
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dendritic polyglycerol anions for the selective targeting of native and inflamed articular cartilage†
Sabine
Reimann
a,
Tobias
Schneider
b,
Pia
Welker
c,
Falko
Neumann
a,
Kai
Licha
a,
Gundula
Schulze-Tanzil
b,
Wolfgang
Wagermaier
d,
Peter
Fratzl
*d and
Rainer
Haag
 *a
*a
aInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: haag@chemie.fu-berlin.de
bInstitute of Anatomy, General Hospital Nuremberg, Paracelsus Medical University, Prof.-Ernst-Nathan-Str. 1, 90419 Nuremberg, Germany
cInstitute of Anatomy and Cell Biology Charité Universitätsmedizin Berlin, Philipp-Str. 12, 10115 Berlin, Germany
dMax Planck Institute of Colloids and Interfaces, Department of Biomaterials, 14424 Potsdam, Germany. E-mail: Peter.Fratzl@mpikg.mpg.de
First published on 26th May 2017
Abstract
The destruction of articular cartilage is a critical feature in joint diseases. An approach to selectively target the damaged tissue is promising for the development of diagnostic and therapeutic agents. We herein present the interaction of dendritic polyglycerol (dPG) anions with native and inflamed cartilage. Confocal laser scanning microscopy revealed the inert character of dPG and low functionalized dPG bisphosphonate (dPGBP7%) toward cartilage in vitro. An enhanced binding was observed for highly functionalized dPG bisphosphonate, sulfate, and phosphate, which additionally showed a higher affinity to IL-1β treated tissue. The mixed anion containing sulfate and bisphosphonate groups exhibited an exceptionally high affinity to cartilage and strongly bound to collagen type II, as shown by a normalized fluorescence-based binding assay. All polyglycerol anions, except dPGBP7%, were taken up by chondrocytes within 24 h and no cytotoxicity was found up to 10−5 M. In a rheumatoid arthritis model, dPGBP7% accumulated in mineralized compartments of inflamed joints and showed an increasing affinity to cartilage with higher clinical scores, as evident from histological examinations. For dPGS no interaction with bone but a strong binding to cartilage, independent of the score, was demonstrated. These results make dPG anions promising candidates for the selective targeting of cartilage tissue.
Introduction
Inflammatory and degenerative joint diseases like rheumatoid arthritis (RA) and osteoarthritis affect more than 200 million people all over the world and therefore are some of the most common global health issues.1 Besides alterations of the subchondral bone, synovium, and synovial fluid, the course of disease is characterized by the degradation of articular cartilage including surface erosion, appearance of cracks, and chondrocyte apoptosis.2–5 Conventional therapies encompass the administration of non-steroidal anti-inflammatory drugs (NSAIDs) or intra-articular injection of hyaluronic acid and corticosteroids.3,6,7 However, these treatments mainly focus on easing symptoms to relief pain while cartilage destruction proceeds. The poor efficacy of the common approaches to prevent tissue degeneration is related to the lack of an efficient and selective drug delivery to cartilage, currently limited by several factors: the low permeability of the synovial membrane, the fast clearance of drugs out of the joint cavity, the avascular structure of cartilage that only permits compounds to enter the tissue by diffusion, and the tight extracellular matrix (ECM) that acts as a barrier for molecules to reach the site of action.8 With our ageing population, the socio-economic impact of arthritis will increase and novel approaches for the efficient and selective targeting of cartilage are clearly needed.Articular cartilage is a unique connective tissue, covering the end of long bones, that shows a low potential of regeneration due to its avascular, alymphatic, and non-innervated structure.3 The tissue mainly consists of glycoproteins, anionic proteoglycans like aggrecan, and noncollagenous proteins that are embedded in a dense network of collagen type II fibers and chondrocytes.9 The latter sequester the hydrated ECM and regulate the formation of the cartilage matrix.9 With a thin layer of fine collagen fibrils parallel to the joint surface, the superficial zone is the uppermost surface with a highly dense structure, which acts like a filter for molecules.10,11 The estimated average pore size of cartilage is ∼6 nm, which lets small molecules penetrate the cartilage tissue without hindrance, whereas larger particles are prevented from diffusing into the ECM, which limits the efficacy of anti-arthritic drugs.10,11 However, before entering, systemically administered agents must first reach the joint space by leaving the blood vessels trough the synovial membrane, which is restricted by its limited permeability showing a threshold of ∼10 nm.12,13 Nevertheless, in arthritis as well as with increasing age, a higher porosity of the ECM and the synovial membrane is observed.12,14 Once in the synovial cavity, molecules are subjected to a rapid clearance from the joint space resulting in poor cartilage penetration.13 However, a screening of multiple drugs and formulations for intra articular injections revealed a molecular weight dependent retention time that showed small particles (<10 kDa) had a residence time of several hours, whereas high molecular weight compounds persisted several weeks to months in the synovial fluid.15 The destruction of the tissue during arthritis is mediated by the interplay of inflammatory cytokines, growth factors, and proteases which lead either directly to the degradation of the ECM by cleaving collagen type II and aggrecan or indirectly by altering cell signaling pathways.15 Particularly IL-1β leads to the induction of various degradative matrix metalloproteinases, release of glycosaminoglycans from the cartilage ECM and induction of inflammatory mediators.16 Although disease-modifying anti-arthritic drugs show a high potential to inhibit these factors and are assumed to have a high impact on the treatment of rheumatoid and osteoarthritis, their application is often very costly or related to numerous side-effects due to immunosuppression if used for long-term therapy.3,17 A promising approach to overcome the latter and additionally reduce clearance from the affected tissue is the conjugation of drugs to polymeric scaffolds that are able to efficiently and selectively bind to cartilage and accumulate in the synovial joint.
In the past, various cationic agents were used as targeting ligands for cartilage due to the high amount of anionic species like chondroitin and keratan sulfate and hyaluronic acid within the tissue.9 The binding affinity of quaternary amines to the ECM based on electrostatic interactions with chondroitin sulfate was already reported in 1975 and further demonstrated for several pyridinium compounds in the following decades.18,19 In ongoing studies radiolabeled quaternary amines were administered in vivo for diagnostic applications and dipicolyamine was conjugated to a fluorescent near infrared dye for the imaging of cartilage degeneration.20–23 Recently, a cationic cartilage-specific NIR fluorophore was developed as contrast agent and amine-functionalized tantalum oxide particles have been applied for in vivo imaging of articular cartilage by X-ray computed tomography, whereas nitroxide-covered poly(propyleneimine) and poly(amido-amine) (PAMAM) dendrimers were used as MRI contrast agents.24–26 Other approaches utilized the collagen type II specific peptide sequence WYRGRL as a targeting ligand. The intra articular injection of peptide-functionalized poly(propylene sulfide) nanoparticles showed a size dependent uptake into the cartilage tissue.27 Whereas particles of 38 nm were able to penetrate into the ECM and accumulated in chondrocytes, those with a size of 96 nm did not enter and were found at the cartilage surface. Moreover, Hu et al. combined collagen type II and proteoglycan targeting properties with a therapeutic agent by conjugation of the WYRGRL peptide and pepstatin A to a DOTAM ligand bearing several amino groups to create a cartilage-targeted drug carrier.28 Furthermore, also anionic systems based on lipid nanoparticles covered with chondroitin sulfate have been developed for the targeted delivery of diacerein and aceclofenac.29,30 However, these systems are still in an early stage of development and suffer from poor efficacy as a result of a fast clearance, lack of selectivity, insufficient enrichment in the cartilage ECM, or, off-target side effects due to their accumulation in the reticuloendothelial system (RES), which is related to their physiochemical properties.
Polymeric structures can be prepared with various functional groups in different sizes by which toxicity, renal excretion, and recognition by the RES can be adjusted.31,32 Moreover, the display of multiple ligands on the surface of a polymeric scaffold allows for multivalent binding which leads to an increased binding affinity along with an enhanced selectivity and is hence a promising approach to improve the targeting properties of a compound.33 Systems such as nanogels,34–36 self-assembled supramolecular structures,37 and particles in the nanometer range derived from biocompatible dendritic polyglycerol (dPG)38 represent an ideal platform to target different tissues due to their tunable physicochemical properties. For instance, these architectures can be adjusted concerning their molecular weight, size, stability, and morphology while surface charges can be controlled simultaneously by the modification with anionic39 or cationic40 groups to obtain multivalent binding sites. In previous studies, dPG-based agents were successfully applied as targeted drug delivery systems,41–43 and the high affinity of multivalent dendritic polyglycerol sulfate (dPGS) to inflamed joints was revealed in a RA rat model44 which might have been related to an enhanced accumulation in cartilage tissue. Moreover, the selective targeting of collagenous compartments within native and demineralized ovine bone was recently demonstrated for sulfated and sulfonated dPG anions, whereas bisphosphonated, phosphated, and phosphonated compounds showed a higher affinity to mineralized areas.45 The overall results indicate a high binding potential of polyglycerol-based anions to cartilage and represent promising candidates for the selective and efficient targeting of the tissue in vitro and in vivo.
We herein present the targeting profiles of dye-labeled dPG anions – in particular sulfates, bisphosphonates, phosphates, and a mixed compound containing sulfate and bisphosphonate groups – toward cartilage in vitro and in vivo as well as their interaction with different ECM components. In vitro binding properties toward cartilage were investigated by confocal laser scanning microscopy (CLSM) with native and IL-1β treated cartilage tissue, the latter to mimic the inflamed situation in arthritis. The degradative effects of IL-1β on the cartilage ECM are well known.46 In a collagen-induced arthritis (CIA) rat model, ex vivo histological examinations of the joints and combined investigations by CLSM and scanning electron microscopy (SEM) revealed the targets of the polyanions on the tissue level. In order to identify molecular binding partners, the affinity of prepared anions toward collagen was evaluated by applying a fluorescence-based binding assay while the cellular uptake and cytotoxicity studies with chondrocytes gave distinct biocompatibility profiles.
Materials and methods
General
Mesyl chloride (MsCl), sodium azide, triphenylphosphine (PPh3), 2-iminothiolane hydrochloride, sucrose, N-hydroxysuccinimide (NHS), bovine serum albumin (BSA), methoxy carboxy polyethylene glycol (mPEG-COOH), sodium ascorbate, Dulbecco's phosphate buffered saline (DPBS) buffer, DMF, pyridine, and methanol were obtained from Sigma-Aldrich (Steinheim, Germany). Chloroform was purchased from VWR (Radnor, PA, USA), celite, sulfur trioxide pyridine complex (SO3 Py), and Tween20 from Acros Organics (Geel, Belgium), THF from Roth (Karlsruhe, Germany), N,N-diisopropylethylamine (DIPEA) from Fluka (Buchs, Switzerland), copper sulfate pentahydrate, N-ethyl-N′-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), and ethylendiamine-tetraacetic acid (EDTA) from Merck (Darmstadt, Germany), deuterated solvents from Deutero (Kastellaun, Germany), soluble bovine collagen type II from abcam (Cambridge, UK), and ICC-labeled collagen II antibody from biorbyt (Cambridge, UK). All chemicals were reagent grade and used without further purification. Reactions sensitive to air or moisture were carried out under argon atmosphere using flame-dried glassware and anhydrous solvents. Ultrafiltration was performed in solvent-resistant stirred cells obtained from Millipore (Billerica, Massachusetts, USA) with PLAC-regenerated cellulose membranes (MWCO 1000 g mol−1). Dialysis was performed in benzoylated cellulose tubing purchased from Sigma-Aldrich (MWCO 2000 g mol−1) changing the solvent at least 5 times over a period of 48 h. SEC was performed with Sephadex™ G-25 superfine (Sigma Aldrich, Steinheim, Germany) in distilled water under room temperature and pressure. 1H-, 13C-, and 31P-NMR spectra were recorded on a Jeol ECX 400 spectrometer or on a Bruker Biospin Avance 700 spectrometer. Chemical shifts δ were reported in ppm using the deuterated solvent peak as the internal standard (CDCl3: δ (1H) = 7.26 ppm, δ (13C) = 77.16 ppm; MeOD-d4: δ (1H) = 3.31 ppm, δ (13C) = 49.00 ppm; D2O: δ (1H) = 4.79 ppm; acetone-D6: δ (1H) = 2.05 ppm, δ (13C) = 29.84, 296.26 ppm; DMSO-D6: δ (1H) = 2.50 ppm, δ (13C) = 39.52 ppm). The average polymer-to-dye ratio was calculated by UV-Vis spectra from a 6 μM solution in DPBS at pH 7.4 recorded on a LAMBDA 950 UV/Vis/NIR spectrometer (PerkinElmer, Waltham, MA, USA) at 25 °C by applying a molar extinction coefficient of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 for ICC λ (552 nm) and of 100000 M−1 cm−1 for 6S-ICG (λ = 785 nm). IR measurements were recorded on a Nicolet Avatar 320 FT-IR equipped with a DTGS detector from 4000 to 650 cm−1 and evaluated with the software EZ OMNIC ESP. Wavenumbers νmax were reported in cm−1; the intensities of the absorption bands were assigned as strong (s), medium (m), and weak (w). Combustion analysis was performed on a VARIO EL III instrument (Elementar, Hanau, Germany) using sulfanilic acid as the standard. DLS and ζ-potential measurements were carried out on a Zetasizer Nano ZS (Malvern Instruments Ltd, Worcestershire, U.K.) equipped with a 4 mW He–Ne laser (λ = 633 nm, NIBS) that operated with a 173° scattering angle (backscatter). Hydrodynamic diameters were determined in UV-transparent disposable cuvettes (UltraVette, 8.5 mm, Brand, Wertheim, Germany) at 25 °C. Samples were dissolved in DPBS (DPBS, 1×, without Ca2+, Mg2+, pH = 7.4, Sigma-Aldrich, Steinheim, Germany) at a concentration of 2 mg mL−1. The stated values are the mean of at least 15 independent measurements. ζ-Potential measurements were conducted at a concentration of 2 mg mL−1 in phosphate buffer (PB, 10 mM, pH 7.4) at 25 °C in folded DTS 1060 capillary cells (Malvern Instruments Ltd, Worcestershire, U.K.). Data evaluation was performed with Malvern Zetasizer Software 6.12. The stated values and standard deviations are the mean of at least five independent measurements with 15 scans each and are based on the Smoluchowski model. ICP-MS measurements were conducted on an Element 2 (Thermo Scientific, Waltham, MA, USA).
000 M−1 cm−1 for ICC λ (552 nm) and of 100000 M−1 cm−1 for 6S-ICG (λ = 785 nm). IR measurements were recorded on a Nicolet Avatar 320 FT-IR equipped with a DTGS detector from 4000 to 650 cm−1 and evaluated with the software EZ OMNIC ESP. Wavenumbers νmax were reported in cm−1; the intensities of the absorption bands were assigned as strong (s), medium (m), and weak (w). Combustion analysis was performed on a VARIO EL III instrument (Elementar, Hanau, Germany) using sulfanilic acid as the standard. DLS and ζ-potential measurements were carried out on a Zetasizer Nano ZS (Malvern Instruments Ltd, Worcestershire, U.K.) equipped with a 4 mW He–Ne laser (λ = 633 nm, NIBS) that operated with a 173° scattering angle (backscatter). Hydrodynamic diameters were determined in UV-transparent disposable cuvettes (UltraVette, 8.5 mm, Brand, Wertheim, Germany) at 25 °C. Samples were dissolved in DPBS (DPBS, 1×, without Ca2+, Mg2+, pH = 7.4, Sigma-Aldrich, Steinheim, Germany) at a concentration of 2 mg mL−1. The stated values are the mean of at least 15 independent measurements. ζ-Potential measurements were conducted at a concentration of 2 mg mL−1 in phosphate buffer (PB, 10 mM, pH 7.4) at 25 °C in folded DTS 1060 capillary cells (Malvern Instruments Ltd, Worcestershire, U.K.). Data evaluation was performed with Malvern Zetasizer Software 6.12. The stated values and standard deviations are the mean of at least five independent measurements with 15 scans each and are based on the Smoluchowski model. ICP-MS measurements were conducted on an Element 2 (Thermo Scientific, Waltham, MA, USA).
Cytotoxicity
Human articular cartilage was removed from femur heads or tibia plateaus of patients receiving hip or knee replacements in accordance with the institutional ethical committee of the Charité-Universitätsmedizin Berlin, Campus Benjamin Franklin (EA4063/06). Chondrocytes were isolated by enzymatic digestion of cartilage ECM and resuspended in chondrocyte growth medium as previously described.47 The metabolic activity of chondrocytes was analyzed by an alamarBlue® assay (Life technologies, Darmstadt, Germany) using cells from three human donors (mean age: 52.7 years, 2 male and 1 female donor) seeded in a 96-well plate at a density of 294.118 cells per mm2. 24 h after seeding, cells were incubated with the respective unlabeled polymer (Table S2, ESI†) or control compound at concentrations of 10−3, 10−5, and 10−6 mol L−1 for 24 h. 10% of alamarBlue® test reagent was added to the culture medium (Biochrom, Berlin, Germany) and the read out was performed by measuring the absorbance at a wavelength of 570 nm and a reference wavelength of 595 nm every hour for 24 h using a Genios microplate reader (Tecan Group, Männedorf, Switzerland). Glycerol served as positive non-toxic and sodium dodecyl sulfate (SDS, Carl Roth, Karlsruhe, Germany) as toxic negative control at a concentration of 10−3 mol L−1 each. Stated cell activity refers to 100% of the reference (culture medium). Experiments were conducted independently as triplicates with error bars representing the standard deviation.Uptake by articular chondrocytes
Human articular chondrocytes were seeded onto poly-L-lysine coated glass cover slides (25000 cells per well) placed in 6 well plates. Cells were allowed to adhere for 24 h, incubated with the respective ICC-labeled polymer (Table S1, ESI†) at a concentration of 10−6 mol L−1 for 24 h, and fixed for 15 min in 4% paraformaldehyde ready to use solution (PFA, Santa Cruz Biotechnology, Heidelberg, Germany). The cytoskeleton was stained using Phalloidin-Alexa488 (Santa Cruz Biotechnology, Heidelberg, Germany) and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma Aldrich, Munich, Germany) was used for counter-staining of the cell nuclei. Images were taken with a DMi8 confocal microscope (Leica Microsystems, Wetzlar, Germany) equipped with a 20× oil immersion microscope objective. Three independent experiments with articular chondrocytes (femoral head cartilage) from three human donors (mean age: 82.5 years, 2 female and 1 male donors) were performed.
Penetration depth into native and IL-1β treated cartilage
Vital cartilage was harvested from knee joints of 4–6 month old pigs (derived from the slaughter house) and chips with a diameter of 6 mm were prepared using a biopsy punch. The probes were pre-incubated for 72 h (37 °C, 5% CO2) in cell culture medium with or without 10 ng mL−1 IL-1β, before treatment with the respective ICC-labeled polymers (Table S1, ESI†) for 24–72 h at a concentration of 10−6 mol L−1, embedded in TissueTek (Sakura Finetek, Staufen, Germany), and sliced using a cryotome. Controls, which were treated with glycerol-ICC were included in some experiments for detection of unspecific background fluorescence (not shown). Cryosections were stained with DAPI to visualize the cell nuclei. Images were taken with the DMi8 confocal microscope (Leica Microsystems, Wetzlar, Germany). In order to mimic inflamed tissue, cartilage chips were treated with IL-1β (10 ng mL−1) for 72 h prior to incubation with the respective ICC conjugate.Collagen type II binding affinity
Binding affinity studies were conducted on a 96-well plate (Nunc FluoroNunc MaxiSorp, Thermo Scientific, Dreieich, Germany). Each well was coated with dopamine containing hPG amine for 30 minutes as previously described.48 Soluble bovine collagen type II (1 mg mL−1 in 0.5 M acetic acid, pH 4.5) was immobilized on the surface over night at 37 °C via a NHS/EDC coupling procedure in DPBS buffer (1×, pH 7.4).48 In order to block remaining amine groups, the wells were incubated with a mixture of mPEG-COOH (2 kDa, 0.1 mg mL−1), NHS (0.01 mg mL−1), and EDC (0.1 mg mL−1) in DPBS (1v/1v/1v) over night at 37 °C, and washed with DPBS over 4 times. To prove the successful immobilization of collagen type II on the surface, a collagen type II antibody was used as a positive control. For that purpose, a blocking solution (DPBS++ buffer, 2% BSA, 5% sucrose) was added for 90 min at room temperature. Any residues were removed by addition of a washing solution (DPBS++ buffer, 1% BSA, 0.05% Tween20) to the wells followed by PBS over 4 times each. Subsequently, wells were incubated using an ICC-labeled collagen type II antibody (1 mg mL−1, 1:500 dilution in DPBS++ buffer and 0.1% BSA) for 1 h at room temperature followed by washing with the washing solution and DPBS over 4 times each. To investigate the binding affinity of the prepared polymers to collagen type II, the respective ICC-conjugated polyanion was added to the wells and incubated for 24 h at 37 °C at a concentration of 10−5 mol L−1 in DPBS directly after immobilization of collagen type II and washed with DPBS buffer over 4 times. The fluorescence intensity signal was measured by an Infinite 200 PRO multimode microplate reader (Tecan, Männedorf, Switzerland) using an excitation wavelength of 525 nm and an emission wavelength of 570 nm. Values were determined with an i-control software from 16 reads per well from three independent measurements.
Animal model
Lewis rats were purchased from Charles Rivers, fed a standard diet and had access to drinking water ad libitum. Animal studies were carried out in accordance with internationally approved standards, have been validated and approved by the State Office of Health and Social Affairs, Berlin (LAGeSo) (no. G0427). Animals were killed in deep anesthesia by intravenous injection of T61 ad us. vet. (Intervet, Germany) at a dose of 2 mL per animal after fluorescence imaging. RA was induced in 15 female Lewis rats (150 ± 10 g on arrival) by subcutaneous injection of soluble bovine collagen type II (collagen induced arthritis (CIA) model) as already published.49–51 Disease scoring was performed 14 days post injection (p.i.) by evaluating the clinical symptoms of the right and left ankle joints separately as previously described.44 Comparable to other studies using the same animal model, rats developed severe to mild RA with half of the joints remaining unaffected by the disease.Imaging device and image processing
The in vivo imaging was performed with a camera-based fluorescence imaging system using an EMCCD Camera (iXon BV887, Andor Technology, Belfast, UK). A light emitting diode (LED) with λmax = 750 nm, a short pass filter (λ = 750 nm, LCLL 750), and an illuminated area of 20 cm × 20 cm was applied as excitation source. Adjusted parameters were: exposure time 100 ms; EM gain 3000; shift time 0.9 ns per line; delay 18.8 ms; preamplifier 4.6; detector temperature −65 °C; binning 2 pixels by 2 pixels; accumulation series with 100 frames, and accumulation cycle time 200 ms. For suppression of excitation light, two fluorescence long pass filters (λ50% = 800 nm, 5 OD, 800 LP, 800 ALP) were applied and the images were analyzed as published by extracting measured values in the regions of interest (ROI).51 Briefly, ROI were set and illumination intensities were corrected for background. Images are normalized to the mean intensity from a ROI placed over the reference standard cube. Mean values are determined in ROI for the right and left ankle.In vivo and ex vivo imaging and fluorescence histology
For in vivo imaging, 6S-ICG conjugates were injected once in sterile PBS at a dose of 2 mg kg−1 intravenously (i.v.) via the tail vein 14 days after inducing RA. Images were taken over 24 h after administration of the dye conjugate. For histological recovery, the rats additionally received the respective ICC-conjugate once i.v. at a dose of 1 mg kg−1. Fluorescence histology was performed ex vivo on cryo sections (5 μm) of tibiotarsal articulations. For this purpose, the entire feet were removed 3 h p.i., toes were cut off, and cryo sections along the longitudinal-sagittal plane of the respective joint were prepared, which were put on cellotape, and fixed with acetone for 10 min. Remaining probes were stored at −80 °C in TissueTek until further usage (see SEM and CLSM imaging). Histology images were taken with a Leica DMRB fluorescence microscope (Leica Microsystems, Wetzlar, Germany) equipped with a Spot digital camera and a Spot 32 v2.1 software (Diagnostic Instruments, Sterling Heights, MI) using the 550 nm channel for excitation. Cell nuclei were stained with DAPI.Sample preparation and scanning electron microscopy (SEM)
For SEM imaging, feet from rats that additionally received the respective ICC-conjugate once i.v. at a dose of 1 mg kg−1 (see ex vivo imaging) were used. For this purpose, TissueTek was removed by hand from the respective foot and the TissueTek-free joint sample was dehydrated over one week by immersion in an ethanol series increasing the amount of ethanol from 50% to 100%. Each foot was embedded in a mixture of methyl methacrylate (MMA), nonylphenyl-polyethyleneglycol acetate and dibenzoyl peroxide, then heated in an oven (1 day at 40 °C, 1 day at 48 °C, 1 day at 55 °C), cut to length, ground, and polished in an automatic polisher (Logitech PM5, Logitech Ltd, Glasgow, UK). SEM images of the embedded joint samples were taken with a FEI-Quanta 600FEG electron microscope (FEICompany, Oregon, USA) at a working distance of 9.6 mm in low vacuum mode (0.75 Torr). A large field detector (LFD) at 5 kV with a high field width of 4.69 mm was used measuring the back-scattered-electron signal from the sample surface. The joint of an untreated rat was used for the adjustment of the fluorescence intensity, which was applied for all other probes.Confocal laser scanning microscopy (CLSM)
For CLSM imaging, feet from rats that additionally received the respective ICC-conjugate once i.v. at a dose of 1 mg kg−1 (see ex vivo imaging) were used. CLSM micrographs were taken with a Leica TCS SP8 (Leica Microsystems, Wetzlar, Germany) equipped with a HCPL Fluotar 5×/0.15 dry microscope objective. The emission was measured at a range from 405 nm to 633 nm and the excitation wavelength was set to 580 nm. Image stacks were measured with a spatial pixel resolution of 1 μm × 1 μm until a penetration depth of 50 μm. All pictures were edited and processed in ImageJ. All micrographs were taken using the same laser- and microscope settings.Results and discussion
Synthesis
In order to monitor their interaction with cartilage and different ECM components, indocarbocyanine (ICC)-labeled dendritic polyglycerol (dPG) anions, which varied in number of functional groups and their anionic moiety, were synthesized. Consequently, binding properties related to the anionic nature of the polymer could be systematically studied to identify the most potential agents to target healthy and malfunctioning cartilage tissue.The polyanions derived from a neutral dPG scaffold (1) with a molecular weight of Mn = 6000 g mol−1, a degree of branching of 60%, and a poly dispersity index (PDI) of ≈1.6 which was synthesized via a ring-opening multi-branching polymerization (ROMBP) as previously described.52 Dye conjugates originated from the respective azide precursor by reduction with triphenylphosphine (PPh3) and subsequent reaction with the dye maleimide and 2-iminothiolane according to a published procedure.38,50 ICC-labeled dPG (3) served as a negative control for in vitro studies and derived from dPG azide (2) with a degree of functionalization (dF) of 17% which was synthesized by partial mesylation and azidation of the neutral scaffold.40,45 Subsequent reduction of azide groups and dye coupling gave dPG-ICC (3). The synthesis of ICC-labeled dPG sulfate (dPGS-ICC 5) was accomplished by sulfation of 1 with SO3 pyridine complex yielding dPGS azide (4) with a degree of sulfation of 83%, subsequent reduction, and dye conjugation (Fig. 1).45,53 For dPG bisphosphonate with a low dF (dPGBP7%6) a partial copper-catalyzed azide–alkyne click reaction was applied on dPG azide (2) giving the compound with a dF of 7%.39,45 Reduction of remaining azide groups and subsequent dye coupling yielded dPGBP7%-ICC (7) (Fig. 1). The mixed anion containing sulfate and bisphosphonate groups (dPGS/BP7%10) was obtained by sulfation of half azide-functionalized dPG (8) giving dPGS azide (9) with a degree of sulfation of 50% as determined by elemental analysis, followed by partial click reaction of the bisphosphonate alkyne (Fig. 2). A higher functionalization with the bisphosphonate (BP) alkyne was either not possible due to electrostatic shielding or led to precipitation of the charged product. However, with a dPGS bearing a lower amount of azide groups, the coupling of BP alkyne was not observed, whereas the sulfation of dPGBP7% led to degradation of the anionic group as evident from absent signals in the 31P-NMR. Subsequent azide reduction of 10 and dye conjugation gave dPGS/BP7%-ICC (11). The synthesis of ICC-functionalized dPG bisphosphonate (dPGBP-ICC 15) and phosphate (dPGP-ICC 16) was accomplished by click coupling of the respective anionic alkyne on fully functionalized dPG azide (12) giving dPGBP-N3 (13) or dPGP-N3 (14), followed by the reduction of remaining azide groups and subsequent dye conjugation (Fig. 2).39,45 The respective alkyne precursor was synthesized according to a published procedure.39,45 In order to maintain their native physicochemical properties and avoid an unspecific interaction with cartilage due to conjugation with ICC, compounds were functionalized with less than 0.2 dye per polymer in average (Table S1, ESI†), as confirmed by UV-Vis spectroscopy in PBS at 25 °C, in order to avoid entities with more than one dye at a polymer molecule. However, for in vivo imaging, a near infrared (NIR) dye was required in addition to a higher dye loading per polymer to obtain an enhanced contrast. For this purpose, a sulfonated indocyanine green derivative (6S-ICG) was introduced into a dPGS (Fig. S2, ESI†) or dPGBP7% (Fig. S3, ESI†) backbone by the common coupling procedure using 6S-ICG-maleimide which gave the conjugate with a dye to polymer ratio of 1.1 (dPGS-6S-ICG 5a) or 0.9 (dPGBP7%-6S-ICG 7a) after purification. The presence of remaining free dye after purification can be ruled out by applying size exclusion chromatography (SEC) (Fig. S13, ESI†). For cytotoxicity studies, determination of the zeta potential, and dynamic light scattering (DLS) measurements, comparable unlabeled compounds were synthesized (Table S2, ESI†). The prepared polyanions showed molecular weights between Mn = 8000 g mol−1 (dPGBP7%7) and Mn = 30000 g mol−1 (dPGBP 15) with a zeta potential of −13 to −27 mV in phosphate buffer (PB) at pH 7.4 and a hydrodynamic diameter of 7–8 nm in PBS which is in accordance to previously reported data.39
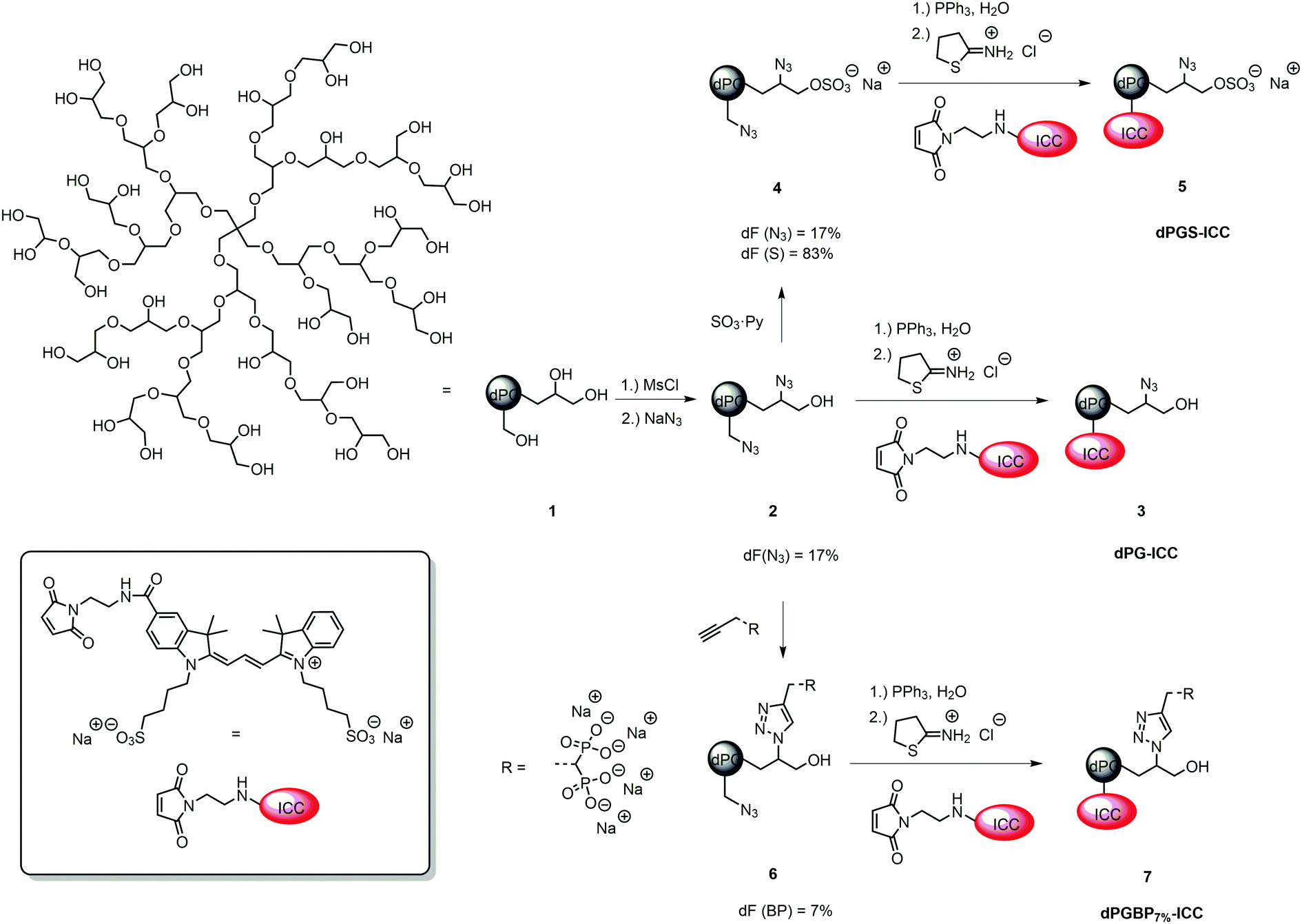 | ||
| Fig. 1 Idealized schematic structure of the dPG scaffold (1) (Mn = 6000 g mol−1) and the reaction pathway for the synthesis of ICC-labeled dPG (3), dPGS (5), and dPGBP7% (7) from low functionalized dPG azide (2). | ||
 | ||
| Fig. 2 (a) Synthesis of dPGS/BP7%-ICC (11) via mesylation, azidation, and sulfation, followed by click reaction of alkyne-functionalized bisphosphonate, reduction, and dye conjugation via maleimide coupling. (b) Reaction pathway for the preparation of dPGBP-ICC (15) and dPGP-ICC (16) via mesylation, azidation, click reaction of the respective alkyne precursor, reduction, and dye conjugation via maleimide coupling. | ||
Penetration depth into native and inflamed articular cartilage in vitro
The binding affinity to explanted native cartilage was already described for dPGS, finding a maximum infiltration after 24 h.47 However, neither binding studies with other dPG anions nor with inflamed cartilage in vitro have been conducted yet. In order to evaluate the penetration depth of the anionic polymers into native tissue in vitro, cartilage chips from porcine joints were incubated for 24 h and 72 h with the respective ICC-labeled polymer in culture medium at a concentration of 10−6 mol L−1. To mimic inflamed tissue as reported previously,16,46 samples were pre-treated with IL-1β for 72 h before incubation with the respective polyanion. Cartilage cryo slices were stained with DAPI (blue) to visualize the cell nuclei. Binding and penetration depth into the ECM were verified by applying CLSM (Fig. 3). The compounds, which were not taken up serve as internal control depicting no unspecific ICC-binding by the cartilage ECM. Previous experiments (not shown), which included also a glycerol-ICC-control, known not to be taken up by cells, revealed no unspecific fluorescence. Neutral dPG (1) did not show any interaction with native or IL-1β treated cartilage, which confirmed the non-targeting properties and the inert character of dendritic polyglycerol towards tissues and surfaces, which was already proven in various in vitro and in vivo studies.45,54–56 In accordance with the previously described uptake study, no internalization into chondrocytes and, moreover, only a very weak binding to the ECM of native cartilage were observed for dPGBP7% (7). In comparison, a slightly stronger binding to the surface and an increased infiltration into subjacent layers were found for dPGBP (15) and dPGP (16), particularly after 72 h. Contrary to our expectations from previous studies,47 also dPGS (5) showed a comparably low binding affinity to the native porcine tissue. However, previous in vitro experiments were conducted on human articular cartilage harvested during joint replacement surgeries in which slight inflammation due to osteoarthritis or age-associated degenerative changes cannot be ruled out, which could result in the presence of molecular targets, absent in young porcine tissue, and presumably led to variations in the binding behavior of dPGS. This assumption is in agreement with the observation that in response to a 72 h lasting dPGS treatment combined with a 72 h pre-incubation with IL-1β, a substantial ECM binding and internalization of dPGS could be observed (Fig. 3). Surprisingly, dPGS/BP7% (11) showed an exceptionally high binding affinity to the ECM, a high penetration depth, and a high accumulation in the cell lacunas. In the absence of IL-1β, for all polyanions a maximum enrichment in native cartilage was determined after 24 h, which decreased over 72 h.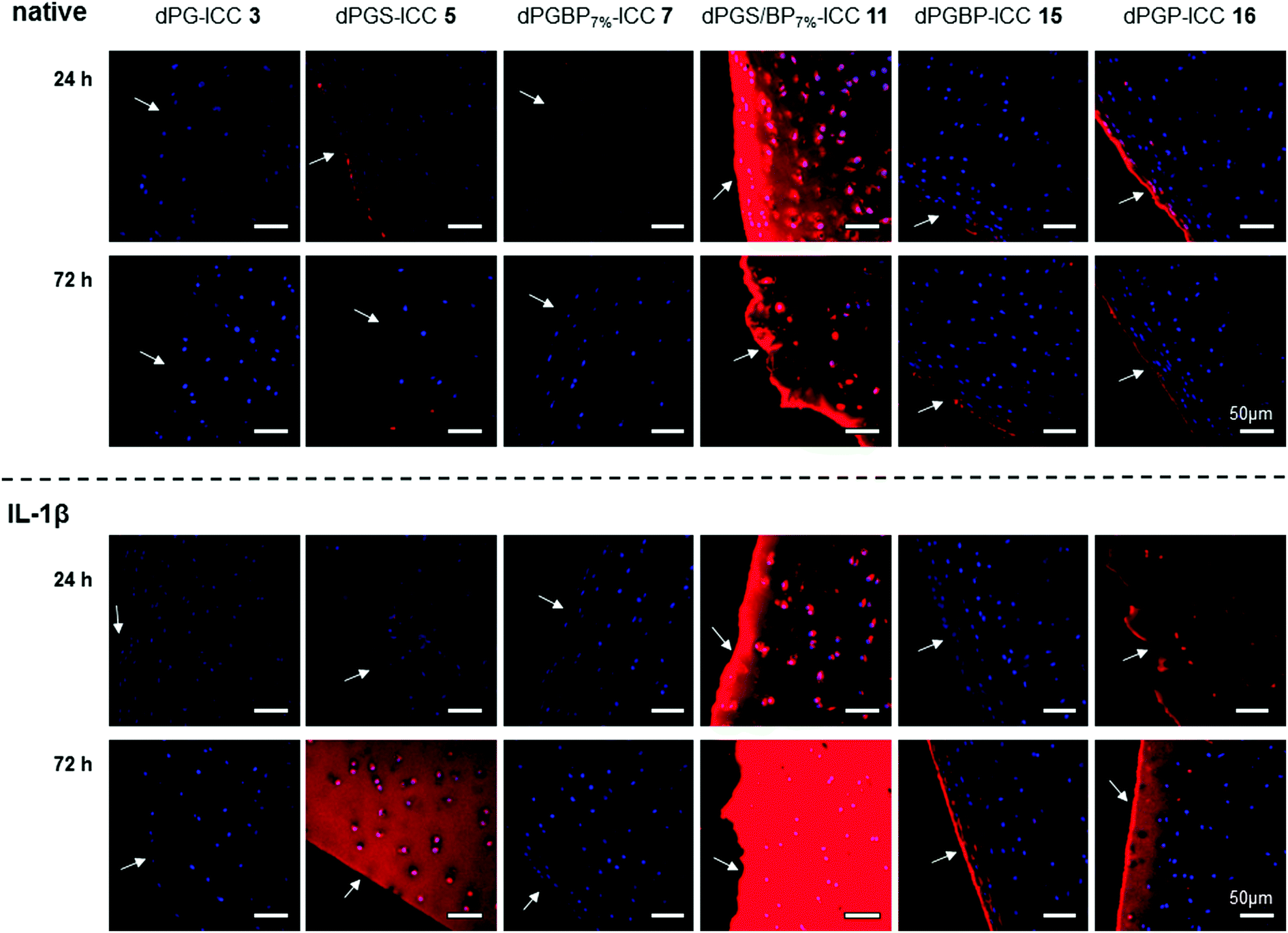 | ||
| Fig. 3 Penetration of ICC-labeled dPG anions (c = 10−6 mol L−1) into native and IL-1β treated porcine cartilage 24 h and 72 h after incubation at 37 °C and 5% CO2. The cell nuclei were stained with DAPI (blue). Arrows highlight the respective cartilage surface. In order to mimic inflamed cartilage, samples were treated with IL-1β (10 ng mL−1) for 72 h prior to incubation with the respective dye conjugate. | ||
Although inflammatory processes were thought to increase the penetration properties of the polyanions, IL-1β treatment resulted in a minor binding of dPGS (5), dPGBP7% (7), and dPGBP (15) within the first 24 h. Surprisingly, 72 h after incubation, a much stronger affinity to the ECM was determined for dPGBP and dPGS with the latter greatly accumulating in deeper areas and cell lacunas. In comparison, for dPGP (16) a strong penetration of the tissue and uptake into articular chondrocytes was already observed after 24 h. However, fluorescence signals within the chondrocytes diminished after 72 h. dPGS/BP7% exhibited an even higher affinity to the ECM of IL-1β treated tissue after 72 h as compared to previous time points and native cartilage. These results indicate that the penetration depth and binding affinity to the ECM of cartilage is not only dependent on the number of anionic moieties but also on the anionic group itself. Moreover, a combination of different anionic functionalities could alter the targeting properties of a polymer. However, the optimal ratio of different moieties would have to be investigated systemically and adjusted for the respective application.
Binding affinity to collagen type II
In recent studies with demineralized bovine bone, dye-labeled dPG anions showed varyingly strong affinities to collagenous compartments depending on the anionic group of the polymer.45 While the main collagen type within the organic matrix of bone is collagen type I, the most abundant component of the ECM in articular cartilage is collagen type II.57–59 Since dPG-based polyanions showed a similar binding behavior to cartilage and demineralized bone dependent on the anionic moiety, collagen represents the most potential molecular binding partner. However, no binding studies with dendritic polyanions toward collagen have been conducted yet. In order to assess quantitative affinities, a fluorescence read out was conducted on a 96-well plate, with immobilized collagen type II, after incubation with ICC-labeled polymers. To ensure a homogenous distribution of the collagen on the surface, a dopamine-containing hyperbranched polyglycerol (hPG) amine was conjugated to the well prior to collagen immobilization.48 The remaining amine groups were blocked with mPEG-COOH using an NHS/EDC coupling procedure, and ICC-conjugated polymers (c = 10−5 mol L−1 in PBS) were incubated for 24 h at 37 °C. To prove the successful coupling of the collagen, an ICC-labeled antibody specific to collagen type II was used as a positive control (Fig. S15, ESI†). In order to compare the binding properties among the polymers, fluorescence signals of the dPG anions were correlated with the dye loading, corrected for the background signal, and are given as percentage of collagen type II binding (Fig. 5). Unspecific binding of the compounds to the non-functionalized well plate as well as the blocked surface can be ruled out since no significant fluorescence signal in both cases was observed (Fig. S15, ESI†).With a binding of less than 5% compared to the mixed anion (11), neutral dPG (3) showed a very weak affinity to collagen, as was already expected from the previous penetration depth experiments with native cartilage. A remarkably low affinity toward collagen was also found for dPGBP (15) which bound with less than 10%. Surprisingly, dPGS (5) and dPGP (16) showed similar results, although they were found to deeply penetrate the ECM of cartilage, accumulating in chondrocyte lacunas of subjacent layers in the inflamed state. In comparison dPGBP7% (7) exhibited a slightly increased affinity to collagen of around 11% whereas dPGS/BP7% (11) provided far greater affinity. Comparable binding patterns are also conceivable for other collagen types, however, experiments to confirm this assumption have not been conducted, as collagen type II is the most abundant type in cartilage. In the previous studies investigating the binding properties of glycosaminoglycans like chondroitin sulfate and heparin to collagen, molecules with a higher charge density showed a higher affinity due to electrostatic forces.60,61 Assuming that electrostatic interactions play a crucial role in these binding events, the exceptionally high affinity to collagen of the mixed anion was rather unexpected since dPGS in comparison showed only minor binding properties, although it provided more potential binding motifs and a comparable charge density (Table S2, ESI†). Moreover, a zeta potential in the same range (∼−20 mV) was also determined for dPGP and dPGBP, while both compounds exhibited a low affinity to collagen. Hence, the surface charge seems to play a minor role, whereas the nature of the anionic moiety is more likely to be important for binding and has to be taken into account, when it comes to designing molecules for targeting collagen or cartilage. However, these findings are in accordance with experiments conducted on the penetration depth into native and inflamed cartilage, which indicate that one of the binding partners of the mixed anion is indeed collagen type II, whereas the molecular targets of all other polyanions still remain unclear. Recent studies with dPGS revealed a high protein absorption profile of the polyanion, probably because of its interaction with basic amino acid residues.62–64 Since the ECM of cartilage contains various non-collagenous proteins, the binding of the polyanions to these components has to be further investigated to identify their molecular targets.57 Moreover, an interaction with proteoglycans, which may lead to an enhanced uptake into the cartilage tissue, cannot be fully excluded. However, binding is considered unlikely due to electrostatic repulsion of the anions by anionic residues of the proteoglycans.
Cellular uptake and cytotoxicity
Although dPG and dPGS have been intensively investigated for their uptake properties by different cell types, only a few studies have been conducted on other anionic polyglycerol derivatives.45,47,65–68 In order to examine the uptake of the prepared polyanions by human articular chondrocytes, cells were incubated with ICC-labeled nanoparticles (Table S1, ESI,†c = 10−6 mol L−1) for 24 h (Fig. 4) and 72 h (not shown) at room temperature. To demonstrate their subcellular location, cell nuclei were stained with DAPI (blue) and actin cytoskeleton was stained with phalloidin-Alexa488 (green). In compliance with previous findings, no internalization of the neutral polymer (3) was observed.45,65,68 Moreover, also dPGBP7% (7) did not show any significant interaction, which indicates that a minimum content of anionic moieties on the polymer is needed for cell recognition. However, to test this assumption, the uptake of nanoparticles, which vary in size, charge, and number of anionic groups, would have to be investigated, since a size-dependent internalization via an endocytotic pathway is also possible, as already described for the neutral polymer.68 In comparison, all other polyanions were incorporated by the cells and were predominately found at the perinuclear region. The strongest uptake was found for dPGP-ICC (16) assuming its internalization into the nucleus due to the strong overlap of the DAPI signal and the fluorescence of the dye-labeled anion. Stainings of the actin-cytoskeleton at 24–72 h revealed no changes due to toxic effects e.g. morphological alterations of the cytoskeleton by the tested compounds. However, it must be noted that no quantitative description can be deduced from these results due to the different dye contents per polymer (Table S1, ESI†). The influence of dendritic polyglycerol anions on the metabolic activity of human chondrocytes was measured by an alamarBlue® assay after incubation of comparable non-labeled polymers (Table S2, ESI†) at concentrations of 10−3, 10−5, and 10−6 mol L−1. The relative metabolic activity was determined 24 h after treatment by UV-Vis spectroscopy measuring the amount of reduced alamarBlue® by colorimetric change. None of the synthesized compounds showed a significant cytotoxicity in chondrocytes within the tested concentration range, except for highly functionalized dPGBP (15) at a concentration of 10−3 mol L−1 (Fig. S14, ESI†). However, a metabolic activity over 80% of the untreated or glycerol treated controls was still found, which was rather unexpected, since dPG anions showed a higher toxicity in murine fibroblasts.45 Surprisingly, in all other cases, a slightly increased metabolic activity was observed after incubation with the respective compound, which indicates a high cell compatibility with the prepared probes. An insufficient cellular uptake of the compounds leading to a low cytotoxic effect can be ruled out since all polyelectrolytes except for dPG and dPGBP7% were internalized by chondrocytes. Moreover, an influence on the cell metabolic activity of remaining copper used as catalyst during the alkyne–azide click coupling can be excluded, because the amount of copper ions was in the ppb range as determined by inductively coupled plasma mass spectrometry (ICP-MS). In conclusion, these results indicate a remarkable high chondrocyte compatibility with the prepared polyanions. Even at 10−3 mol L−1 concentration, the compounds did not show any severe cytotoxicity, although they underwent cellular uptake except for the neutral polymer and dPGBP7%. However, non-toxic long-term consequences remain to be proved in vivo for all compounds tested here.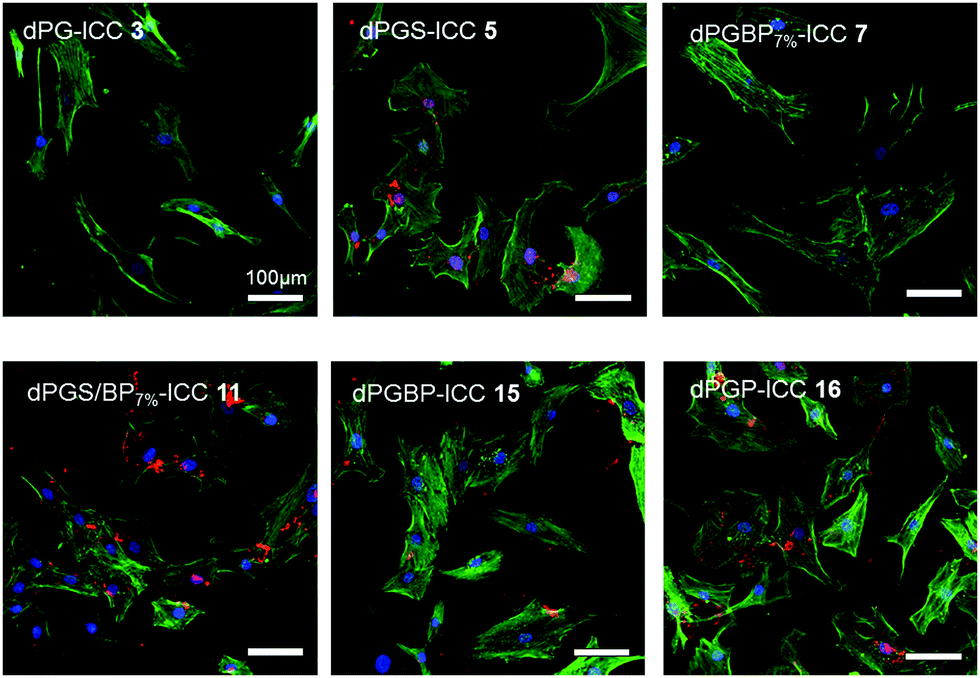 | ||
| Fig. 4 Uptake of ICC-labeled dPG derivatives at a concentration of 10−6 mol L−1 by cultured human articular chondrocytes 24 h after incubation at 37 °C and 5% CO2. The cell nuclei were stained with DAPI (blue) and the actin cytoskeleton was stained with phalloidin-Alexa488 (green). | ||
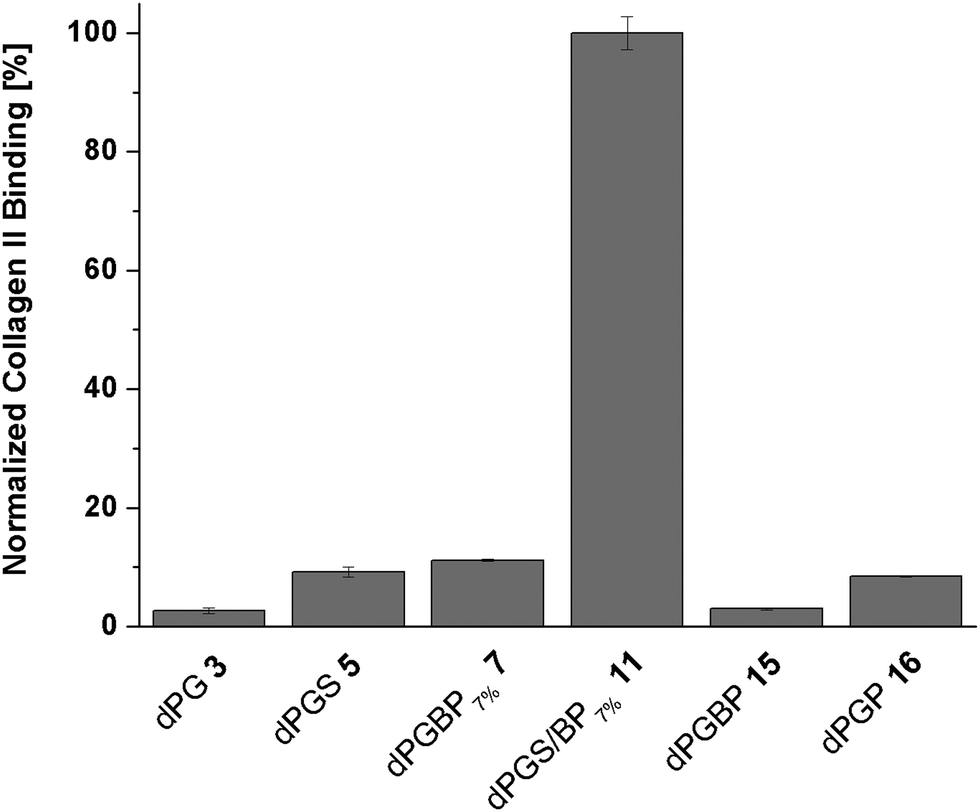 | ||
| Fig. 5 Normalized collagen type II binding of ICC-labeled dPG anions to collagen type II after incubation over 24 h at 37 °C (c = 10−6 mol L−1) in PBS pH 7.4 determined by a fluorescence read out. The binding was corrected for the background signal and unspecific binding to the non-functionalized surface. Normalization was performed due to different amounts of dyes per polymer. | ||
In vivo fluorescence imaging and fluorescence histology of rat joints
The insufficient selectivity between different tissues and undesired toxic side effects are one of the major drawbacks for the efficient drug delivery to a certain target. With a hydrodynamic diameter of 7–8 nm, the prepared dendritic polyanions were below the threshold for renal clearance32 and were expected to be safely and rapidly excreted from the body. In a previous study, highly functionalized dPGBP was found to exhibit a remarkably high affinity to mineralized compartments in bone by complexation of calcium ions.45 However, applied in vivo, highly functionalized BP polymers as well low-molecular weight compounds bear the risk of severe side effects such as osteonecrosis or esophageal cancer.69 The utilization of bisphosphonated polymers with a low degree of functionalization is a promising approach to overcome this issue, and we assume that the selective targeting of bone in vivo can be realized with dPGBP7% since this compound has a minor affinity to cartilage in vitro. In contrast, dPGS was successfully applied as dye conjugate for the imaging of inflamed joints in a CIA rat model, which might also be related to an enhanced affinity toward articular cartilage besides binding L-selectin and complement factors.44,70 However, no experiments have been done yet to clarify this assumption. Although dPGS/BP7% showed the highest affinity to cartilage in vitro, bisphosphonate groups could influence the binding properties to bone in vivo and could result in a decreased selectivity. Hence, dPGS was used for the further studies, which also showed a high affinity toward IL-1β treated cartilage.In order to investigate their targeting properties of bone and cartilage in vivo, 6S-ICG-conjugates of dPGS and dPGBP7% were applied as imaging agents in healthy and CIA rats, the latter induced by subcutaneous injection of soluble bovine collagen type II. After 14 days 6S-ICG-conjugates were administered intravenously and near infrared (NIR) fluorescence imaging was performed at 800 nm over 24 h. Fluorescence intensities were quantified in vivo by normalization with a standard fluorescent cube (Fig. 6). In healthy animals, undergoing no treatment with bovine collagen type II, both compounds were found exclusively at sites of injection, whereas an enrichment in inflamed joints was observed in CIA rats depending on the clinical score (Fig. S16, ESI†). For dPGS-6S-ICG, an accumulation was already demonstrated after 10 min p.i. with a maximum enrichment after 1 h (data not shown), which is comparable to previously reported studies.44 In contrast, dPGBP7%-6S-ICG (7a) slowly accumulated (Fig. S17, ESI†) in the joints and had a maximum enrichment 24 h after injection. However, fluorescence signals were comparably weak and not only occurred in the inflamed articulations but throughout the whole foot and the knee joints (Fig. 6).
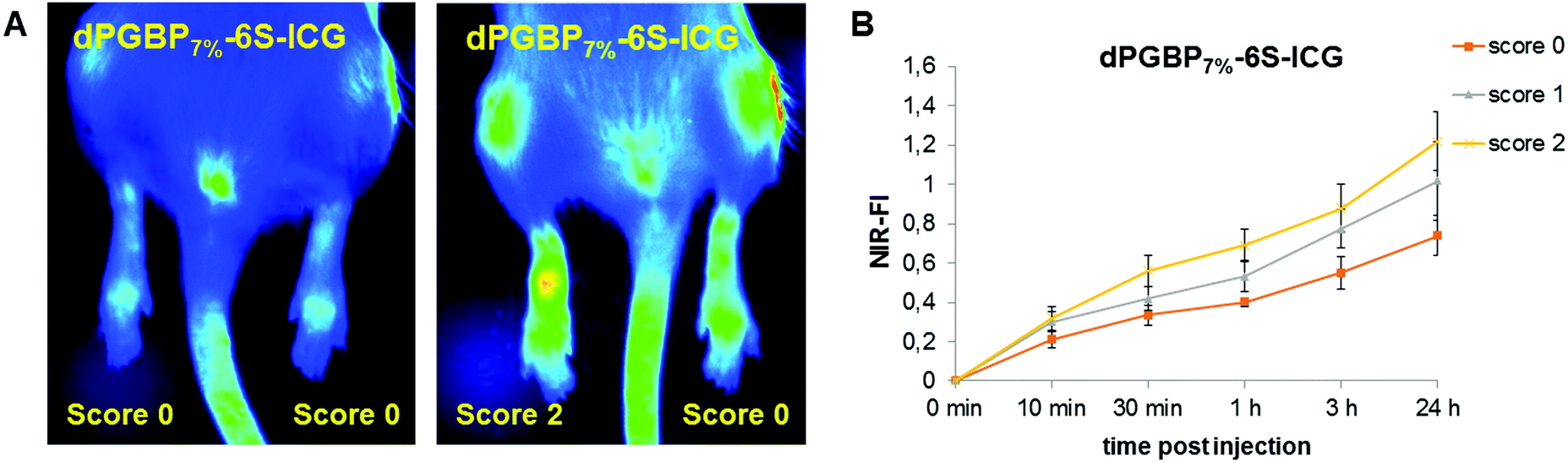 | ||
| Fig. 6 (A) Near infrared (NIR) images of rats with collagen induced rheumatoid arthritis @ 800 nm showing different scores, 24 h after intravenous injection of dPGBP7%-6S-ICG (7a). (B) NIR-fluorescence intensity (NIR-FI) of rat joints with different clinical scores normalized at a standard cube at different time points p.i. of dPGBP7%-6S-ICG (7a). | ||
For their histological recovery within CIA joints, rats additionally received the respective ICC-conjugate, and tibiotarsal articulations were analyzed ex vivo 3 h p.i. by fluorescence microscopy using the 550 nm channel for excitation. Whereas dPGS-ICC (5) did not show any interaction with bone, a high accumulation was observed in chondrocytes and cartilage independent of the CIA score (Fig. 7), which demonstrates the high sensitivity of dPGS for inflammatory and degenerative processes at early stages of joint diseases. In comparison, dPGBP7%-ICC (7) was exclusively found in the bone and around the bone marrow taken up by osteoblasts in joints with a score 0, whereas no enrichment in the connective tissue or cartilage occurred (Fig. S18, ESI†). So far, bisphosphonate compounds have been described to mainly interact with bone-resorbing osteoclasts either interfering in the mevalonic acid pathway or being incorporated as nonhydrolyzable adenosine triphosphate analogs leading to apoptosis or cell inactivation.71–74 Nevertheless, BPs were also reported to influence the cell proliferation of osteoblasts in vitro and in vivo due to their cellular uptake which is in accordance with the observed findings.75–78 The main affinity of bisphosphonates to bone, however, is related to their strong complexation of calcium cations which explains the accumulation of dPGBP7% in the mineralized matrix compared to its absence in organic compartments.79 In CIA joints with a score 2 and higher, an enrichment of ICC-functionalized dPGBP7% (7) was also observed in cartilage (Fig. 7), which was rather unexpected, because the compound was only found in calcified areas in joints with no clinical symptoms. There was, however, no uptake by chondrocytes. The higher affinity to articular cartilage in malfunctioning joints could be either related to a mineralization of cartilage in RA or an enhanced destruction of the tissue which would probably lead to more accessible binding partners and has to be further investigated. Nevertheless, the data presented here do not prove long-term biocompatibility.
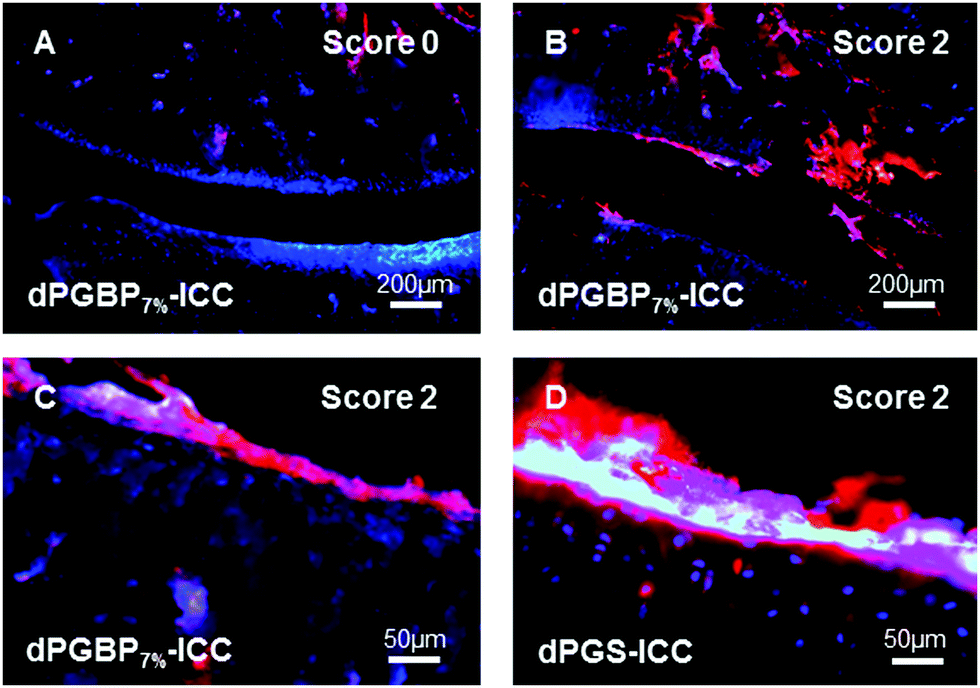 | ||
| Fig. 7 Fluorescence microscopy images of tissue slides from tibiotarsal articulations of rats with collagen-induced rheumatoid arthritis (CIA) 3 h after intravenous injection of (A–C) ICC-labeled dPGBP7% (7) or (D) ICC-labeled dPGS (5) (both red) with clinical scores of 0 (A) or 2 (B–D). The cell nuclei were stained with DAPI (blue). | ||
SEM and CLSM
Histological examinations of tibiotarsal articulations from CIA rats revealed the affinity of dPGS and dPGBP7% to cartilage in inflamed joints to different extents. However, since the bisphosphonate only binds to mineralized compartments in healthy tissue, it remains unclear whether the polyanion accumulates in native or degraded cartilage or if calcified areas are addressed which would indicate a mineralization of cartilage in RA. To clarify this issue, scanning electron microscopy (SEM) images of the rat joints embedded in PMMA were taken to distinguish between highly and less mineralized areas and were superimposed with CLSM pictures until the combined stacked areas fitted perfectly. Superimposed images of dPGS (5) were used for comparison. To minimize background signals due to autofluorescence of the tissue, an untreated joint was used for the adjustment of the fluorescence intensity (Fig. S20, ESI†).In accordance with the histological examinations, the bisphosphonated compound (7) was found to accumulate in the bone of inflamed ankles as well as joints with no clinical symptoms (Fig. 8). An enrichment of the polyanion was particularly found around the bone marrow, osteocyte lacunas, and the epiphyseal plate, where metabolic processes proceed and bone growth occurs,80 but also around blood vessels. Surprisingly, the compound was not found in compartments such as the epiphyseal line, the inner part of osteocyte lacunas, and the calcified cartilage. Whereas only minor accumulation in non-calcified cartilage was observed in joints with score 0, a stronger enrichment was found in the cartilage of rats with higher scores showing a high accumulation in specific areas (Fig. 8). However, these areas did not show any sign of calcification, which becomes obvious in the SEM pictures. One possibility for the strong binding could be an advanced destruction of the cartilage ECM during RA resulting in more accessible binding partners. However, it remains unclear what molecular targets the dPG bisphosphonate is binding to. A general calcification of cartilage during the course of the disease can be ruled out, since no changes in the grey values and therefore in the mineral content of cartilage were found (Fig. S19, ESI†). Nevertheless, a tendency towards higher enrichment of dPGBP7% in cartilage with increasing CIA scores was clearly observed (Fig. S21, ESI†).
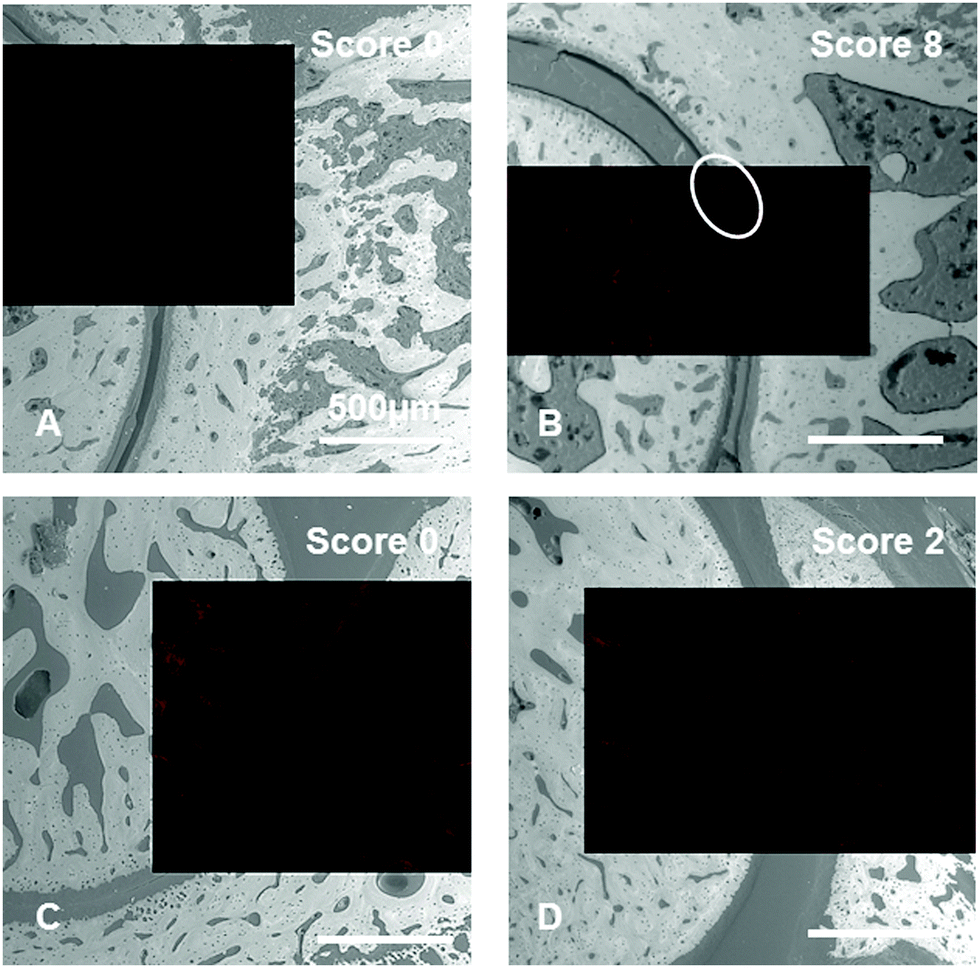 | ||
| Fig. 8 Superimposed images taken by confocal laser scanning microscopy (CLSM) (red) and scanning electron microscopy (SEM) (grey) of CIA rat joints embedded in PMMA showing different scores. Rats once received ICC-labeled dPGBP7% (7) (A and B) or dPGS (5) (C and D) intravenously prior to ex vivo imaging. Images show tibiotarsal articulations along the longitudinal-sagittal plane. Feet were removed 3 h p.i. and stored at −80 °C in TissueTek until further usage. | ||
In contrast, dPGS (5) shows a strong affinity to blood vessels and cartilage independent of the CIA score (Fig. 8), although the sulfated compound did not accumulate in joints of untreated rats. These findings might be related to a high sensitivity of dPGS towards molecular changes at very early stages of RA, where no clinical symptoms are observed. In summary, dPG anions showed differently pronounced affinities to bone, cartilage, and other joint compartments, depending on the severity of the destruction, which makes them interesting scaffolds for diagnostic and therapeutic applications for arthritis at different stages. With its high sensitivity toward cartilage destruction, dPGS and its derivatives, for instance, could be used to selectively target degraded cartilage at very early stages, whereas dPGBP7% could be applied in more severe cases to identify cartilage lesions.
Conclusions
We herein demonstrated the synthesis of highly water-soluble, dye-labeled dendritic polyglycerol anions, in particular sulfated, phosphated, and bisphosphonated derivatives with a low and a high degree of functionalization, as well as a mixed anion containing sulfate and bisphosphonate groups. By applying CLSM, differently pronounced affinities of the polyanions toward articular cartilage and its matrix components were verified and an enhanced binding to the ECM was proven in the inflammatory state. This study revealed that the interaction of polyanions with cartilage is highly dependent on the anionic moiety, the number of anionic groups, and the progression of tissue destruction. Moreover, it was shown that the combination of different moieties on a polymer could alter its targeting properties. These results make dendritic polyglycerol anions promising candidates for therapeutic and diagnostic applications in inflammation-driven diseases, where selective targeting of cartilage tissue is required. Taking into account that they also vary in binding affinities toward calcified tissue as previously published by our group, these anions can also be used for the differentiation between bone and cartilage. Moreover, hybrid structures combining different anionic moieties could be used to address compartments that show features of both tissues such as calcified cartilage. However, further studies have to be conducted to identify the molecular targets of the polyanions in order to understand their interaction with the ECM.Acknowledgements
The authors thank the IMPRS on Multiscale Bio-Systems, the DRS graduate school “Molecular Science”, and the collaborative research center SFB 765 of the Deutsche Forschungsgemeinschaft (DFG) for financial support. We thank Christoph Schlaich for providing us dopamine containing hPG amine, Andreas Roschger for the support with the evaluation of grey scales, and Dr Pamela Winchester for proofreading the manuscript. Open Access funding provided by the Max Planck Society.References
- T. Vos, A. D. Flaxman, M. Naghavi, R. Lozano, C. Michaud, M. Ezzati, K. Shibuya, J. A. Salomon, S. Abdalla, V. Aboyans, J. Abraham, I. Ackerman, R. Aggarwal, S. Y. Ahn, M. K. Ali, M. A. AlMazroa, M. Alvarado, H. R. Anderson, L. M. Anderson, K. G. Andrews, C. Atkinson, L. M. Baddour, A. N. Bahalim, S. Barker-Collo, L. H. Barrero, D. H. Bartels, M.-G. Basáñez, A. Baxter, M. L. Bell, E. J. Benjamin, D. Bennett, E. Bernabé, K. Bhalla, B. Bhandari, B. Bikbov, A. B. Abdulhak, G. Birbeck, J. A. Black, H. Blencowe, J. D. Blore, F. Blyth, I. Bolliger, A. Bonaventure, S. Boufous, R. Bourne, M. Boussinesq, T. Braithwaite, C. Brayne, L. Bridgett, S. Brooker, P. Brooks, T. S. Brugha, C. Bryan-Hancock, C. Bucello, R. Buchbinder, G. Buckle, C. M. Budke, M. Burch, P. Burney, R. Burstein, B. Calabria, B. Campbell, C. E. Canter, H. Carabin, J. Carapetis, L. Carmona, C. Cella, F. Charlson, H. Chen, A. T.-A. Cheng, D. Chou, S. S. Chugh, L. E. Coffeng, S. D. Colan, S. Colquhoun, K. E. Colson, J. Condon, M. D. Connor, L. T. Cooper, M. Corriere, M. Cortinovis, K. C. de Vaccaro, W. Couser, B. C. Cowie, M. H. Criqui, M. Cross, K. C. Dabhadkar, M. Dahiya, N. Dahodwala, J. Damsere-Derry, G. Danaei, A. Davis, D. De Leo, L. Degenhardt, R. Dellavalle, A. Delossantos, J. Denenberg, S. Derrett, D. C. Des Jarlais, S. D. Dharmaratne, M. Dherani, C. Diaz-Torne, H. Dolk, E. R. Dorsey, T. Driscoll, H. Duber, B. Ebel, K. Edmond, A. Elbaz, S. E. Ali, H. Erskine, P. J. Erwin, P. Espindola, S. E. Ewoigbokhan, F. Farzadfar, V. Feigin, D. T. Felson, A. Ferrari, C. P. Ferri, E. M. Fèvre, M. M. Finucane, S. Flaxman, L. Flood, K. Foreman, M. H. Forouzanfar, F. G. R. Fowkes, R. Franklin, M. Fransen, M. K. Freeman, B. J. Gabbe, S. E. Gabriel, E. Gakidou, H. A. Ganatra, B. Garcia, F. Gaspari, R. F. Gillum, G. Gmel, R. Gosselin, R. Grainger, J. Groeger, F. Guillemin, D. Gunnell, R. Gupta, J. Haagsma, H. Hagan, Y. A. Halasa, W. Hall, D. Haring, J. M. Haro, J. E. Harrison, R. Havmoeller, R. J. Hay, H. Higashi, C. Hill, B. Hoen, H. Hoffman, P. J. Hotez, D. Hoy, J. J. Huang, S. E. Ibeanusi, K. H. Jacobsen, S. L. James, D. Jarvis, R. Jasrasaria, S. Jayaraman, N. Johns, J. B. Jonas, G. Karthikeyan, N. Kassebaum, N. Kawakami, A. Keren, J.-P. Khoo, C. H. King, L. M. Knowlton, O. Kobusingye, A. Koranteng, R. Krishnamurthi, R. Lalloo, L. L. Laslett, T. Lathlean, J. L. Leasher, Y. Y. Lee, J. Leigh, S. S. Lim, E. Limb, J. K. Lin, M. Lipnick, S. E. Lipshultz, W. Liu, M. Loane, S. L. Ohno, R. Lyons, J. Ma, J. Mabweijano, M. F. MacIntyre, R. Malekzadeh, L. Mallinger, S. Manivannan, W. Marcenes, L. March, D. J. Margolis, G. B. Marks, R. Marks, A. Matsumori, R. Matzopoulos, B. M. Mayosi, J. H. McAnulty, M. M. McDermott, N. McGill, J. McGrath, M. E. Medina-Mora, M. Meltzer, Z. A. Memish, G. A. Mensah, T. R. Merriman, A.-C. Meyer, V. Miglioli, M. Miller, T. R. Miller, P. B. Mitchell, A. O. Mocumbi, T. E. Moffitt, A. A. Mokdad, L. Monasta, M. Montico, M. Moradi-Lakeh, A. Moran, L. Morawska, R. Mori, M. E. Murdoch, M. K. Mwaniki, K. Naidoo, M. N. Nair, L. Naldi, K. M. V. Narayan, P. K. Nelson, R. G. Nelson, M. C. Nevitt, C. R. Newton, S. Nolte, P. Norman, R. Norman, M. O'Donnell, S. O'Hanlon, C. Olives, S. B. Omer, K. Ortblad, R. Osborne, D. Ozgediz, A. Page, B. Pahari, J. D. Pandian, A. P. Rivero, S. B. Patten, N. Pearce, R. P. Padilla, F. Perez-Ruiz, N. Perico, K. Pesudovs, D. Phillips, M. R. Phillips, K. Pierce, S. Pion, G. V. Polanczyk, S. Polinder, C. A. Pope III, S. Popova, E. Porrini, F. Pourmalek, M. Prince, R. L. Pullan, K. D. Ramaiah, D. Ranganathan, H. Razavi, M. Regan, J. T. Rehm, D. B. Rein, G. Remuzzi, K. Richardson, F. P. Rivara, T. Roberts, C. Robinson, F. R. De Leòn, L. Ronfani, R. Room, L. C. Rosenfeld, L. Rushton, R. L. Sacco, S. Saha, U. Sampson, L. Sanchez-Riera, E. Sanman, D. C. Schwebel, J. G. Scott, M. Segui-Gomez, S. Shahraz, D. S. Shepard, H. Shin, R. Shivakoti, D. Silberberg, D. Singh, G. M. Singh, J. A. Singh, J. Singleton, D. A. Sleet, K. Sliwa, E. Smith, J. L. Smith, N. J. C. Stapelberg, A. Steer, T. Steiner, W. A. Stolk, L. J. Stovner, C. Sudfeld, S. Syed, G. Tamburlini, M. Tavakkoli, H. R. Taylor, J. A. Taylor, W. J. Taylor, B. Thomas, W. M. Thomson, G. D. Thurston, I. M. Tleyjeh, M. Tonelli, J. A. Towbin, T. Truelsen, M. K. Tsilimbaris, C. Ubeda, E. A. Undurraga, M. J. van der Werf, J. van Os, M. S. Vavilala, N. Venketasubramanian, M. Wang, W. Wang, K. Watt, D. J. Weatherall, M. A. Weinstock, R. Weintraub, M. G. Weisskopf, M. M. Weissman, R. A. White, H. Whiteford, S. T. Wiersma, J. D. Wilkinson, H. C. Williams, S. R. M. Williams, E. Witt, F. Wolfe, A. D. Woolf, S. Wulf, P.-H. Yeh, A. K. M. Zaidi, Z.-J. Zheng, D. Zonies, A. D. Lopez and C. J. L. Murray, Lancet, 2012, 380, 2163–2196 CrossRef.
- S. Frisenda, C. Perricone and G. Valesini, Autoimmun. Rev., 2013, 12, 591–598 CrossRef CAS PubMed.
- J. Clouet, C. Vinatier, C. Merceron, M. Pot-vaucel, Y. Maugars, P. Weiss, G. Grimandi and J. Guicheux, Drug Discovery Today, 2009, 14, 913–925 CrossRef CAS PubMed.
- R. J. Lories and F. P. Luyten, Nat. Rev. Rheumatol., 2011, 7, 43–49 CrossRef CAS PubMed.
- I. B. McInnes and G. Schett, N. Engl. J. Med., 2011, 365, 2205–2219 CrossRef PubMed.
- X. Ayral, Best Pract. Res., Clin. Rheumatol., 2001, 15, 609–626 CrossRef CAS PubMed.
- D. S. Pisetsky and E. St. Clair, J. Am. Med. Assoc., 2001, 286, 2787–2790 CrossRef CAS PubMed.
- C. H. Evans, BioDrugs, 2005, 19, 355–362 CrossRef CAS PubMed.
- K. E. Kuettner, Clin. Biochem., 1992, 25, 155–163 CrossRef CAS PubMed.
- J. A. Buckwalter, H. J. Mankin and A. J. Grodzinsky, Instr. Course Lect., 2005, 54, 465–480 Search PubMed.
- A. Maroudas and P. Bullough, Nature, 1968, 219, 1260–1261 CrossRef CAS PubMed.
- J. R. Levick, Arthritis Rheum., 1981, 24, 1550–1560 CrossRef CAS PubMed.
- M. Bottini, K. Bhattacharya, B. Fadeel, A. Magrini, N. Bottini and N. Rosato, Nanomedicine, 2016, 12, 255–268 CrossRef CAS PubMed.
- V. C. Mow, A. Ratcliffe and A. Robin Poole, Biomaterials, 1992, 13, 67–97 CrossRef CAS PubMed.
- N. Gerwin, C. Hops and A. Lucke, Adv. Drug Delivery Rev., 2006, 58, 226–242 CrossRef CAS PubMed.
- N. N. Pathak, M. C. Lingaraju, V. Balaganur, V. Kant, A. S. More, D. Kumar, D. Kumar and S. K. Tandan, Inflammation Res., 2015, 64, 161–169 CrossRef CAS PubMed.
- M. P. Hellio Le Graverand-Gastineau, Osteoarthritis Cartilage, 2009, 17, 1393–1401 CrossRef PubMed.
- G. D. Olsen, E. M. Chan and W. K. Riker, J. Pharmacol. Exp. Ther., 1975, 195, 242–250 CAS.
- H. Garrigue, J. C. Maurizis, J. C. Madelmont, C. Nicolas, J. M. Meyniel, A. Louvel, P. Demerseman, H. Sentenac-Roumanou and A. Veyre, Xenobiotica, 1991, 21, 583–595 CrossRef CAS PubMed.
- J.-C. Maurizis, M. Rapp, C. Nicolas, M. Ollier, M. Verny and J.-C. Madelmont, Drug Metab. Dispos., 2000, 28, 418–422 CAS.
- C. Nicolas, M. Verny, I. Giraud, M. Ollier, M. Rapp, J.-C. Maurizis and J.-C. Madelmont, J. Med. Chem., 1999, 42, 5235–5240 CrossRef CAS PubMed.
- M. Ollier, J.-C. Maurizis, C. Nicolas, J. Bonafous, M. de Latour, A. Veyre and J.-C. Madelmont, J. Nucl. Med., 2001, 42, 141–145 CAS.
- X. Hu, Q. Wang, Y. Liu, H. Liu, C. Qin, K. Cheng, W. Robinson, B. D. Gray, K. Y. Pak, A. Yu and Z. Cheng, Biomaterials, 2014, 35, 7511–7521 CrossRef CAS PubMed.
- J. D. Freedman, H. Lusic, B. D. Snyder and M. W. Grinstaff, Angew. Chem., Int. Ed., 2014, 53, 8406–8410 CrossRef CAS PubMed.
- C. S. Winalski, S. Shortkroff, E. Schneider, H. Yoshioka, R. V. Mulkern and G. M. Rosen, Osteoarthritis Cartilage, 2008, 16, 815–822 CrossRef CAS PubMed.
- H. Hyun, E. A. Owens, H. Wada, A. Levitz, G. Park, M. H. Park, J. V. Frangioni, M. Henary and H. S. Choi, Angew. Chem., Int. Ed., 2015, 54, 8648–8652 CrossRef CAS PubMed.
- D. A. Rothenfluh, H. Bermudez, C. P. O'Neil and J. A. Hubbell, Nat. Mater., 2008, 7, 248–254 CrossRef CAS PubMed.
- H.-Y. Hu, N.-H. Lim, D. Ding-Pfennigdorff, J. Saas, K. U. Wendt, O. Ritzeler, H. Nagase, O. Plettenburg, C. Schultz and M. Nazare, Bioconjugate Chem., 2015, 26, 383–388 CrossRef CAS PubMed.
- A. Jain, S. K. Mishra, P. R. Vuddanda, S. K. Singh, R. Singh and S. Singh, Nanomedicine, 2014, 10, 1031–1040 CrossRef CAS PubMed.
- M. Bishnoi, A. Jain, P. Hurkat and S. K. Jain, J. Drug Targeting, 2014, 22, 805–812 CrossRef CAS PubMed.
- H. Ringsdorf, J. Polym. Sci., Polym. Symp., 1975, 51, 135–153 CrossRef CAS.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- D. Steinhilber, S. Seiffert, J. A. Heyman, F. Paulus, D. A. Weitz and R. Haag, Biomaterials, 2011, 32, 1311–1316 CrossRef CAS PubMed.
- D. Steinhilber, M. Witting, X. Zhang, M. Staegemann, F. Paulus, W. Friess, S. Küchler and R. Haag, J. Controlled Release, 2013, 169, 289–295 CrossRef CAS PubMed.
- D. Steinhilber, A. L. Sisson, D. Mangoldt, P. Welker, K. Licha and R. Haag, Adv. Funct. Mater., 2010, 20, 4133–4138 CrossRef CAS.
- H. Jin, W. Huang, X. Zhu, Y. Zhou and D. Yan, Chem. Soc. Rev., 2012, 41, 5986–5997 RSC.
- M. A. Quadir and R. Haag, J. Controlled Release, 2012, 161, 484–495 CrossRef CAS PubMed.
- M. Weinhart, D. Gröger, S. Enders, J. Dernedde and R. Haag, Biomacromolecules, 2011, 12, 2502–2511 CrossRef CAS PubMed.
- S. Roller, H. Zhou and R. Haag, Mol. Diversity, 2005, 9, 305–316 CrossRef CAS PubMed.
- A. F. Hussain, H. R. Krüger, F. Kampmeier, T. Weissbach, K. Licha, F. Kratz, R. Haag, M. Calderón and S. Barth, Biomacromolecules, 2013, 14, 2510–2520 CrossRef CAS PubMed.
- A. Sousa-Herves, P. Wurfel, N. Wegner, J. Khandare, K. Licha, R. Haag, P. Welker and M. Calderon, Nanoscale, 2015, 7, 3923–3932 RSC.
- F. S. Mehrabadi, J. Adelmann, S. Gupta, S. Wedepohl, M. Calderon, U. Brinkmann and R. Haag, Curr. Cancer Drug Targets, 2016, 16, 639–649 CrossRef CAS PubMed.
- K. Licha, P. Welker, M. Weinhart, N. Wegner, S. Kern, S. Reichert, I. Gemeinhardt, C. Weissbach, B. Ebert, R. Haag and M. Schirner, Bioconjugate Chem., 2011, 22, 2453–2460 CrossRef CAS PubMed.
- D. Gröger, M. Kerschnitzki, M. Weinhart, S. Reimann, T. Schneider, B. Kohl, W. Wagermaier, G. Schulze-Tanzil, P. Fratzl and R. Haag, Adv. Healthcare Mater., 2014, 3, 375–385 CrossRef PubMed.
- J. D. Kisiday, C. W. McIlwraith, W. G. Rodkey, D. D. Frisbie and J. Steadman, Cartilage, 2012, 3, 245–254 CrossRef PubMed.
- T. Schneider, P. Welker, R. Haag, J. Dernedde, T. Hug, K. Licha, B. Kohl, S. Arens, W. Ertel and G. Schulze-Tanzil, Inflammation Res., 2015, 64, 917–928 CrossRef CAS PubMed.
- Q. Wei, K. Achazi, H. Liebe, A. Schulz, P.-L. M. Noeske, I. Grunwald and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 11650–11655 CrossRef CAS PubMed.
- D. E. Trentham, A. S. Townes and A. H. Kang, J. Exp. Med., 1977, 146, 857–868 CrossRef CAS PubMed.
- T. Fischer, I. Gemeinhardt, S. Wagner, D. v. Stieglitz, J. Schnorr, K.-G. A. Hermann, B. Ebert, D. Petzelt, R. MacDonald, K. Licha, M. Schirner, V. Krenn, T. Kamradt and M. Taupitz, Acad. Radiol., 2006, 13, 4–13 CrossRef PubMed.
- S. Vollmer, A. Vater, K. Licha, I. Gemeinhardt, O. Gemeinhardt, J. Voigt, B. Ebert, J. Schnorr, M. Taupitz, R. Macdonald and M. Schirner, Mol. Imaging, 2009, 8, 330–340 CAS.
- A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS.
- H. Türk, R. Haag and S. Alban, Bioconjugate Chem., 2004, 15, 162–167 CrossRef PubMed.
- K. Pant, D. Gröger, R. Bergmann, J. Pietzsch, J. Steinbach, B. Graham, L. Spiccia, F. Berthon, B. Czarny, L. Devel, V. Dive, H. Stephan and R. Haag, Bioconjugate Chem., 2015, 26, 906–918 CrossRef CAS PubMed.
- C. Siegers, M. Biesalski and R. Haag, Chem. – Eur. J., 2004, 10, 2831–2838 CrossRef CAS PubMed.
- M. Weinhart, I. Grunwald, M. Wyszogrodzka, L. Gaetjen, A. Hartwig and R. Haag, Chem. – Asian J., 2010, 5, 1992–2000 CrossRef CAS PubMed.
- P. J. Roughley, Arthritis Res., 2001, 3, 342–347 CrossRef CAS PubMed.
- D. Eyre, Arthritis Res., 2002, 4, 30–35 CrossRef CAS PubMed.
- S. A. Nesbitt and M. A. Horton, Science, 1997, 276, 266–269 CrossRef CAS PubMed.
- U. Lindahl and M. Hook, Annu. Rev. Biochem., 1978, 47, 385–417 CrossRef CAS PubMed.
- E. C. Tsilibary, G. G. Koliakos, A. S. Charonis, A. M. Vogel, L. A. Reger and L. T. Furcht, J. Biol. Chem., 1988, 263, 19112–19118 CAS.
- M. Weinhart, D. Gröger, S. Enders, S. B. Riese, J. Dernedde, R. K. Kainthan, D. E. Brooks and R. Haag, Macromol. Biosci., 2011, 11, 1088–1098 CrossRef CAS PubMed.
- S. Staufenbiel, C. Weise and R. H. Müller, Macromol. Symp., 2014, 345, 42–50 CrossRef CAS.
- A. Boreham, J. Pikkemaat, P. Volz, R. Brodwolf, C. Kuehne, K. Licha, R. Haag, J. Dernedde and U. Alexiev, Molecules, 2016, 21, 22–34 CrossRef PubMed.
- J. Khandare, A. Mohr, M. Calderón, P. Welker, K. Licha and R. Haag, Biomaterials, 2010, 31, 4268–4277 CrossRef CAS PubMed.
- D. Gröger, F. Paulus, K. Licha, P. Welker, M. Weinhart, C. Holzhausen, L. Mundhenk, A. D. Gruber, U. Abram and R. Haag, Bioconjugate Chem., 2013, 24, 1507–1514 CrossRef PubMed.
- D. Maysinger, D. Gröger, A. Lake, K. Licha, M. Weinhart, P. K. Y. Chang, R. Mulvey, R. Haag and R. A. McKinney, Biomacromolecules, 2015, 16, 3073–3082 CrossRef CAS PubMed.
- S. Reichert, P. Welker, M. Calderón, J. Khandare, D. Mangoldt, K. Licha, R. K. Kainthan, D. E. Brooks and R. Haag, Small, 2011, 7, 820–829 CrossRef CAS PubMed.
- M. McClung, S. T. Harris, P. D. Miller, D. C. Bauer, K. S. Davison, L. Dian, D. A. Hanley, D. L. Kendler, C. K. Yuen and E. M. Lewiecki, Am. J. Med., 2013, 126, 13–20 CrossRef CAS PubMed.
- J. Dernedde, A. Rausch, M. Weinhart, S. Enders, R. Tauber, K. Licha, M. Schirner, U. Zügel, A. von Bonin and R. Haag, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19679–19684 CrossRef CAS PubMed.
- J. D. Bergstrom, R. G. Bostedor, P. J. Masarachia, A. A. Reszka and G. Rodan, Arch. Biochem. Biophys., 2000, 373, 231–241 CrossRef CAS PubMed.
- J. E. Fisher, M. J. Rogers, J. M. Halasy, S. P. Luckman, D. E. Hughes, P. J. Masarachia, G. Wesolowski, R. G. G. Russell, G. A. Rodan and A. A. Reszka, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 133–138 CrossRef CAS.
- J. C. Frith, J. Mönkkönen, S. Auriola, H. Mönkkönen and M. J. Rogers, Arthritis Rheum., 2001, 44, 2201–2210 CrossRef CAS PubMed.
- J. C. Frith, J. Mönkkönen, G. M. Blackburn, R. G. G. Russell and M. J. Rogers, J. Bone Miner. Res., 1997, 12, 1358–1367 CrossRef CAS PubMed.
- A. I. Idris, J. Rojas, I. R. Greig, R. J. van’t Hof and S. H. Ralston, Calcif. Tissue Int., 2008, 82, 191–201 CrossRef CAS PubMed.
- L. I. Plotkin, R. S. Weinstein, A. M. Parfitt, P. K. Roberson, S. C. Manolagas and T. Bellido, J. Clin. Invest., 1999, 104, 1363–1374 CrossRef CAS PubMed.
- L. I. Plotkin, S. C. Manolagas and T. Bellido, Bone, 2006, 39, 443–452 CrossRef CAS PubMed.
- J. H. Tobias, J. W. M. Chow and T. J. Chambers, Bone, 1993, 14, 619–623 CrossRef CAS PubMed.
- R. G. G. Russell, Pediatrics, 2007, 119, 150–162 CrossRef PubMed.
- M. Pines and S. Hurwitz, Poult. Sci., 1991, 70, 1806–1814 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, analytical data, collagen binding assay, calcification of cartilage, CLSM and SEM images, specification of compounds. See DOI: 10.1039/c7tb00618g |
| This journal is © The Royal Society of Chemistry 2017 |