembly approach using gold nanoparticles (Au NPs) as a structural core. The nanocarriers feature a multilayer shell of POEGMA–PDPA–PMPC triblock copolymers with a chain-end thiol functional group for anchoring to the Au NP surface. This water-soluble triblock copolymer was synthesised via atom transfer radical polymerisation (ATRP) from a bi-functional initiator containing a disulphide bridge. The resultant nanocarriers exhibit high biocompatibility plus excellent colloidal stability and antifouling capability in bio-media (50% PBS/FBS). Encapsulation and release of a hydrophobic drug can be effectively triggered by a pH-stimulus. Meanwhile drug-loaded nanocarriers show enhanced efficacy towards cancer cells compared to plain drug.
Compartmentalised micro and nanostructures are abundant in nature and are essential to many processes in living organisms, such as intercellular signaling mediated by exosomes,1 and storage and release of materials by vacuoles. Inspired by nature, different kinds of artificial compartmentalised nanostructures have been developed. A large group of these artificial nanostructures have been employed for drug delivery purposes, including micelles,2,3 vesicles,4,5 metal–organic frameworks6 and inorganic nanoparticles.7 Encapsulating drugs within nanostructures increases their solubility and circulation time and can reduce side effects by selective release into unhealthy areas, limiting damage to healthy tissues.8,9 For example, pH responsive nanostructures are often exploited for release of drugs into areas of low pH through protonation and acid catalysed bond cleavage.10,11
For practical applications, chemical composition of the substituent materials of artificial nanostructures have to be biocompatible. In this context, block copolymers based on poly(oligo(ethylene glycol)methyl ether methacrylate) POEGMA, poly(2-(diisopropyl amino)ethyl methacrylate) PDPA and poly(2-(methacryloyloxy)ethyl phosphorylcholine) PMPC show particular advantages because of their high biocompatibility and ease of synthesis.12 Polymersomes of PMPC–PDPA and POEGMA–PDPA have been used for delivery of anticancer drugs,13,14 proteins,15 DNA16 and antibiotics.17 In these systems, the pH-responsive hydrophobicity of PDPA has been exploited for rapid disassembly of nanocarriers in mildly acidic environments within cells endosomes.16,18 In addition PMPC has been shown to interact with scavenger B receptors on cell membranes, which are overexpressed on cancer cells, facilitating entry of polymersomes into the cells.19 Nevertheless, the above and similar types of nanocarriers can be demanding to make and handle owing to the dynamic nature of these systems.
Anchoring functional polymers onto the surface of gold nanoparticles (Au NPs) via self-assembly has been considered a powerful strategy for producing multifunctional drug carriers.20 In addition to their low cytotoxicity, ease of synthesis and unique plasmonic properties that can be exploited for theranostic and synergistic therapies,21,22 Au NPs can serve as a structural core for forming well-defined, stable nanostructures, in contrast to some other self-assembled structures. For instance, non-crosslinked, low molecular weight micelles are prone to dissociation below their critical micelle concentration, resulting in premature release of their payload.23 In the context of biomedical applications, Au NPs are conventionally coated by polyethylene glycol (PEG) which may suffer from fouling at high protein concentration.24
Herein we use Au NPs as a structural core for anchoring triblock copolymers of POEGMA–PDPA–PMPC via a facile assembly approach to form antifouling, pH-responsive nanocarriers (Fig. 1a). POEGMA is a less-coiled block that favours the formation of a dense inner shell on the Au NPs. It also enhances the solubility of the intermediate polymer chains during polymerisation. The PDPA layer can serve as a pH-responsive sponge for hydrophobic drug encapsulation and release (Fig. 1b and c). Doxorubicin, a highly toxic cancer drug that has serious side effects such as cardiotoxicity,25 is used as a model drug. The undesirable side effects can be reduced by incorporating the drug into a wide variety of nanostructures including hydrogels, liposomes and polymeric micelles, for improved selectivity.26 The zwitterionic PMPC outer corona is designed to enhance the colloidal stability across different pH and ionic strength. In addition, PMPC is also known to be antifouling owing to its high degree of hydration in aqueous media.27 We demonstrate that these nanocarriers indeed exhibit high biocompatibility, notable colloidal stability plus antifouling capability in concentrated bio-media, and enhancement in drug efficacy towards cancer cells. To the best of our knowledge, this represents the first example of anchoring POEGMA–PDPA–PMPC based polymer onto Au NPs and of studying the resultant drug delivery performance in cancer cells.
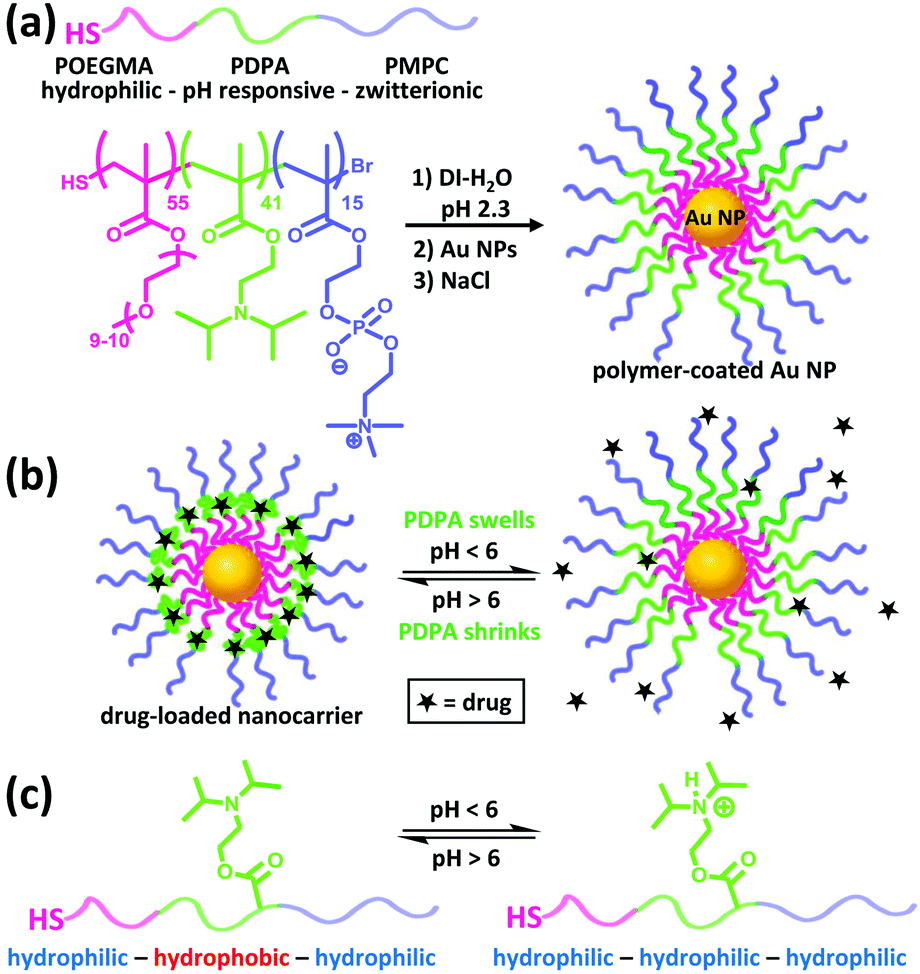 | ||
| Fig. 1 (a) Formation of compartmentalised nanocarriers via self-assembly of POEGMA–PDPA–PMPC triblock copolymers on the surface of Au NPs. (b) Encapsulation and release of hydrophobic drugs by the nanocarriers can be gated by the pH level of surrounding media, via the (c) change in hydrophobicity of the PDPA block upon (de)protonation. | ||
POEGMA–PDPA–PMPC triblock copolymers were first prepared as outlined in Fig. 2a based on previously reported methods.28 Atom transfer radical polymerisation (ATRP) via sequential addition from a bi-functional initiator containing a disulphide bridge was used. OEGMA (Mn = 500 g mol−1) was polymerised first, followed by addition of DPA and then MPC. Block ratio and purity were confirmed by nuclear magnetic resonance (NMR) spectroscopy and gel permeation chromatography (GPC), using conversion measurements and comparison of peak integrals (Fig. S1, ESI†). Reducing conditions during purification directly cleaved the disulphide bridge in the initiator as confirmed by GPC (Fig. S2, ESI†), leaving free thiol groups on the POEGMA termini of the polymer.
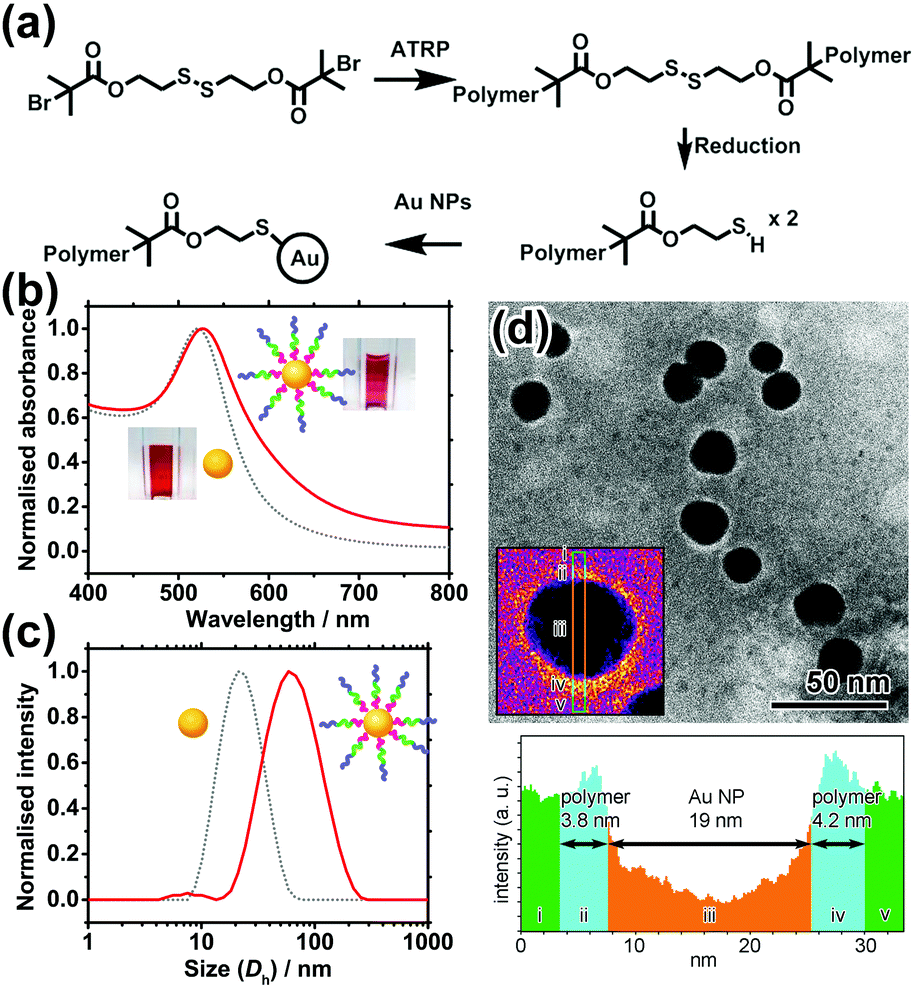 | ||
| Fig. 2 (a) Synthetic scheme of thiolated triblock polymers and their conjugation to Au NPs. (b) UV-vis spectra and (c) hydrodynamic diameter (Dh as measured by DLS) of Au NPs before (grey dot) and after (red solid) polymer conjugation. (d) Top: TEM image of polymer-coated Au NPs. Inset showing a false-colour image of a selected NP. Bottom: Corresponding line profile of the inset. | ||
Citrate-coated Au NPs (d = 15.5 ± 2.5 nm), prepared via the Turkevich method, were then coated with the polymer by displacement of citrate ions with thiol groups on the end of the polymer chains. Au NPs were added to a polymer solution containing a large excess of polymer at pH 2.3, where the pH-sensitive amino groups of the PDPA block are fully protonated and the polymer chains fully extended to avoid micellisation of the polymer. NaCl was subsequently added to enhance the packing of the polymer shell by reducing the charge repulsion between protonated amines on the PDPA block. The resultant core–shell nanocarriers can be readily purified by centrifugation because of the high-density Au NP core. Following removal of excess polymer, NaCl and displaced citrate by centrifugation and redispersion, the polymer coating was characterised and confirmed by UV-vis spectroscopy, dynamic light scattering (DLS) measurements and transmission electron microscopy (TEM) as shown in Fig. 2b–d.
The UV-vis spectrum shows a 4 nm red shift of the localised surface plasmon resonance (LSPR) peak of Au NPs upon polymer conjugation (Fig. 2b). The shift can be attributed to the increase in refractive index of the media (represented by the polymer coating) immediately surrounding the Au NP. Meanwhile, DLS reveals an increase in hydrodynamic diameter (Dh) of 46 nm upon polymer conjugation at pH 2.3. This implies the thickness of the fully protonated polymer shell is 23 nm, consistent with the theoretical, fully extended polymer length of 27.9 nm. In TEM images (Fig. 2d) of a sample prepared using phosphotungstic acid (PTA) negative staining, the polymer shell is visible as a white halo around the dark Au NPs, with a thickness of 4.0 ± 0.9 nm. The grafting density is estimated to be ∼0.11 chains per nm2 (i.e. polymer footprint ∼8.8 nm2 and ∼86 chains per NP). In line with our expectation, the shell thickness determined by TEM is smaller than that given by DLS because the former was measured in a dry state in which the polymer shell shrank significantly upon dehydration, while the latter was determined in a fully hydrated state (Tables S1 and S2, ESI†).
Colloidal stability of nanocarriers in bio-media is an important property as it affects the uptake efficiency by cells.7 Citrate-stabilised Au NPs are prone to aggregation upon an increase in ionic strength of the media owing to the corresponding decrease in charge repulsion between them.29 Similarly, they are also prone to protein fouling, which can cause aggregation and change in surface properties, potentially leading to immune response of patients.30 Zwitterionic materials, in particular PMPC, have been shown to effectively reduce protein fouling at high serum concentrations.24,27,29 To investigate the colloidal stability of the polymer-coated Au NPs in these conditions, solutions of the NPs were mixed at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with phosphate buffered saline (PBS) and fetal bovine serum (FBS). PBS has a high salt concentration which causes citrate-coated NPs to aggregate31 (Fig. S5 and S6, ESI†) and FBS contains large amounts of proteins such as bovine serum albumin. UV-vis and DLS were used to monitor the solutions for aggregation and protein fouling. If aggregation occurs, the LSPR peak of the Au NPs will broaden and red shift due to plasmonic coupling between aggregated Au NPs. If aggregation is severe, the optical extinction decreases because of precipitation of NPs out of solution. In contrast to citrate-stabilised Au NPs, polymer-coated nanocarriers show no significant change in the LSPR peak position in either medium (Fig. 3a). Antifouling of the nanocarriers is further verified by the DLS data which shows no significant change in hydrodynamic diameter when the NPs are in 50% FBS (Fig. S7a, ESI†). In contrast, citrate-coated Au NPs show a size increase of 39.5 nm in 50% FBS (Fig. S7b, ESI†), indicating the occurrence of protein fouling as expected.31 It is noted that this does not quantitatively discard protein interactions with the nanocarriers.
:1 ratio with phosphate buffered saline (PBS) and fetal bovine serum (FBS). PBS has a high salt concentration which causes citrate-coated NPs to aggregate31 (Fig. S5 and S6, ESI†) and FBS contains large amounts of proteins such as bovine serum albumin. UV-vis and DLS were used to monitor the solutions for aggregation and protein fouling. If aggregation occurs, the LSPR peak of the Au NPs will broaden and red shift due to plasmonic coupling between aggregated Au NPs. If aggregation is severe, the optical extinction decreases because of precipitation of NPs out of solution. In contrast to citrate-stabilised Au NPs, polymer-coated nanocarriers show no significant change in the LSPR peak position in either medium (Fig. 3a). Antifouling of the nanocarriers is further verified by the DLS data which shows no significant change in hydrodynamic diameter when the NPs are in 50% FBS (Fig. S7a, ESI†). In contrast, citrate-coated Au NPs show a size increase of 39.5 nm in 50% FBS (Fig. S7b, ESI†), indicating the occurrence of protein fouling as expected.31 It is noted that this does not quantitatively discard protein interactions with the nanocarriers.
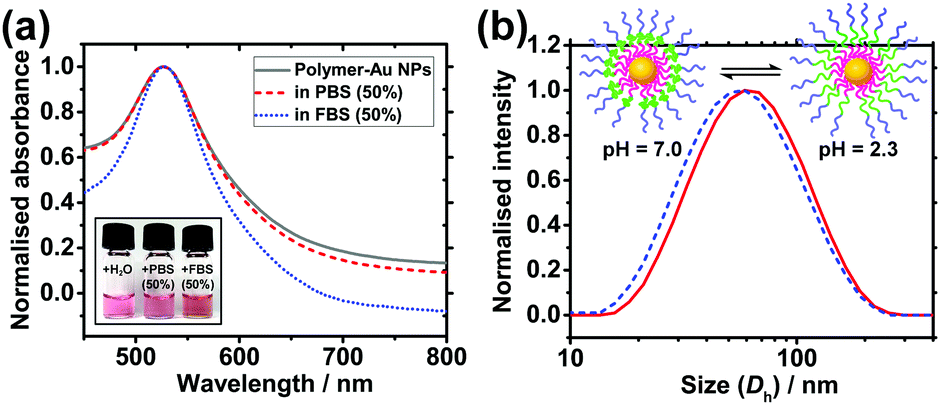 | ||
| Fig. 3 (a) Colloidal stability and antifouling of nanocarriers in bio-media. UV-vis spectra of polymer-coated NPs in deionised water (grey solid), 50% PBS (red dash), and 50% FBS (blue dot, FBS background subtracted). Inset shows a photo of the samples. (b) pH-Responsiveness of the nanocarriers. Hydrodynamic diameter of polymer-coated Au NPs at pH 2.3 (red solid) and pH 7.0 (blue dot) at 25 °C. | ||
The response of the nanoparticles to pH changes was then investigated. The hydrodynamic diameter of the polymer-coated NPs was measured at pH 2.3 and pH 7.0 at 25.0 °C. A size decrease of 3.4 nm was observed upon an increase in pH from 2.3 to 7.0 (Fig. 3b). The pKa of the PDPA is approximately 6 at 37 °C with values varying from 5.75 at 50 °C to 7.6 at 5 °C.18 The positive charges along the chain repel each other and this is sufficient to overcome the entropic tendency of the polymer to coil. Hence PDPA chains are expected to be stretched. An increase in pH leads to deprotonation of the amino groups and the PDPA segment of the polymer chain becomes hydrophobic as it is no longer charged (Fig. 1c). This means that PDPA chains can now relax to a more coiled configuration with a consequent decrease in size. Attachment of polymer on Au NPs via the numerous amino groups of the PDPA block is unlikely because the PDPA chains would then be wrapping around the Au NP as opposed to extending from the Au NP surface. Therefore, upon deprotonation, no or very little contraction would be observed in this scenario.
Drug encapsulation and triggered release by the nanocarriers were investigated using the cancer drug doxorubicin (DOX). DOX is a hydrophobic drug (logP = 1.27) and its water solubility increases slightly in the hydrochloric acid form. DOX-loading was performed by introducing DOX during the polymer coating step at pH 2.3, followed by incubation at pH 7.2. Excess DOX and polymer were then removed by centrifugation and redispersion. By measuring the decrease in fluorescence of the resultant supernatant, the encapsulation efficiency, i.e. (amount of DOX encapsulated/amount added) × 100%, is calculated to be ∼37%, and the drug loading is estimated to be ∼47%. Triggered release experiments were performed by incubating 3 sets of DOX-loaded nanoparticles (NP-DOX) in the dark at 37 °C, pH 7.2 for 24 hours. Periodically, samples were taken from each vial and centrifuged. Fluorescence of the supernatant was then measured (Fig. 4a). At pH = 7.2, no release of DOX was observed for the first 24 hours. This is attributed to the long length and highly hydrophobic nature of the PDPA block. After 24 hours, the pH of the solution was decreased to ∼4.0 so that the PDPA shell would be protonated and become hydrophilic, triggering the release of DOX as shown by the fluorescence data.
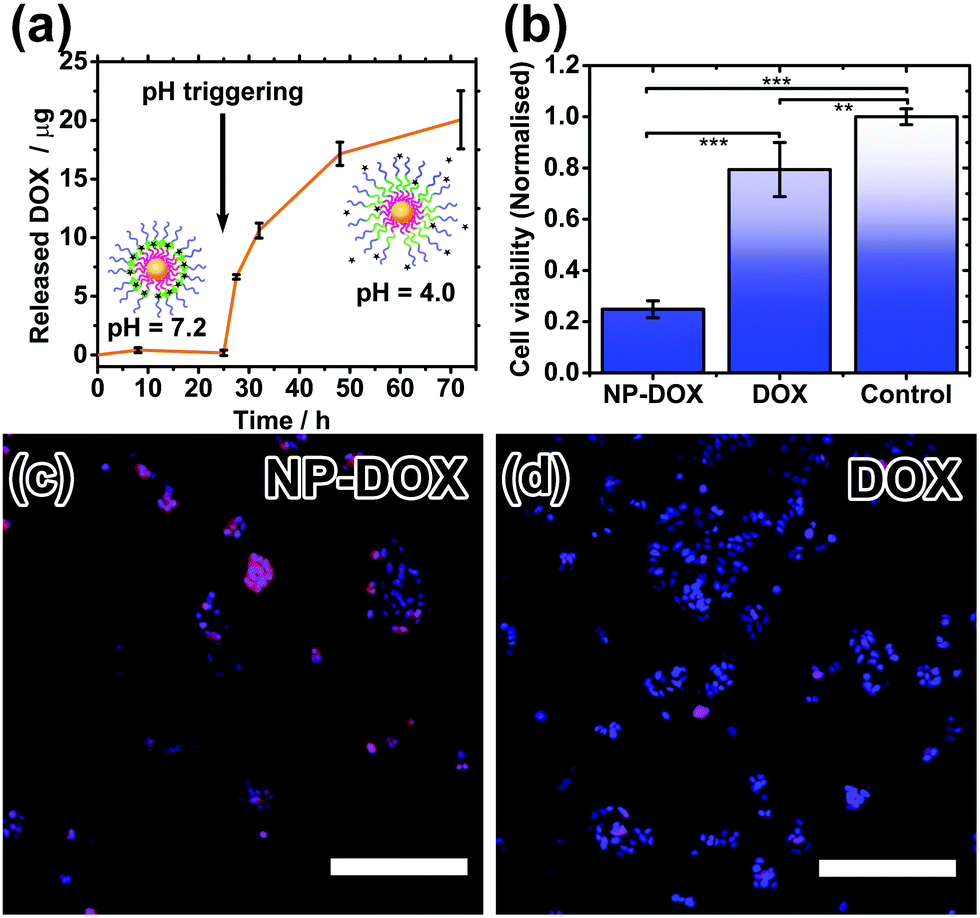 | ||
| Fig. 4 (a) pH-Triggered drug release profile. (b) Cell viability of MCF-7 breast cancer cells after 24 h treatment with NP-DOX and DOX. Fluorescence readings from resazurin assay were normalised to the average of control cells without any treatment (significance **p < 0.001; ***p < 0.0001). (c and d) Qualitative observation of intracellular doxorubicin release in MCF-7 breast cancer cells after 1 h incubation. NP-DOX can result in effective drug uptake as indicated by the red fluorescence. Blue fluorescence is indicative of the cell nuclei (magnification 10× | scale bar: 250 microns). | ||
Finally, cell studies were carried out to investigate the biocompatibility of the nanocarriers and their ability to kill cancer cells. As a control, a biocompatibility assay verifies that the nanocarriers alone are not toxic to healthy cells (Fig. S8, ESI†). MCF-7 breast cancer cells were then treated with DOX-loaded nanocarriers and their cell-killing efficiency was compared to free doxorubicin at the same concentration. The cell viability after 24 hours was significantly lower for the NP-DOX than for plain DOX (Fig. 4b). Hence, these safe nanocarriers have the potential to increase the efficacy of chemotherapy. Drug uptake by cells was verified by fluorescence microscopy. Both NP-DOX and DOX resulted in drug internalisation by MCF-7 after one-hour incubation (red fluorescence in Fig. 4c and d). This shows that DOX in the NP-DOX construct remains active and efficacious.
In conclusion, drug nanocarriers with a multifunctional polymer shell have been synthesised via a facile self-assembly approach using Au NPs as a structural core. The POEGMA–PDPA–PMPC triblock copolymers were synthesised via sequential ATRP from a disulphide initiator. These nanocarriers are notably stable, antifouling and biocompatible owing to the highly-hydrated, zwitterionic outer corona. The pH-responsive middle layer allows encapsulation and triggered release of a hydrophobic drug in a controllable manner. Proof-of-concept experiments show that the nanocarriers can enhance the efficacy of hydrophobic drug towards cancer cells, resulting in a significant decrease in cell viability compared to the control using unencapsulated drug. Following further studies, the nanocarriers have potential to be used for co-delivery of hydrophobic and hydrophilic drugs by utilising both the PDPA and the POEGMA layers of the shell. In light of theranostics, functional properties of the inorganic NP core can be simultaneously exploited, for instance in photothermal therapy, and in the case of hybrid nanoparticles, refractive index32 and rheology33 sensing at the nanoscale.
Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
The research is funded by the UCL BEAMS Future Leader Award through the EPSRC 2016 Institutional Sponsorship Award (EP/P511262/1) and by the Royal Society Research Grant (RG150551). EE and TCL are grateful to the Studentship funded by the A*STAR-UCL Research Attachment Programme through the EPSRC Centre for Doctoral Training in Molecular Modelling and Materials Science (EP/L015862/1). AP thanks the NC3Rs for sponsoring his fellowship while GB acknowledges both the ERC (MEViC) starting grant and the EPSRC (EP/N026322/1) Established Career Fellowship to cover part of his salary. We finally thank the BTG ltd for donating the MPC monomer.Notes and references
- C. Corrado, S. Raimondo, A. Chiesi, F. Ciccia, G. De Leo and R. Alessandro, Int. J. Mol. Sci., 2013, 14, 5338–5366 CrossRef CAS PubMed.
- X. J. Loh, M.-H. Tsai, J. D. Barrio, E. A. Appel, T.-C. Lee and O. A. Scherman, Polym. Chem., 2012, 3, 3180–3188 RSC.
- X. J. Loh, J. del Barrio, P. P. C. Toh, T.-C. Lee, D. Jiao, U. Rauwald, E. A. Appel and O. A. Scherman, Biomacromolecules, 2012, 13, 84–91 CrossRef CAS PubMed.
- D. Jiao, J. Geng, X. J. Loh, D. Das, T.-C. Lee and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 9633–9637 CrossRef CAS PubMed.
- X. J. Loh, J. del Barrio, T.-C. Lee and O. A. Scherman, Chem. Commun., 2014, 50, 3033–3035 RSC.
- R. C. Huxford, J. Della Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS PubMed.
- X. J. Loh, T.-C. Lee, Q. Dou and G. R. Deen, Biomater. Sci., 2016, 4, 70–86 RSC.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X.-J. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS PubMed.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS PubMed.
- J. Gaitzsch, V. Chudasama, E. Morecroft, L. Messager and G. Battaglia, ACS Macro Lett., 2016, 5, 351–354 CrossRef CAS.
- C. Pegoraro, D. Cecchin, L. S. Gracia, N. Warren, J. Madsen, S. P. Armes, A. Lewis, S. MacNeil and G. Battaglia, Cancer Lett., 2013, 334, 328–337 CrossRef CAS PubMed.
- L. Simón-Gracia, H. Hunt, P. Scodeller, J. Gaitzsch, V. R. Kotamraju, K. N. Sugahara, O. Tammik, E. Ruoslahti, G. Battaglia and T. Teesalu, Biomaterials, 2016, 104, 247–257 CrossRef PubMed.
- X. Tian, S. Nyberg, P. S. Sharp, J. Madsen, N. Daneshpour, S. P. Armes, J. Berwick, M. Azzouz, P. Shaw, N. J. Abbott and G. Battaglia, Sci. Rep., 2015, 5, 11990 CrossRef CAS PubMed.
- H. Lomas, I. Canton, S. MacNeil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Adv. Mater., 2007, 19, 4238–4243 CrossRef CAS.
- K. Wayakanon, M. H. Thornhill, C. W. I. Douglas, A. L. Lewis, N. J. Warren, A. Pinnock, S. P. Armes, G. Battaglia and C. Murdoch, FASEB J., 2013, 27, 4455–4465 CrossRef CAS PubMed.
- R. T. Pearson, N. J. Warren, A. L. Lewis, S. P. Armes and G. Battaglia, Macromolecules, 2013, 46, 1400–1407 CrossRef CAS.
- H. E. Colley, V. Hearnden, M. Avila-Olias, D. Cecchin, I. Canton, J. Madsen, S. MacNeil, N. Warren, K. Hu, J. A. McKeating, S. P. Armes, C. Murdoch, M. H. Thornhill and G. Battaglia, Mol. Pharmaceutics, 2014, 11, 1176–1188 CrossRef CAS PubMed.
- R. A. Sperling, P. Rivera Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- L. Vigderman and E. R. Zubarev, Adv. Drug Delivery Rev., 2013, 65, 663–676 CrossRef CAS PubMed.
- A. J. Mieszawska, W. J. M. Mulder, Z. A. Fayad and D. P. Cormode, Mol. Pharmaceutics, 2013, 10, 831–847 CrossRef CAS PubMed.
- A. Sosnik and M. Menaker Raskin, Biotechnol. Adv., 2015, 33, 1380–1392 CrossRef CAS PubMed.
- W. Yang, L. Zhang, S. Wang, A. D. White and S. Jiang, Biomaterials, 2009, 30, 5617–5621 CrossRef CAS PubMed.
- O. Tacar, P. Sriamornsak and C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157–170 CrossRef CAS PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- J. Park, S. Kurosawa, J. Watanabe and K. Ishihara, Anal. Chem., 2004, 76, 2649–2655 CrossRef CAS PubMed.
- J. Gaitzsch, M. Delahaye, A. Poma, F. Du Prez and G. Battaglia, Polym. Chem., 2016, 7, 3046–3055 RSC.
- L. L. Rouhana, J. A. Jaber and J. B. Schlenoff, Langmuir, 2007, 23, 12799–12801 CrossRef CAS PubMed.
- A. M. Alkilany and C. J. Murphy, J. Nanopart. Res., 2010, 12, 2313–2333 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, A. K. Patri, J. Zheng, J. D. Clogston, N. Ayub, P. Aggarwal, B. W. Neun, J. B. Hall and S. E. McNeil, Nanomedicine, 2009, 5, 106–117 CrossRef CAS PubMed.
- H.-H. Jeong, A. G. Mark, M. Alarcón-Correa, I. Kim, P. Oswald, T.-C. Lee and P. Fischer, Nat. Commun., 2016, 7, 11331 CrossRef CAS PubMed.
- H.-H. Jeong, A. G. Mark, T.-C. Lee, M. Alarcón-Correa, S. Eslami, T. Qiu, J. G. Gibbs and P. Fischer, Nano Lett., 2016, 16, 4887–4894 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including synthesis, experimental procedure and supporting data. See DOI: 10.1039/c7tb00654c |
| This journal is © The Royal Society of Chemistry 2017 |