
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective co-delivery of nutlin-3a and p53 genes via core–shell microparticles for disruption of MDM2–p53 interaction and reactivation of p53 in hepatocellular carcinoma†
Pooya
Davoodi
a,
Madapusi P.
Srinivasan
ab and
Chi-Hwa
Wang
 *a
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117585, Singapore. E-mail: chewch@nus.edu.sg; Fax: +65-67791936; Tel: +65-65165079
bCivil, Environmental and Chemical Engineering, RMIT University, GPO Box 2476, Melbourne, VIC 3001, Australia
First published on 26th June 2017
Abstract
The tumor suppressor protein p53 is the most frequently inactivated, mutated, or deleted transcriptional factor in tumor cells. Recent studies have shown that the negative regulation of p53 by the murine double minute 2 (MDM2) protein in human cells interrupts the p53 apoptotic pathway and causes tumorigenesis. Therefore, the disruption of the MDM2–p53 complex by small molecules such as nutlin-3a and the administration of the active p53 protein can effectively restore the apoptotic activity of the p53 protein in tumor cells. This study aims to introduce a unique combined p53-based gene and chemotherapy approach using core–shell polymeric microparticles for the localized treatment of cancers. Core–shell microparticles were successfully fabricated in a single step using a modified electrohydrodynamic atomization (EHDA) technique, where the core and shell layers were loaded with nutlin-3a and β-cyclodextrin-g-chitosan/p53 nanoparticles, respectively. The grafting of β-cyclodextrin (β-CD) onto chitosan chains demonstrated remarkable cellular uptake (∼5-fold) compared to pure chitosan at N/P = 6, attributed to a strong interaction and temporary disruption of the lipid bilayer in the cell membrane by the synthesized copolymer. The therapeutic efficiencies of single- and dual-agent loaded microparticle formulations were also evaluated and compared against free-drug treatment in terms of cell viability and intracellular expression of p53, caspase 3, and MDM2 proteins via an MTS assay, an enzyme-linked immunosorbent assay, and an immunostaining assay. The results revealed that the controlled and sustained release of both agents from the microparticles synergistically enhanced the anti-proliferative efficacy of the agents via the continuous overexpression of p53 and caspase 3 proteins over 5 days. However, MDM2 protein expression remained at the basal level over that period. The findings also indicated that nutlin-3a could impose excessive oxidative stress on cancer cells, where the overproduction of reactive oxygen species (ROS) with irreversible destructive effects on subcellular organelles such as the nucleus (DNA) and mitochondria could be considered as a secondary apoptotic pathway induced by nutlin-3a. Inspired by the observations, the proposed drug delivery system can serve as a unique and powerful drug and gene delivery system with a far-reaching application in human cancer therapy.
1. Introduction
Cancer is an abnormal growth of cells within an organ tissue caused by mutagenesis including inactivation of the p53 protein.1,2 p53 is a potent tumor suppressor protein that plays a pivotal role in cellular resistance to malignant transformation, cell cycle regulation, and cell death.3–5 The p53 signal transduction pathway is activated in response to a range of physiological stresses to protect cells against tumorigenesis.4 In normal cells, the intracellular activity of p53 is precisely controlled by the murine double minute 2 (MDM2) protein through an auto-regulatory feedback loop.6–8 However, the overexpression of the p53 negative regulator (i.e. MDM2) and the epigenetic inactivation of MDM2 (i.e. ARF10) may abrogate the p53 regulatory pathway and accelerate cellular malignant transformation. Recent studies have revealed that approximately 50% of all human tumors harbor inactivated or mutated p53 genes, which consequently attenuate the essential functions of p53.9–11The interaction of the MDM2 protein with the C-terminal region12,13 of the p53 blocks transcriptional activities of p53 through three different mechanisms, including (i) direct inactivation of p53 transcriptional activities, (ii) p53 relocation into the cytoplasm, and (iii) proteasomal degradation of p53 facilitated by E3 ubiquitin ligase.14–16 Therefore, the disruption of p53–MDM2 interaction using small molecules would be a promising strategy for the restoration of p53 functions in cells retaining wild-type p53. The reactivation of the p53 pathway not only induces p53 protein accumulation in cancer cells to promote the apoptosis of tumor cells, but also increases the sensitivity of drug resistant tumors to conventional treatments such as chemotherapy and radiotherapy.17 In fact, a successful cancer treatment is directly correlated with the damage of DNA and the activity of p53, which are initiated by a majority of chemotherapeutic agents as the main mechanism of action (MOA).17–19
Nutlin-3a is a non-genotoxic MDM2 inhibitor which can effectively bind to MDM2 and suppress its activities in cells with wild-type p53 and those with mutated or deleted p53.20 However, the recommended dose of nutlin-3a is considerably lower in cells expressing wild-type p53.8 The increase in the intracellular level of p53 is detectable 1 to 3 h after receiving nutlin-3a. However, the expression of p53 exhibits a downward trend after ∼4–6 h due to the significant reduction of nutlin-3a concentration.8 Therefore, the use of an appropriate drug delivery system (DDS) for the controlled and sustained release of nutlin-3a over a prolonged period may significantly improve its therapeutic outcome.
Although a DDS can somewhat alleviate the toxicity induced to normal cells by the repeated administration of MDM2 inhibitors,1,21,22 the non-specific cellular activation of p53 via MDM2 inhibitors may still activate the apoptotic pathway and induce neoplasm in normal tissues. The tissue distribution analysis of MDM2 inhibitors in mice showed a remarkable activation of the p53 protein in all tissues after injection.20 Mendrysa et al. (2003) demonstrated that genetically reduced MDM2 expression (∼30%) and the subsequent increase in the p53 level had no contradictory influence on the animal lifespan, while mice with higher p53 levels showed smaller size, mild disturbances in hematopoiesis, and amplified apoptosis in the small intestine as compared with control groups. The results of another study on animals treated with nutlin-3a revealed that the higher p53 activity led to efficient suppression in cancer cells, while normal human fibroblast cells underwent cell-cycle arrest, but preserved their viability for a prolonged period.9,23,24 Hence, to achieve a more optimistic therapeutic outcome, an efficient drug delivery system will be essential to locally provide a high concentration of an anti-MDM2 drug around tumor cells through a controlled release manner. Despite the disruption of MDM2–p53 interaction by nutlin-3a, the missense mutations of the DNA-binding domain in inactivated p53 can potentially decline tumor responses to chemotherapy. However, recent findings have shown that the successful restoration of p53 gene functions via anticancer gene delivery can improve tumor sensitivity towards chemotherapy and potentially overcome the poor therapeutic outcomes after the sole form of therapy. Although recombinant viruses have presented excellent transfection efficiency which is essential for successful gene therapy, their application as gene delivery vectors has been extremely confined due to serious concerns about their toxicity, immunogenicity, and large-scale production. Therefore, numerous efforts have been directed towards producing non-viral gene carriers (artificial viruses) for the safe delivery of DNA and RNA interference (RNAi).25–30 For instance, Kumar et al. prepared new lipopolymers in which low molecular weight and branched polyethylene amine (LMW PEI) molecules hydrophobically modified with ferrocene-terminated alkyl tails. Their results showed a greater transfection efficiency as compared to high molecular weight PEI (25 kDa) and commercially available transfection agents.27 In another study, a new multifunctional gene carrier was prepared via crosslinking LMW PEI with lactitol diacrylate (LDA) which caused higher cellular uptake, cell targeting, and a rapid intracellular release of genes in vitro and in vivo.28
To date, chitosan (CS), a natural linear cationic polysaccharide with a random distribution of N-acetylglucosamine (GlcNAc) and glucosamine (GlcN) units, has gained much attention as a gene delivery vector owing to its biocompatibility, low immunogenicity, ease of modification, and biodegradability.31,32 Although CS primary amino groups can be readily protonated at low pH, its poor solubility under physiological conditions (pH ∼ 7.0) and rather low transfection efficiency in vitro and in vivo have significantly confined its extensive applications in the biomedical domain.33 To overcome these dilemmas, the conjugation of cell-permeable molecules and/or targeting ligands such as β-cyclodextrin (β-CD) has been proposed to enhance the CS transfection efficiency and improve the stability of biomolecules under physiological conditions.
Based on the above considerations, we designed and fabricated monodispersed double-wall microparticles comprising a poly(lactic-co-glycolic acid) (PLGA) core surrounded by a poly(D,L-lactic acid) (PDLLA) shell using a coaxial electrohydrodynamic atomization (CEHDA) technique. We hypothesized that the simultaneous delivery of nutlin-3a and p53 plasmid DNA from core–shell microparticles can address the limitations mentioned above. β-CD-graft-chitosan (β-CD-g-CS) was synthesized to efficiently encapsulate p53 plasmid DNA into positively charged nanoparticles and improve their transfection efficiency. Subsequently, the nanoparticles and nutlin-3a were loaded into the microparticles using the CEHDA technique. The new microparticle formulations were examined on hepatocellular carcinoma (HepG2) cells in vitro.
2. Materials and methods
2.1. Materials
Chitosan (CS) (Mw = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–190000 Daltons based on viscosity) and p-toluenesulfonyl chloride (>98%) were purchased from Sigma-Aldrich. The degree of deacetylation determined by 1H-NMR spectroscopy was ∼85%. β-Cyclodextrin (β-CD, 98% purity) was acquired from Sinopharm Chemical Reagent Co. Ltd (China). PLGA 50:50 (inherent viscosity in hexafluoroisopropanol: 0.55–0.75 dL g−1) and PDLLA (inherent viscosity in chloroform: 0.55–0.75 dL g−1) were purchased from LACTEL Absorbable Polymers (DURECT Corporation, USA). Sodium hydroxide (NaOH) was purchased from Merck Pte Ltd. Nutlin-3a (>98%) was purchased from ChemScene, LLC (New Jersey, USA). All other reagents used were of analytical grade and commercially available.
000–190000 Daltons based on viscosity) and p-toluenesulfonyl chloride (>98%) were purchased from Sigma-Aldrich. The degree of deacetylation determined by 1H-NMR spectroscopy was ∼85%. β-Cyclodextrin (β-CD, 98% purity) was acquired from Sinopharm Chemical Reagent Co. Ltd (China). PLGA 50:50 (inherent viscosity in hexafluoroisopropanol: 0.55–0.75 dL g−1) and PDLLA (inherent viscosity in chloroform: 0.55–0.75 dL g−1) were purchased from LACTEL Absorbable Polymers (DURECT Corporation, USA). Sodium hydroxide (NaOH) was purchased from Merck Pte Ltd. Nutlin-3a (>98%) was purchased from ChemScene, LLC (New Jersey, USA). All other reagents used were of analytical grade and commercially available.
2.2. Synthesis and characterization of β-CD-g-CS
The mass percentage of four key elements (carbon (C), hydrogen (H), nitrogen (N), and sulfur (S)) in the plain CS, 6-OTs-β-CD, and β-CD-g-CS samples were measured using an Elementar vario MICRO cube. All the samples were freeze-dried for 48 h prior to analysis. The substitution of –NH2 groups on chitosan chains with 6-OTs-β-CD was calculated as follows:
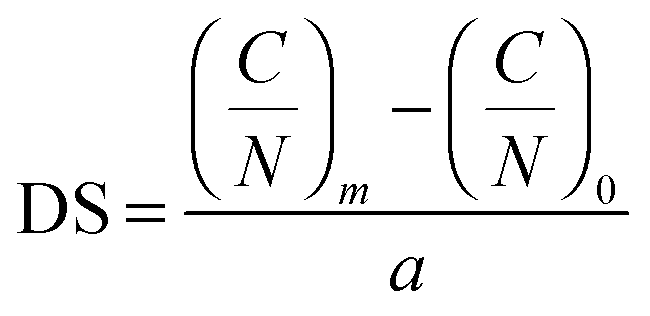 | (1) |
The crystallinity of plain CS and β-CD-g-CS were determined by X-ray diffraction (XRD) using a CuKα radiation lamp at λ = 1.5405 Å. Data were recorded at a step speed of 0.1°/5 s in the 3°–40° range at 25 °C.
2.3. Preparation and characterization of pDNA/β-CD-g-CS
The pCMV-p53 plasmid DNA was amplified in Escherichia coli (E. coli) DH5α cells and extracted and purified using a HiSpeed® Plasmid Maxi Kit (Qiagen, Inc.). The concentration of the pDNA in the final solution was measured using a Nanodrop spectrophotometer (ND1000, Thermo Fisher) at wavelengths of 260 and 280 nm. The quality of the pDNA was determined with the ratio of absorbance at 260 nm to 280 nm, where the ratio between 1.8 and 2.0 was considered as pure pDNA.Plasmid DNA-NPs were prepared according to a previously reported procedure.34 A solution of chitosan or β-CD-g-CS in sodium acetate buffer (5 mM, pH ∼ 5.5) and a pDNA solution (Tris–EDTA buffer) were preheated to 50–55 °C, separately. Then, equal volumes (<200 μl) of the solutions were quickly mixed, vortexed for 30 s, and kept at 45 °C for 30 min to form stable and uniform nanoparticles.
:1 v/v ratio were loaded into pre-cast wells in the 1% agarose gel. The electrophoresis was performed at 100 V for 1 h in Tris acetate–EDTA (1× TAE) buffer. The gel was subsequently immersed in SYBR® Gold Nucleic Acid Gel Stain solution for 15 min and was de-stained in DI water for 15 min. DNA bands were visualized under a UV transilluminator (G:BOX, Syngene).
In order to investigate the shielding effect of β-CD-g-CS on the integrity of pDNA upon particle formation, pDNA was extracted from nanoparticles and tested by gel electrophoresis. The nanoparticles (100 μl, 6.4 μg DNA) were mixed with 80 μl of chitosanase (0.2–0.25 U ml−1) and 20 μl of lysozyme (100 U ml−1) in sodium acetate buffer (pH ∼ 5.5). In order to complete the digestion process, the nanoparticles were incubated for 4 h at 37 °C. The gel electrophoresis experiment was conducted according to the process explained above and pDNA bands were visualized under a UV transilluminator (G:BOX, Syngene).
2.4. Fabrication of core–shell microparticles
:10 v/v to form a homogeneous mixture.
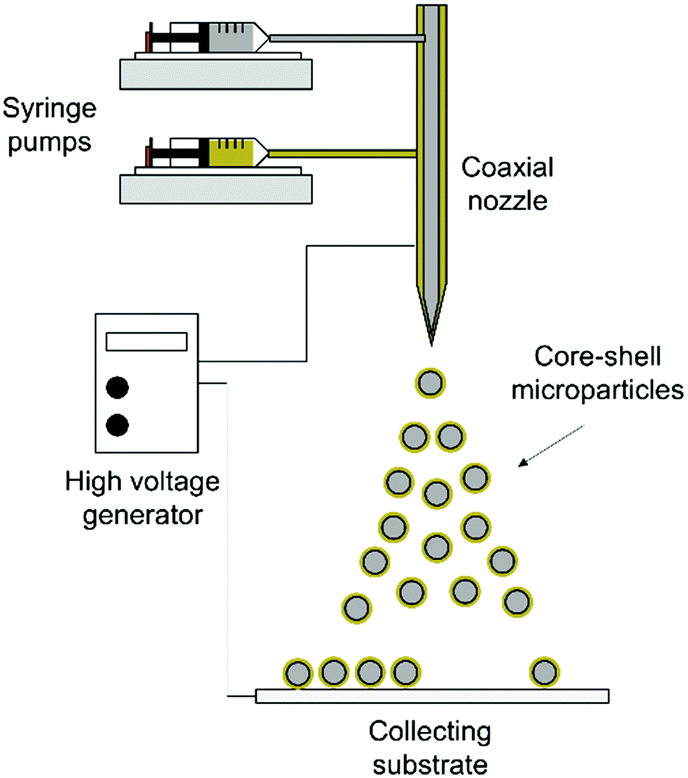 | ||
| Scheme 1 Schematic illustration of coaxial electrohydrodynamic atomization (CEHDA). | ||
The inner morphology of the microparticles was investigated using laser scanning confocal microscopy (LSCM) (FV1000, Olympus, Tokyo, Japan) equipped with an argon laser. To distinguish the core from shell compartments inside the microparticles, coumarin 6 fluorescent dye (λex.: 490 nm and λem.: 500–540 nm) was added to the core solution prior to the particle fabrication.
:50 v/v) and analyzed using a high-performance liquid chromatography (HPLC) machine equipped with a C18 reverse-phase (RP) column and a UV-2487 UV-detector, where acetonitrile:H2PO4 buffer (50:50 v/v) at 1.0 ml min−1 was utilized as a mobile phase. The concentration of pDNA in the aqueous phase was quantified using a Quant-iT™ PicoGreen® dsDNA Assay Kit (Thermo Fisher Scientific Inc., Rockford, IL). The fluorescence intensity was measured using a TeCAN Infinite®-200PRO microplate reader (TeCAN Trading AG, Switzerland) at excitation and emission wavelengths of 480 and 520 nm, respectively.
2.5. In vitro cellular uptake and cell transfection experiment
Fluorescein isothiocyanate (FITC)-labeled chitosan was synthesized, as previously reported37 with slight modifications. Briefly, 1 g of chitosan was suspended in 40 ml of DI water, followed by the addition of 60 ml of HCl (0.1 M) and 25 ml of methanol. Next, 6 mg of FITC dissolved in 50 ml of methanol was added dropwise to the chitosan solution and stirred for 3 h at room temperature. Finally, the reaction mixture was diluted in 600 ml of DI water and the pH was adjusted to 7.5 with 0.1 M sodium hydroxide. The precipitate was subsequently collected using vacuum filtration and washed with an ethanol:water (70:30 v/v) mixture three times, followed by lyophilization to obtain FITC–chitosan.
To measure the cellular uptake of nanoparticles, HepG2 cells were cultivated in a 12-well plate culture dish (1 × 105 cells per well) and allowed to adhere and grow overnight. The cells were incubated with nanoparticles containing 0.5 μg of pDNA for 4 h. Then, the medium was removed and the cells were gently rinsed with 1× PBS three times to remove residual free particles from the cells. The cells were subsequently fixed with 4% formaldehyde for 15 min, followed by one-step washing using PBS and staining with Hoechst 33342 (1 μg ml−1, Thermo Fisher Scientific Inc., Rockford, IL) for 10 min. The samples were subsequently analyzed using a TeCAN microplate reader in fluorescence mode at the following excitation (λex) and emission (λem) wavelengths: Hoechst (λex = 360 nm, λem = 461 nm) and FITC (λex = 495 nm, λem = 519 nm).
For the qualitative assessment of cellular uptake, the cells were treated under the conditions explained above. After 4 h of incubation, the cells were washed three times with 1× PBS and fixed with 4% formaldehyde for 15 min at room temperature. Subsequently, the nuclei of the cells were stained with Hoechst 33342 (1 μg ml−1) for 10 min at room temperature. The samples were imaged using a confocal microscope equipped with a 20× objective lens at the wavelengths indicated above.
The cell transfection efficiency was evaluated under the conditions explained above. However, in these experiments, p53–pDNA was substituted with Enhanced Green Fluorescent Protein (EGFP) plasmid DNA which emitted green fluorescent light upon excitation at ∼485 nm. To quantify the transfection efficiency, cells were cultured in a brown 96 well-plate at 6 × 103 cells per well overnight and then were treated with the formulations for 6 h before incubating in fresh medium for 42 h. The cells were subsequently washed with 1× PBS and stained with Hoechst 33342 (1 μg ml−1) for 10 min at room temperature before the commencement of the experiment. The percentage of positive EGFP cells was calculated via normalizing the intensity of the EGFP green light to the intensity of blue light emitted from cell nuclei measured using a TeCAN Infinite®-200PRO microplate reader (TeCAN Trading AG, Switzerland). The cells were visualized under a fluorescence microscope equipped with green and blue filters.
2.6. Biocompatibility evaluation of β-CD-g-CS
The biocompatibility of the synthetic polymers was evaluated against HepG2, C6 glioma, and HeLa cell lines in vitro. The cells were seeded into a 96-well plate and allowed to adhere and grow overnight. The polymers were incubated with Dulbecco's Modified Eagle Medium (DMEM, HyClone™, GE Healthcare Life Sciences) at 37 °C for 48 h and then injected into the cells at predetermined concentrations (0–500 μg ml−1). Chitosan and branched-polyethyleneimine (bPEI-25k) were employed as controls. After 24 h, the medium was discarded and the cells were rinsed with pre-warmed 1× PBS twice. Next, 100 μl of fresh medium and 20 μl of Cell Titer 96® Aqueous One Solution Cell Proliferation Assay Reagent (Promega, USA) were added to each well and the plate was incubated at 37 °C for 4 h. As the formation of the formazan product was directly correlated with the number of living cells, the absorbance of the solutions was recorded at 490 nm using a TeCAN microplate reader and the cell viability was determined by eqn (2):| Cell viability (%) = [(Abs)sample − (Abs)blank]/[(Abs)control − (Abs)blank] × 100 | (2) |
2.7. Nutlin-3a: free-drug tests
In order to verify the involvement of ROS, the cells were pretreated with a ROS scavenger agent (N-acetyl-L-cysteine, NAC, Sigma Aldrich) at a final concentration of 10 mM. After 1 h, the pre-treatment medium was removed and the cells were incubated with a NAC supplemented medium containing nutlin-3a. At the end of the experiment, cells were stained with H2DCFDA and Hoechst 33342 and fluorescence intensities were measured using a TeCAN microplate reader at the corresponding wavelengths indicated above.
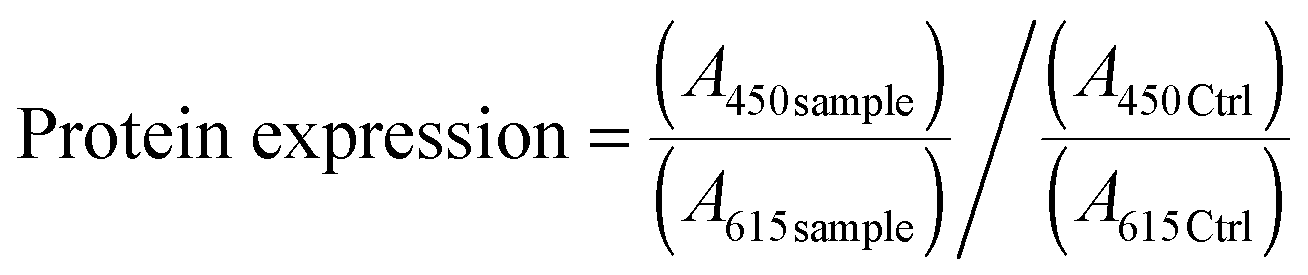 | (3) |
2.8. In vitro toxicity of drug loaded microparticles
To examine the long-term cytotoxicity effect of the drugs (i.e. nutlin-3a and pDNA-NPs) released from core–shell microparticles, cells were cultivated in 24-well plates at an initial density of 5 × 104 cells per well. After 48 h, the medium was replaced with fresh medium containing different microparticle formulations. The blank microparticles served as the control group. As the release of nutlin-3a from the formulations was relatively slow, nutlin-3a loaded microparticles were first incubated in PBS solution at 37 °C with shaking at 240 rpm for one week and then were transferred into the well plates for cytotoxicity tests. The equivalent amounts of nutlin-3a and pDNA-NPs released within five days were added to cells as free drug groups. In order to simulate the real chemotherapy conditions, the medium containing the therapeutic agents (free drugs) was removed after 4 h and the cells were incubated with fresh medium. For nutlin-3a + pDNA–NP free drug tests, the cells were first incubated with pDNA/nanoparticles for 4 h, followed by removal of the medium and addition of medium containing the equivalent amount of nutlin-3a released over 5 days. The medium was replaced with the fresh one after 4 h. Untreated cells were considered as a control group. All the samples were subjected to an MTS cell viability assay as explained in Section 2.6.2.9. Expression of p53, caspase 3, and MDM2 proteins (microparticle treatment)
The expression of p53, caspase 3 and MDM2 proteins after 5 days of treatment with different formulations was quantitatively and qualitatively determined as described in Sections 2.7.5 and 2.7.6. The cells were treated with the different formulations as explained in Section 2.8.2.10. Statistical analysis
One-way analysis of variance (ANOVA) and Student’s t-test were used to analyze data. Differences among the groups were statistically significant at a confidence level of 95% (p < 0.05).3. Results and discussion
3.1. Fabrication and characterization of microparticles
Fig. 1a shows the size and morphology of the microparticles fabricated using CEHDA at various concentrations and core:shell flow rate ratios. The size of the microparticles was around ∼14–17 μm at a flow rate ratio of 1:2 and showed a gradual increase with increase of the shell flow rate. However, the increase of the shell flow rate beyond 1:4 yields particles with broad size distribution which were not suitable for drug delivery applications. On the other hand, the shell solution concentration had a marginal impact on particle size, while it significantly affected the surface morphology of the microparticles. Particles had porous surfaces at low shell concentrations, while less porous and smoother surfaces formed at higher concentrations. Dichloromethane, a chlorohydrocarbon with a very low boiling temperature (40 °C), is a highly volatile solvent that evaporates quickly when particles fly toward the collecting substrate. Therefore, polymer chains do not have sufficient time for rearrangement inside solidifying polymer droplets and form rough surfaces (panels A–C). However, when the shell concentration is high (panels D–F), there are considerable entanglement forces among polymer chains, which reduce the effect of solvent evaporation on the surface morphology of the final products. It is worth noting that a high shell concentration and flow rate may yield fiber + microparticle products, which are more suitable for tissue engineering applications. The inner structure of the microparticles was visualized using a confocal microscope after adding coumarin-6 to the core solution. As seen in Fig. 1b, the core compartments were completely surrounded by a polymeric shell. The average fluorescence intensity profile at the cross section of the microparticles also revealed that the shell layer was uniformly distributed around the core compartment. Although the shell thickness can be determined using confocal images, the actual thickness is larger as coumarin-6 may prematurely release into the shell solution at the solidification stage.
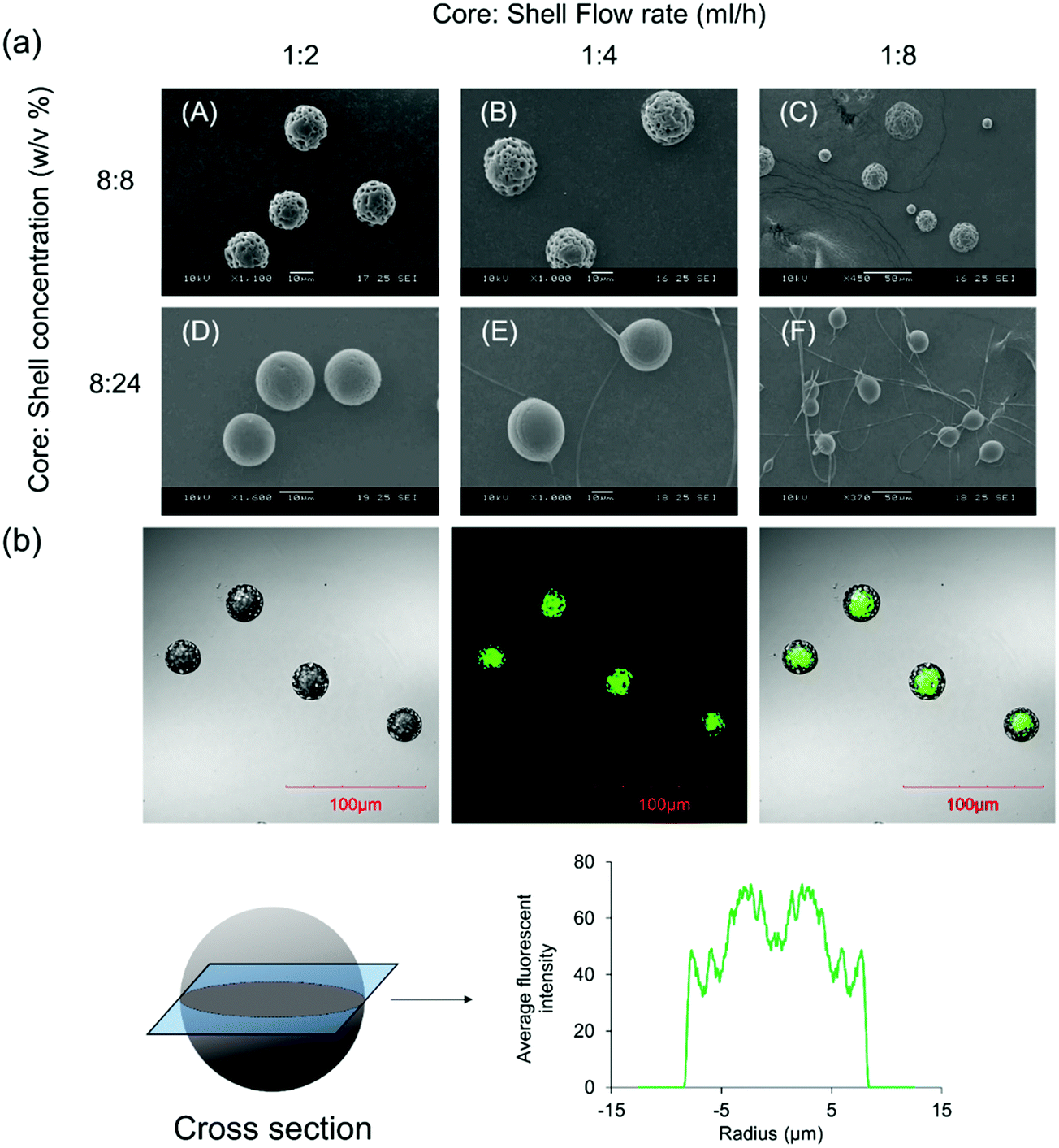 | ||
| Fig. 1 Core–shell microparticles fabricated using the CEHDA technique. (a) Size and morphology of the microparticles at different flow rates and shell concentrations. (b) Core and shell compartments were distinguished after adding coumarin-6 to the core solution. Images were captured using LSCM. The distribution of coumarin-6 inside the microparticles was calculated from fluorescence intensities. Scale bar = 100. | ||
PLA and PLGA are two U.S. Food and Drug Administration (FDA) approved polymers enormously used for drug delivery. They can be dissolved in organic solvents such as DCM and subsequently can be electrosprayed using a CEHDA set-up, where the conductivity of the polymer solutions and their surface tensions play pivotal roles in the successful formation of core–shell micro-structures. In addition, these polymers show a moderate degradation rate compared to others (e.g. polycaprolactone). Although choosing a lower molecular weight may accelerate a degradation process, the formation of microparticles after electrospraying is compromised due to the lower polymer chain entanglement and instability of the cone-jet spraying mode.38,39
3.2. Synthesis and characterization of β-CD-g-CS
The chemical structures of pure chitosan (CS) and β-CD-g-CS were examined by FTIR and 1H-NMR spectroscopy. As shown in Fig. 2a, CS spectra displayed an absorbance at 3417 cm−1 due to the stretching of –OH and –NH groups. The deformation of N–H (amine) and the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N (amide group) groups in CS demonstrated absorption peaks at 1598 cm−1, 1639 cm−1, and 1373 cm−1, respectively. CS spectra showed more absorptions at 1145, 1072, and 1022 cm−1 corresponded to the symmetric stretching of C–O–C and the vibration of C–O groups. β-CD spectra exhibited an absorption at 3379 cm−1 corresponding to –OH stretching and three absorption peaks at 1180, 1076, and 1022 cm−1 assigned to C–O–C and C–O groups. The characteristics peaks at 1597 cm−1 and 1149 cm−1 in 6-OTs-β-CD spectra corresponded to the CO stretching of the aromatic ring and SO, respectively. Lastly, β-CD-g-CS exhibited all characteristics peaks of β-CD and CS at the wavenumbers indicated above.
O and C–N (amide group) groups in CS demonstrated absorption peaks at 1598 cm−1, 1639 cm−1, and 1373 cm−1, respectively. CS spectra showed more absorptions at 1145, 1072, and 1022 cm−1 corresponded to the symmetric stretching of C–O–C and the vibration of C–O groups. β-CD spectra exhibited an absorption at 3379 cm−1 corresponding to –OH stretching and three absorption peaks at 1180, 1076, and 1022 cm−1 assigned to C–O–C and C–O groups. The characteristics peaks at 1597 cm−1 and 1149 cm−1 in 6-OTs-β-CD spectra corresponded to the CO stretching of the aromatic ring and SO, respectively. Lastly, β-CD-g-CS exhibited all characteristics peaks of β-CD and CS at the wavenumbers indicated above.
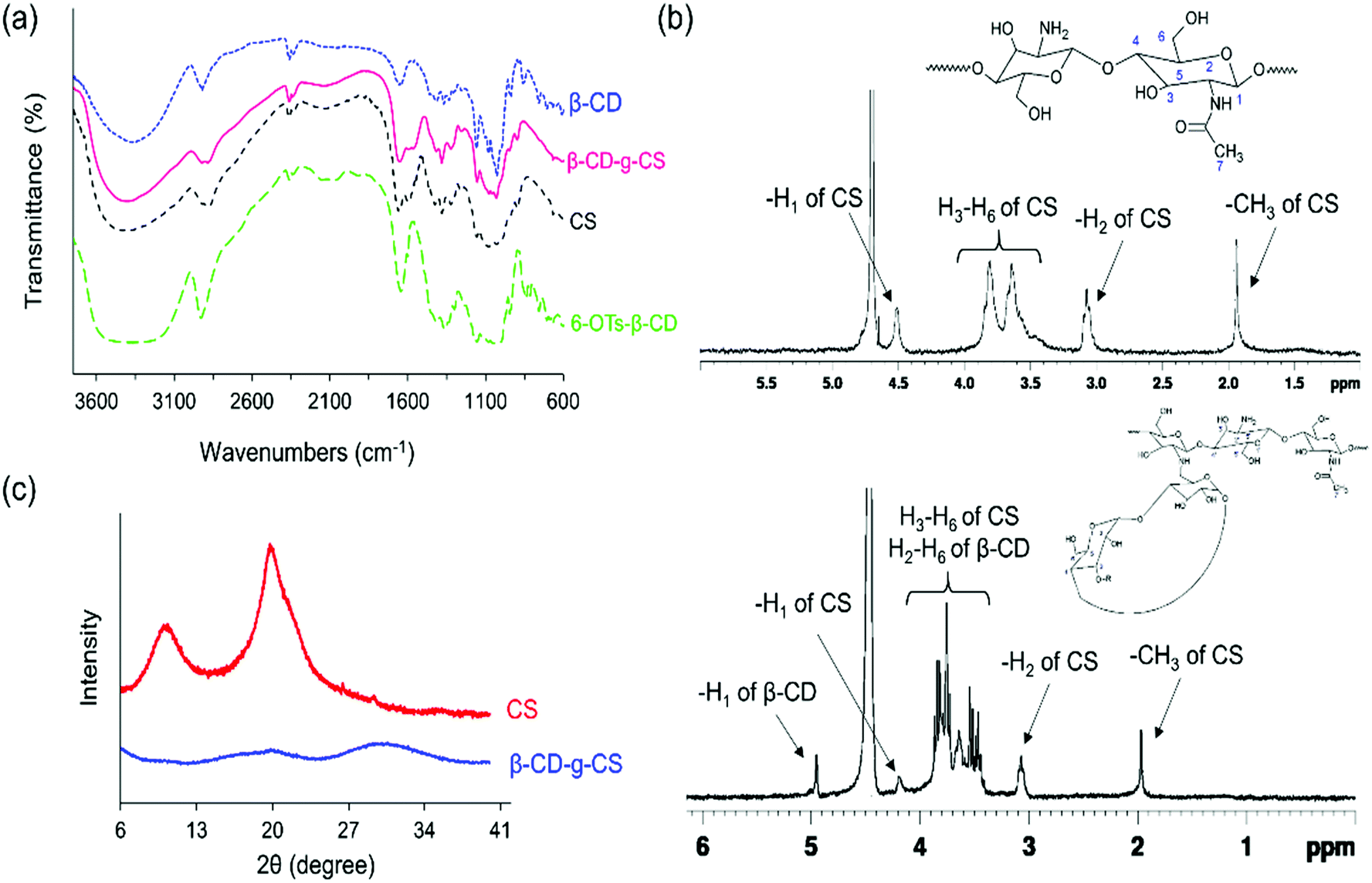 | ||
| Fig. 2 FTIR, 1H NMR, and XRD spectra of pure CS, CS, and β-CD-g-CS at a β-CD:CS molar ratio of 2:1. | ||
The degree of deacetylation and the number of β-CDs grafted onto the CS backbone were estimated using 1H NMR spectra (Fig. 2b). The CS spectrum displayed a single peak at 1.96 ppm and multiple peaks between 3 and 4 ppm, which were assigned to the –CH3 of GlcNAc groups and H2–H6 of the CS chain, respectively. Thus, the degree of deacetylation was around 87%. After conjugation of β-CD onto the CS chains, the 1H NMR spectrum of β-CD-g-CS demonstrated proton peaks at δ = 1.96 ppm, 3.2 ppm, and 4.9 ppm corresponding to GlcNAc, H2 protons in CS, and H1 protons in β-CD, respectively. The multiple protons at δ = 3.8–3.3 ppm were assigned to β-CD protons and new peaks at δ = 8.1 ppm and 7.5 ppm indicated the protons of tosyl groups (aromatic rings).
Pure chitosan is a crystalline biodegradable polymer. The crystallinity of chitosan depends on the degree of deacetylation of chitosan, where the higher the degree of deacetylation, the higher the degree of crystallinity. To evaluate the physical state of β-CD-g-CS, an X-ray diffraction test was conducted for chitosan and β-CD-g-CS. The XRD pattern of chitosan exhibits two well-defined peaks at 2θ = 11° and 2θ = 20° (Fig. 2c) corresponding to crystal forms I and II, respectively.40 Despite the crystalline structure of native CS, the XRD pattern of β-CD-g-CS did not exhibit CS characteristic peaks. The amorphous structure of β-CD-g-CS can be attributed to the reduction of hydrogen bonds among the CS chains upon substitution of amino groups with β-CD.
3.3. Preparation and characterization of pDNA/β-CD-g-CS
:CS ratios were approximately non-toxic in HepG2, HeLa, and C6 glioma cells, even at high concentrations. In contrast, PEI-25k exhibited concentration-dependent cytotoxicity, where the cell viability reached ∼10% at 500 μg ml−1. Overall, the cytotoxicity results show the great biocompatibility of β-CD-g-CS products for gene delivery applications.
:CS molar ratio enhanced the size of the NPs, even at low N/P ratios. The formation of larger NPs at high β-CD:CS molar ratios can be assigned to spatial distances among polymer chains after conjugation of β-CD molecules onto CS chains that yields looser and larger polyplexes. Moreover, compared to CS NPs, more β-CD-g-CS is required for the formation of NPs at a specific N/P ratio that results in larger particles.
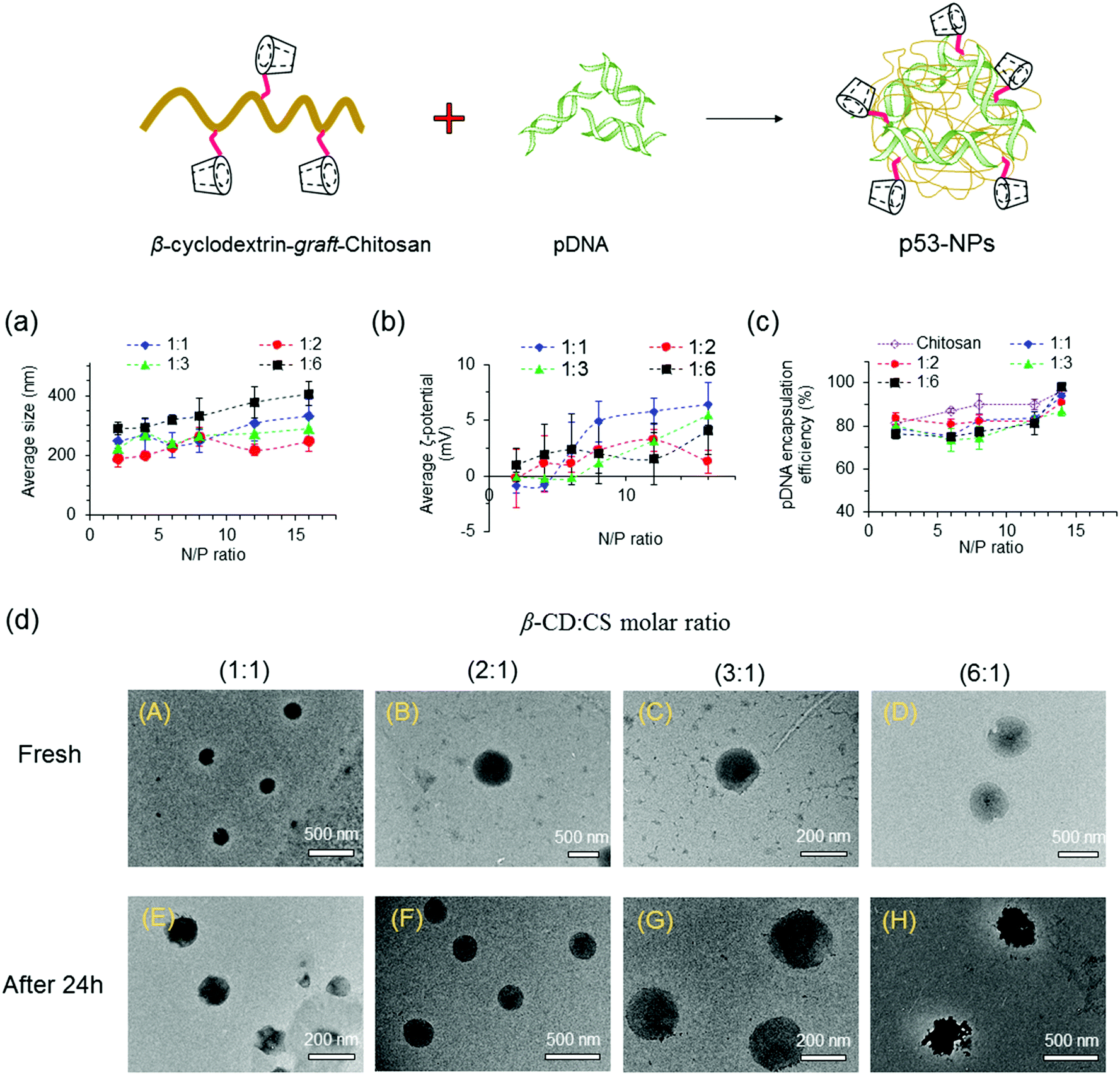 | ||
| Fig. 3 Sizes and zeta potentials of polyplexes. (a) Size distribution and (b) surface charge density of nanoparticles after forming pDNA/β-CD-g-CS complexes at various N/P ratios. (c) Encapsulation efficiency of pDNA at various N/P ratios. (d) FETEM images of NPs at N/P = 6: fresh nanoparticles (top), after 24 h (bottom). NPs were prepared using β-CD-g-CS synthesized at different β-CD:CS molar ratios (1:1, 2:1, 3:1, 6:1). | ||
The zeta (ζ)-potentials of NPs showed positively charged surfaces at N/P ≥ 4 (Fig. 3b). At low N/P ratios, NPs demonstrated negative, neutral, or slightly positive surfaces due to the presence of pDNA molecules near the surface of the NPs. On the other hand, the observation of a positive zeta-potential at high N/P ratios for all NPs can be attributed to the increase of positively charged polymer chains on the particle surfaces.
NPs were prepared via mixing equal volumes of p53–DNA with β-CD-g-CS solutions and p53 DNA solutions. To determine the encapsulation efficiency, NPs were centrifuged and the amount of DNA in the supernatant solution (un-encapsulated pDNA) was determined via a PicoGreen assay. Fig. 3c shows that the EE was greater than 75% for all NPs. However, β-CD-g-CS NPs had generally lower EE compared to their chitosan NPs due to the presence of β-CD and formation of looser and larger polyplexes.
FETEM micrographs of NPs are presented in Fig. 3d. NPs showed a spherical shape with diameters ranging from 200 to 250 nm at N/P ratios ≥ 6 (panels A–D). FETEM images also revealed that retaining NPs in buffer solution for 24 h had no destructive effect on the particle structure (panels E and G). However, the morphology of the NPs slightly changed at high β-CD:CS ratios (6:1) due to the lower stability of the complexes (panel H).
:1) and β-CD-g-CS (3:1) at N/P ratios of 4 and 6, respectively, were selected for cellular uptake tests, as they showed the smallest particle size in Fig. 3. The cellular uptake results demonstrated a FBS-dependent behavior (Fig. 4). pDNA/β-CD-g-CS (3:1) NPs were significantly taken up by HepG2 cells in a medium containing FBS, while pDNA/β-CD-g-CS (2:1) NPs presented a greater cellular uptake in a FBS-free medium. In general, size and surface charge density play critical roles in the transfection efficiency of NPs. The size of the NPs usually increases in serum-supplemented media, as there are considerable electrostatic interactions between positively charged particles and negatively charged serum proteins. Moreover, the adsorption of proteins on the surface of the particles can reduce the surface charge density.41–43 Interestingly, the results of previous studies showed that cell activity increased in the presence of serum, but the medium supplemented with >50% serum could damage cells.44,45 Therefore, the higher transfection ability of pDNA/β-CD-g-CS (3:1) NPs in complete medium can be attributed to the greater stability and moderate charge density of the particles. In contrast, pDNA/β-CD-g-CS (2:1) NPs had positive surfaces that improved cellular uptake compared with pDNA/β-CD-g-CS (3:1) NPs.
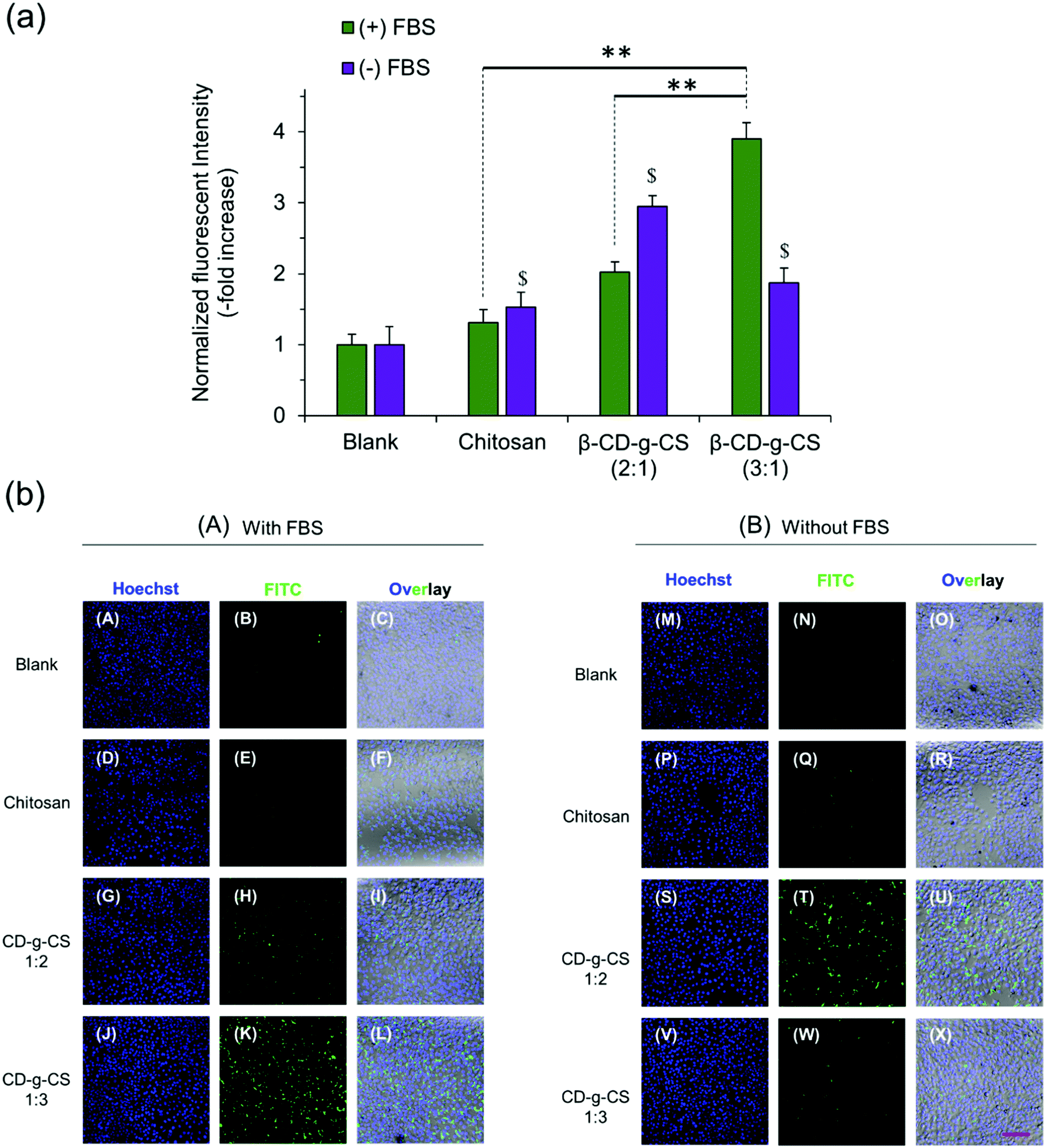 | ||
| Fig. 4 NP cellular uptake in HepG2 cells. (a) Normalized fluorescence intensity and (b) confocal micrographs of polymer/pDNA NPs at N/P ratios of 4 and 6 corresponding to β-CD-g-CS (2:1) and β-CD-g-CS (3:1), respectively. Cells were cultivated in DMEM supplemented with (A) and without (B) serum (FBS). Pure CS/pDNA NPs at an N/P ratio of 5 were used as the control. Data represent the mean ± standard deviation of n = 4. | ||
Interestingly, the cellular uptake of CS NPs in both cases was lower than β-CD-g-CS NPs. Therefore, we may conclude that the presence of β-CD on the NP surface and its interactions with the cell lipid membrane can significantly promote the cellular uptake of particles. The confocal images of cells incubated with NPs also qualitatively approved our previous data, where β-CD-g-CS (3:1) and β-CD-g-CS (2:1) NPs were significantly taken up by HepG2 cells in media with and without FBS, respectively.
The transfection efficiency of plasmid DNA/β-CD-g-CS nanoparticles was also evaluated, where the intracellular expression of the EGFP protein was calculated via the intensity of the green fluorescent light emitted from transfected cells. As shown in Fig. S3 (ESI†), pDNA/β-CD-g-CS had a significantly greater transfection efficiency compared with the pDNA/CS. In addition, nanoparticles released from microparticles could transfect the cells while the light intensity was not so strong, which was attributed to the lower concentration of EGFP–pDNA nanoparticles released from core–shell microparticles over the experimental period.
3.4. Cytotoxicity of nutlin-3a
Fig. S4 (ESI†) shows the growth inhibition of HepG2 cells after 24 h and 48 h treatment with nutlin-3a at different concentrations. The IC50 (inhibitory concentration at which 50% of the cells are killed) value of nutlin-3a was ∼0.1 mM on the first day, while it decreased to 0.05 mM after 48 h. Therefore, the longer incubation of the cells with nutlin-3a (as the main hypothesis of this study) could improve the treatment outcome and provide a strong justification for localized delivery of nutlin-3a to cancer cells in a controlled and sustained manner.3.5. Intracellular reactive oxygen species (ROS) measurements
Fig. S5a (ESI†) shows the normalized intracellular reactive oxygen species (ROS) level in HepG2 cells after being incubated with nutlin-3a for 6 h. The intracellular ROS level showed a dose-dependent trend, which was in agreement with the results of the free drug tests. However, the pretreatment of cells with NAC (ROS scavenger) significantly prevented the intracellular generation of ROS. The induction of oxidative stress upon treatment with nutlin-3a was further supported by confocal micrographs depicted in Fig. S5b (ESI†). Cells were stained with H2DCF–DA, a ROS fluorescent probe with a green light, where the intensity of the green light increased with the increase of ROS. The oxidative stress originates from the overproduction of oxidizing agents (e.g. O2˙−, OH˙, and H2O2) or depletion of intracellular antioxidant substances including enzymes and non-enzyme components.46,47 The accumulation of oxidizing agents in cells can impose serious damage on subcellular species such as mitochondria, amino acids, proteins, and DNA which eventually activate intracellular mechanisms for cell apoptosis.3.6. Wound closure and colony formation assays
Metastasis from a tumor site to the surrounding tissue constitutes the most life-threatening aspect of cancer.48,49 Therefore, the potential of HepG2 cells for migration and cancer metastasis was evaluated by wound closure and colony formation assays. Fig. 5a shows the results of the wound closure assay after 48 h of incubation with complete medium containing nutlin-3a at various concentrations. The results show that nutlin-3a was able to stop the cell proliferation, but cannot cause cell death at very low concentrations. While the gaps of the control samples were approximately closed after 48 h, the groups treated with nutlin-3a showed marginal cell proliferations and their gaps were still distinguishable after 48 h. In line with the cell migration results, the colony formation ability of HepG2 before and after treatment with nutlin-3a was also evaluated. As seen in Fig. 5b and c, the number and the size of colonies formed after 10 days were dose-dependent. The low concentrations of nutlin-3a had a marginal effect on the number of colonies formed. However, the size of the colonies is smaller than that of blank samples. In contrast, the number of colonies formed after 24 h treatment with 0.1 mM nutlin-3a (24 h – IC50 ∼ 0.1 mM) significantly decreased to 77 ± 34 colonies, which confirmed the inhibitory effect of nutlin-3a at a moderate concentration. Cancer metastasis mostly occurs after degradation of extracellular matrix components by proteolytic enzymes expressed by tumor cells.50 Recent findings have shown that intracellular rearrangement and multi-protein activities have undesirable impacts on cellular migration and invasion.51 In fact, the inactivation of wild-type p53 promotes actin cytoskeletal rearrangement52 and increases the cell migration ability. As the results showed, nutlin-3a as a non-genotoxic agent was able to reduce the metastasis incidence rate that might be due to the reactivation of wild-type p53 in HepG2 cells.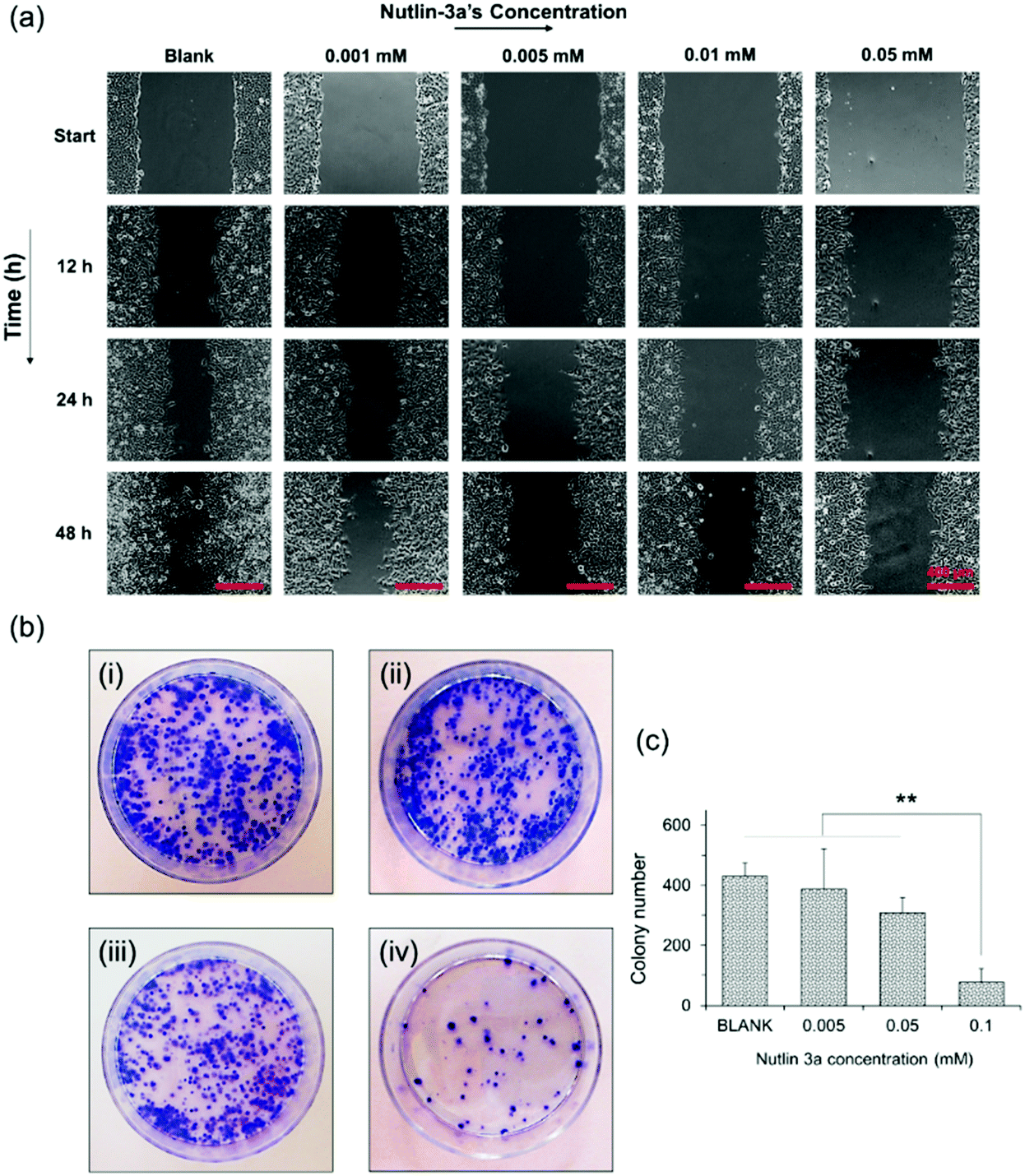 | ||
| Fig. 5 (a) In vitro wound closure experiments. HepG2 cells incubated with nutlin-3a solutions for 1 day. The cell monolayers were scraped in a straight line using a sterile 10 μl pipette tip. Images were acquired 0, 12, 24, and 48 h after scratching via a bright field inverted microscope. The control wound closure was set at 100% for cells without treatment (after 48 h). Scale bar = 100 μm. (b) Digital images of the colony formation assay. Colonies were produced by HepG2 cells following plating of 1000 cells and 10 days of incubation: (i) cells without treatment (blank); and cells were treated with nutlin-3 at (ii) 0.005 mM, (iii) 0.05 mM, and (iv) 0.1 mM at 37 °C for 24 h, and subsequently were plated in 50 mm Petri dishes. (c) Average number of colonies formed after 10 days. Data represent the mean ± standard deviation of n = 5. Statistical significance: **p < 0.01. | ||
3.7. In vitro expressions of p53, caspase 3 and MDM2 proteins (free-drug tests)
HepG2 cells express the wild-type p53 tumor suppressor gene that consequently leads to the activation of caspase 3 proteins and cell apoptosis. To investigate the molecular responses of HepG2 cells to anti-MDM2 agents, the intracellular expressions of p53, caspase 3, and MDM2 proteins were evaluated after treatment with nutlin-3a (0–0.25 mM) for 4 h. After treatment, elevated p53 and caspase 3 expressions were detected at nutlin-3a concentrations >0.05 mM (Fig. 6). However, the MDM2 level remained at the basal level due to the ability of nutlin-3a in the inhibition and/or deactivation of the MDM2 protein. The immunofluorescence staining of p53, caspase 3, and MDM2 proteins was also performed after 4 h treatment. As shown in Fig. 7, minimal p53 (panels P1–P4) and caspase 3 (panels Ca1–Ca4) were expressed by control (untreated) cells, where no fluorescent light was observed.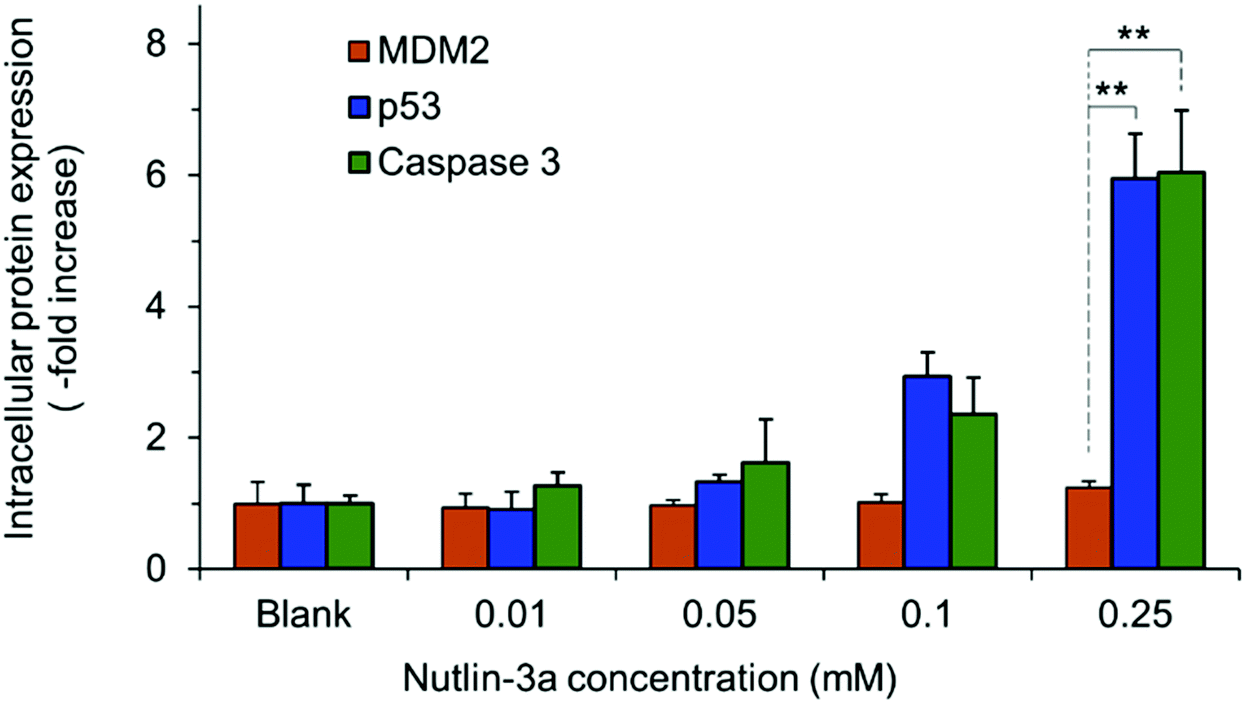 | ||
| Fig. 6 The intracellular expressions of p53, caspase 3, and MDM2 proteins in HepG2 cells at 4 h after commencement of the treatment. Cells were treated with 100 μl of media containing nutlin-3a at different concentrations. Data represent the average ± standard deviation of four independent measurements (n = 4). Statistical differences: **p < 0.01. | ||
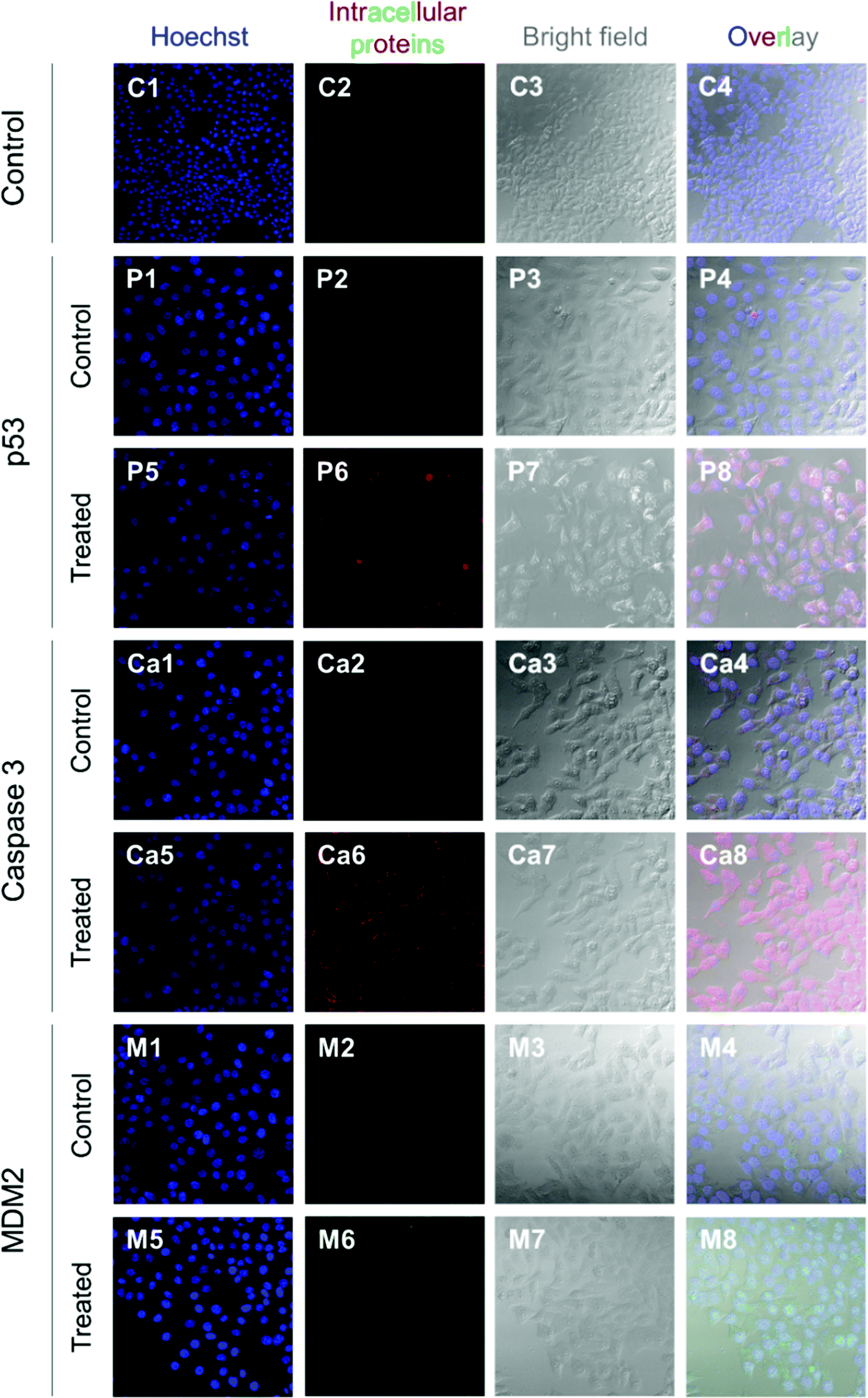 | ||
| Fig. 7 Immunofluorescence staining of p53, caspase 3, and MDM2 proteins in HepG2 cells at 4 h after commencement of the treatment. The p53 and caspase 3 proteins were stained with DyLight™ 550 conjugated goat anti-rabbit IgG (H + L) secondary antibodies (red) and MDM2 proteins were stained with DyLight™ 488 conjugated goat anti-mouse IgG (H + L) secondary antibodies (green). The cell nuclei were stained with Hoechst 33342 (blue). | ||
In contrast, the light intensity was significantly increased after treatment due to the elevated expression of p53 (panels P5–P8) and caspase 3 (panels Ca5–Ca8) by the cells. Since the green fluorescent light intensity remained unchanged for cells before and after treatment (panels M1–M8), it was concluded that the cells did not express the MDM2 protein or the active sites of the protein were blocked by nutlin-3a molecules such that primary antibodies could not bind to MDM2.
3.8. In vitro release tests
The release profiles of three different microparticle formulations are presented in Fig. 8. Nutlin-3a (a small molecule drug) and p53–pDNA NPs were loaded into core and shell compartments using CEHDA, respectively. The release of nutlin-3a exhibited an initial burst of ∼12% and ∼18% for formulations MPs1 and MPs3, respectively. The initial phases were followed by lag phases (7–10 days), in which the cumulative release of the drugs was around 1–2%. Eventually, the release profiles showed a near zero-order trend over the last 40 days. In contrast, NPs exhibited an initial burst release followed by continuous but very slow upward trends for formulations MPs2 and MPs3 (Fig. 8a). Typically, the release of small molecule drugs from drug delivery devices is dominated by an initial burst release followed by a relatively upward trend, indicating a diffusion-controlled process. On the other hand, NPs usually are released through an erosion process due to their larger size (Fig. 8b). The degradation process of microparticles begins upon contacting with aqueous solutions (e.g. PBS or body fluids). Initially, drug molecules that either are placed on the surface of the particles or encapsulated close to the surfaces are quickly released into the surrounding medium. However, the release of drugs from interior parts requires large concave indentations and water penetration. After ∼10 days of incubation, the degradation of the shell layer and the formation of larger pores on the shell layer facilitate the dissolution and diffusion of drug molecules in the medium while concurrently accelerating the degradation of the shell layer and the core compartment. In addition to the changes in the surface morphology, the surface erosion and bulk erosion are extended during the lag phase, which causes an upward trend in the release profiles during the last 40 days. Therefore, surface pore formation, water penetration, and degradation/erosion rates of polymer layers may be the critical factors extensively affecting the release profiles of the drug and NPs.53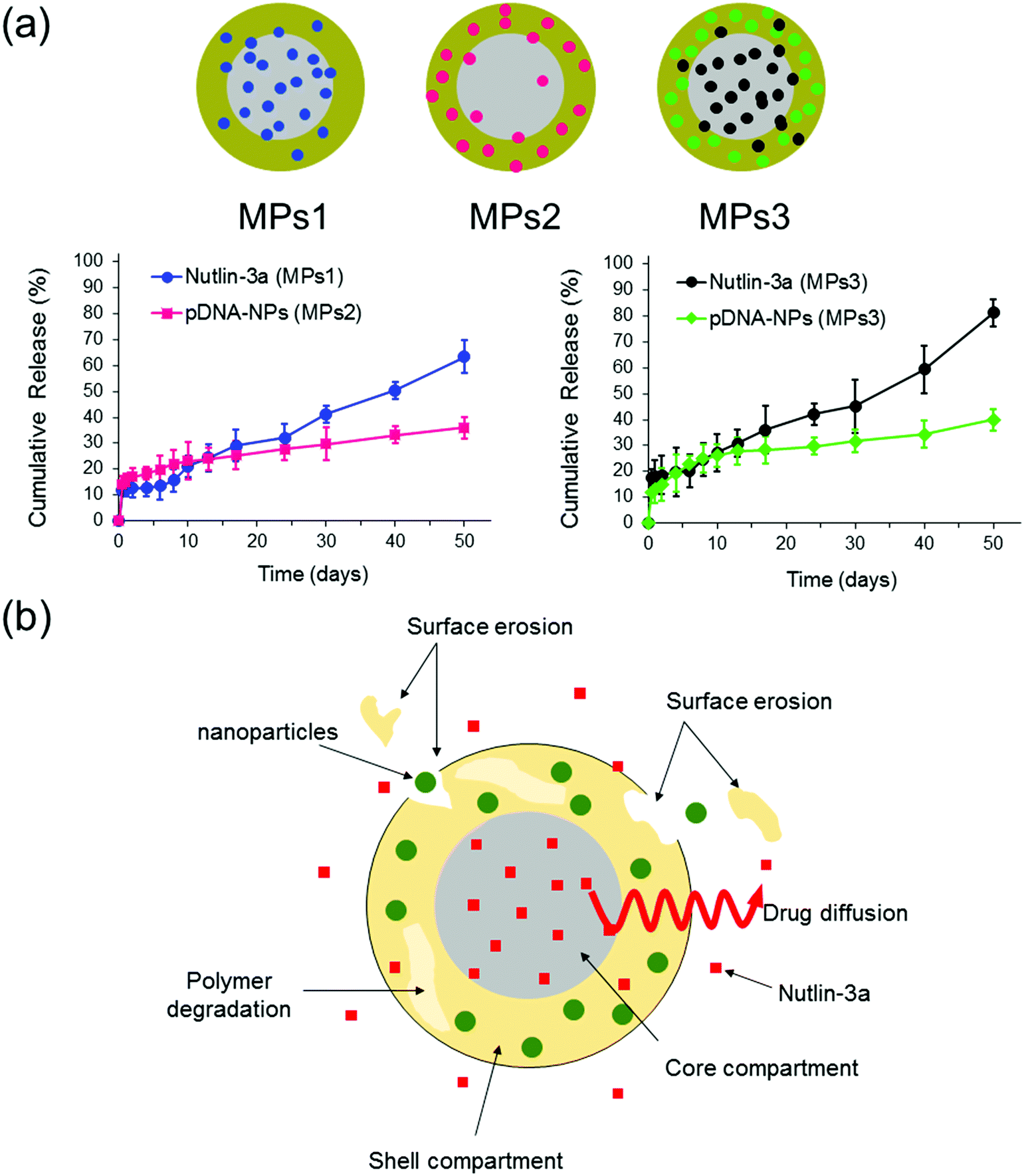 | ||
| Fig. 8 In vitro release profiles of nutlin-3a and p53–pDNA NPs for different core–shell microparticle formulations. MPs1: nutlin-3a (core), MPs2: pDNA–p53 NPs (shell), MPs3: nutlin-3a (core) and pDNA–p53 NPs (shell). (The drug loading contents (DLC) and encapsulation efficiencies (EE) of the different formulations are indicated in Table S2, ESI†.) | ||
As the particles are in the micron size range, they cannot be administered intravenously and must be injected either locally after tumor resection or directly within a tumor tissue, which depends on clinicians and cancer therapists. The long-term (over 50 days) drug and gene release provided using microparticles (Fig. 8a) can also reduce the number of injections required for a specified therapeutic regimen, while other drug administration routes may require multiple injections over the same period.
3.9. Cytotoxicity test (combined treatment)
After the successful encapsulation of nutlin-3a and pDNA–p53 NPs in core–shell microparticles fabricated by CEHDA, the particles were examined against HepG2 cells over five days. As explained in the previous section, the release of nutlin-3a from the microparticles delayed for ∼7 days. Therefore, the microparticles were incubated in 1 ml of sterilized 1× PBS for 7 days, before being transferred into a 96-well plate containing HepG2 cells. In order to mimic in vivo conditions, the equivalent amounts of drugs and genes released over five days (nutlin-3a: ∼13 μg per 0.5 ml, pDNA–p53 NPs: 0.22 μg per 0.5 ml) were added as free-drugs (FD) to cells and removed after 4 h. The results of cell viability experiments after 1 and 5 days of treatment are presented in Fig. 9. Cells treated with nutlin-3a (free-drug) exhibited the lowest viability (∼81%) compared to the other groups at the end of day one. After 5 days, the cell viability of the groups treated with nutlin-3a (FD) and NPs (FD) slightly decreased to ∼75% and ∼83%, respectively. However, greater antiproliferative activity was observed for the groups incubated with drug-loaded microparticles. The cell viability decreased below 40% for cells incubated with nutlin-3a loaded microparticles. In contrast, a combined therapy via core–shell microparticles demonstrated greater cytotoxicity in HepG2 cells (∼31%) compared to the single-agent loaded counterparts. The cell viability difference between the last two groups was statistically significant (p < 0.05). However, the difference was much greater for the other groups (p < 0.01). On day one, cells were exposed to high doses of nutlin-3a, where it showed a moderate antiproliferative effect in the respective group. Meanwhile, the drugs released from the microparticles did not impose severe toxicity on the cells. p53–pDNA NPs were approximately non-toxic to the cells as overexpression of MDM2 proteins (a p53 inhibitor) severely suppressed p53 activities. However, the sustained release of nutlin-3a from the core–shell microparticles over five days could markedly reduce cell viability. The antiproliferative activity of nutlin-3a could be further improved through the simultaneous delivery of p53–pDNA NPs and nutlin-3a using the core–shell microparticles. The greater therapeutic efficiency of the dual-agent loaded microparticles could be attributed to the release of fresh p53 genes and deactivation of MDM2 proteins by nutlin-3a, simultaneously. Moreover, the sustained release of nutlin-3a could continuously generate ROS that impose irreversible and detrimental damage on sub-cellular species. These indicate that there is an apparent advantage of combined p53–pDNA NP and nutlin-3a treatment.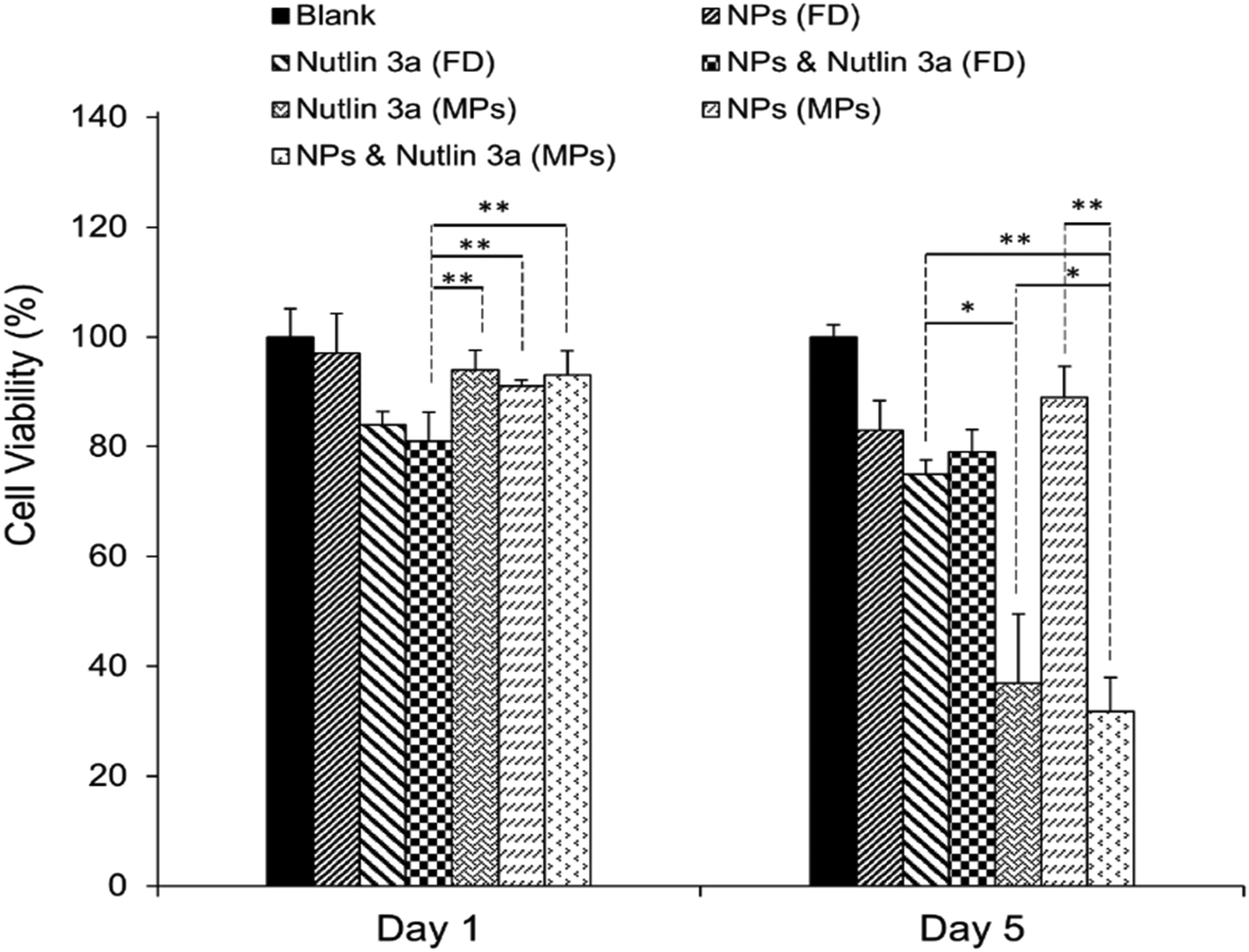 | ||
| Fig. 9 Long-term cell viability experiments. Cells were treated with free drugs or microparticles loaded with nutlin-3a and/or β-CD-g-CS/pDNA NPs for five days. The cell viability was determined using an MTS assay. Data represent the average ± standard deviation of four independent measurements (n = 4). Statistical differences: *p < 0.05, **p < 0.01. | ||
3.10. Intracellular expression of p53, caspase 3 and MDM2 proteins
Fig. 10 presents the normalized expression levels of p53, caspase 3 and MDM2 proteins after five days of treatment with nutlin-3a and β-CD-g-CS/pDNA NPs either as free drugs or as microparticle formulations. While cells treated with NPs (FD) and nutlin-3a (FD) showed a marginal increase in the p53 and caspase 3 expression, elevated levels of the p53 and caspase 3 expressions were detected in the cells treated with the dual-agent loaded microparticles. The nutlin-3a (FD)-treated cells showed at most ∼1.9-fold and ∼1.7-fold higher intracellular p53 and caspase 3 expressions than the control group by the fifth day, while the protein levels remained almost unchanged in the NP (FD)-treated cells. The significant expression of p53 and caspase 3 proteins in the cells treated with the microparticles can be attributed to the simultaneous accumulation of nutlin-3a and fresh p53 genes within the cells. Therefore, the combined nutlin-3a and NP (MP) formulation can induce greater apoptosis as demonstrated by long-term cell viability analysis. Interestingly, the MDM2 expression level did not change significantly and remained at the basal activity level in all groups.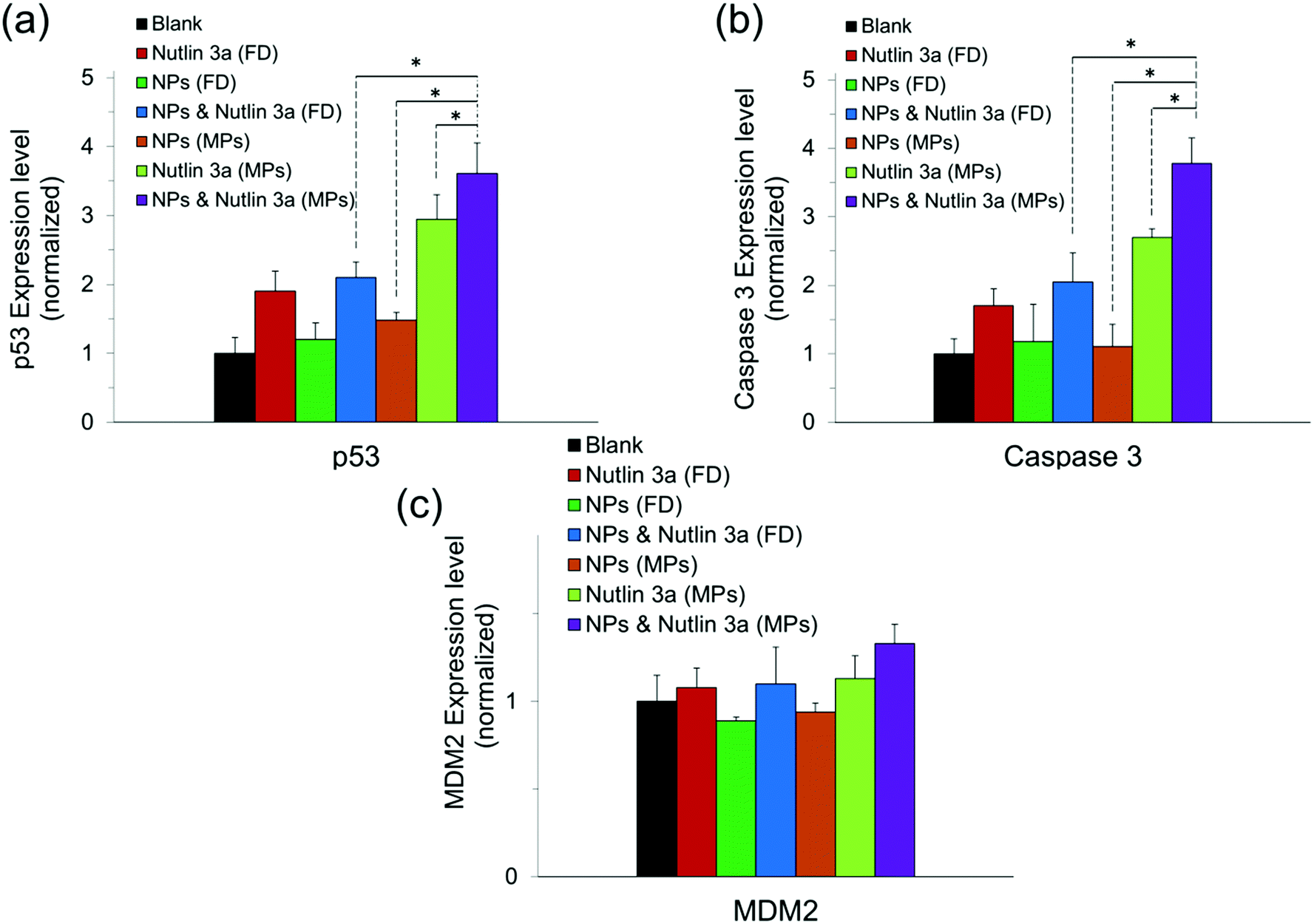 | ||
| Fig. 10 Intracellular expression of p53, caspase 3, and MDM2 proteins in HepG2 cells treated with free drugs and microparticles loaded with nutlin-3a and/or β-CD-g-CS /pDNA five days after commencement of the treatment. Data represent the average ± standard deviation of four independent measurements (n = 4). Statistical differences: *p < 0.05. | ||
Fig. 11 shows the immunofluorescence staining of p53, caspase 3, and MDM2 proteins in HepG2 cells treated with the combined formulation (MPs) at the end of day 5. The intensity of p53 and caspase 3 in combined NP and nutlin-3a (MP) treated cells was brighter and spread uniformly throughout the cells. The results were consistent with the colorimetric measurement of p53 and caspase 3 expression profiles presented in Fig. 10a and b. Moreover, the immunofluorescence staining of the MDM2 protein in the control and treated groups showed relatively similar intensities, which were in line with data shown in Fig. 10c.
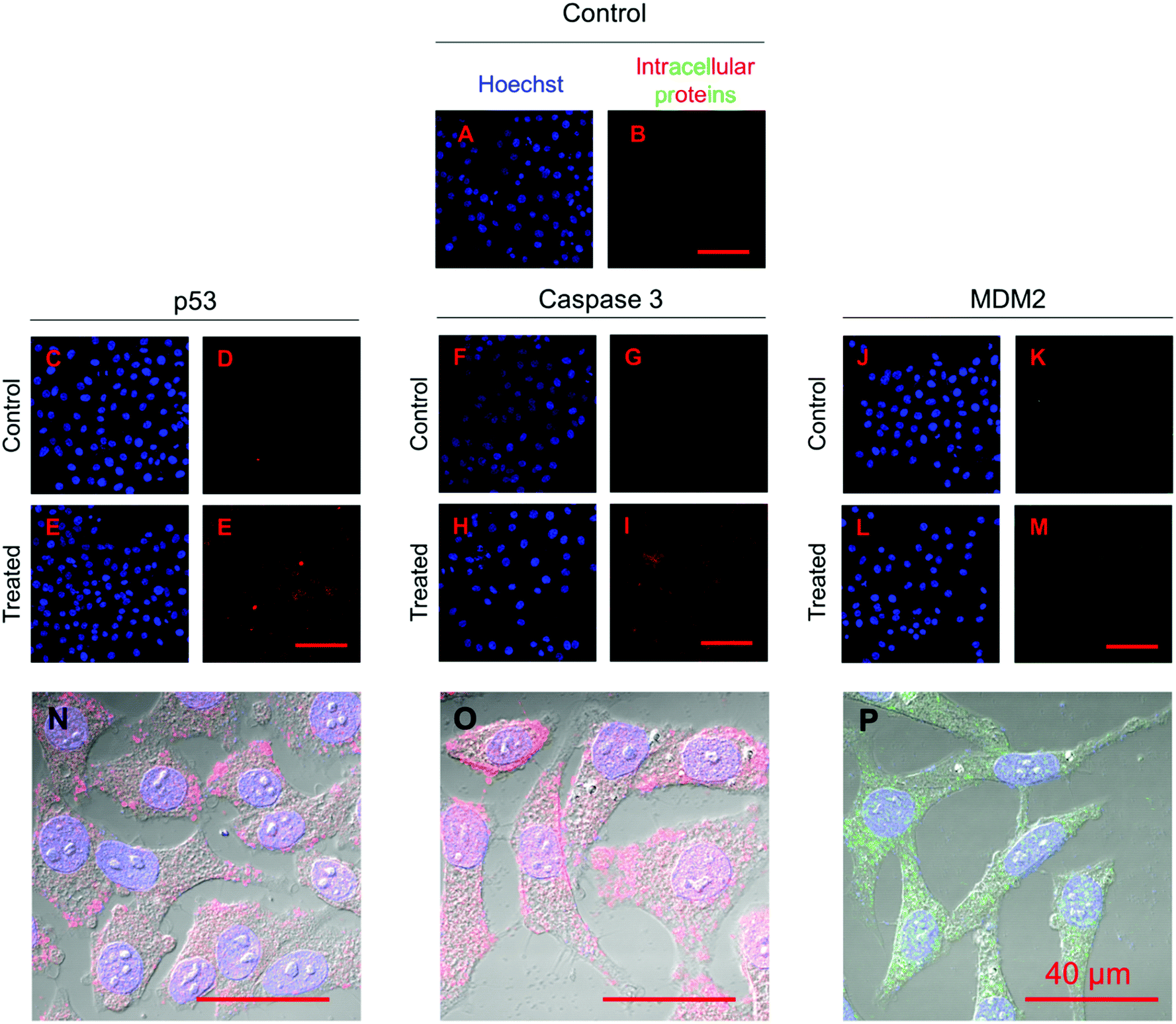 | ||
| Fig. 11 Immunofluorescence staining of p53, caspase 3, and MDM2 proteins in HepG2 cells treated with microparticles loaded with nutlin-3a and β-CD-g-CS/pDNA 5 days after commencement of the treatment. The p53 and caspase 3 proteins were stained with DyLight™ 550 conjugated goat anti-rabbit IgG (H + L) secondary antibodies (red) and MDM2 proteins were stained with DyLight™ 488 conjugated goat anti-mouse IgG (H + L) secondary antibodies (green). The cell nuclei were stained with Hoechst 33342 (blue). | ||
p53 is a transcription factor that plays a key role in the tumor suppression pathway, the regulation of cell cycles, apoptosis, DNA repair, the cellular mechanism, and innate immunity. Therefore, the inactivation or mutation of the p53 protein is believed to be responsible for downregulating p53 activities, mainly in cell-cycle arrest, apoptosis, and/or senescence.11,18,54 In the absence of p53 mutation, the activities of p53 proteins may be suppressed by an oncoprotein, MDM2, overexpressed in malignant cells. Therefore, the disruption of the MDM2–p53 interaction is one of the potential strategies in cancer therapy. In the present study, we hypothesized that the disruption of the p53–MDM2 interaction and providing fresh p53 genes could simultaneously reactivate the p53 regulatory pathway, which results in cell death. However, the intravenous administration of MDM2 inhibitors may cause serious side effects in normal cells, where the upregulation of p53 activities leads to cell cycle arrest in healthy cells. To address this problem and to provide higher drug concentrations at tumor sites, core–shell microparticles were employed as depots that release the MDM2 inhibitor agent and p53–pDNA NPs in a controlled and sustained manner. Nutlin-3a, one of the first small molecules discovered for blocking MDM2 active sites, and p53–pDNA loaded nanoparticles were successfully encapsulated in microparticles. The combination of nanoparticles & nutlin-3a in a microparticle formulation can reduce the number of injections, provide superior control over the release of the agents through controlling particle size, polymer degradation rates, and encapsulation of the agents into appropriate compartments (i.e. core or shell). The latter provides a unique opportunity for the controlled release of the drugs in a concurrent or sequential manner.36,38
It is evident that free-drug administration can provide acute toxicity lasting for a limited duration (Fig. 10), whereas the incubation of cells with both agents at lower doses over an extended period is capable of reducing cell viability and greatly improving treatment efficiency. The controlled and sustained release of the agents showed the extent of apoptosis from caspase 3 expression. In addition, our finding revealed the ability of nutlin-3a to induce ROS in cells and activate caspase 3 upon the release of cytochrome c from damaged mitochondria.47,55,56 Although the second mechanism has been approved for other anticancer agents (e.g. cisplatin and paclitaxel), it must be evaluated for new agents such as nutlin-3a.
4. Conclusions
In this study, core–shell microparticles loaded with nutlin-3a and p53–NPs were successfully fabricated using the CEHDA technique. The microparticle formulations could release the agents with near zero-order kinetics over 50 days, after a small initial burst release (∼15–20%). The results showed that the conjugation of β-CD onto chitosan could remarkably improve the cellular uptake of nanoparticles through the strong interaction of β-CD molecules (on the surface of NPs) with the lipid bilayer in the cell membrane. The in vitro cell culture demonstrated that the proposed formulation was capable of improving the anticancer efficacy of nutlin-3a and p53 proteins in a synergistic manner through the activation of p53 and the elevation of intracellular ROS. Although the expression of p53 and caspase 3 proteins was significantly increased and maintained at elevated levels during treatment with the combined formulation, the MDM2 expression level was maintained at the basal level due to the blockage of MDM2 binding sites. Owing to these advantages mentioned above, the proposed formulation holds great promise for the treatment of cancers with alterations in wild-type p53 genes, while enhancing the bioavailability of the agents and significantly reducing their adverse side effects on healthy tissues through localized drug delivery.Conflict of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was financially supported by A*STAR and the National University of Singapore under project/grant numbers APG2013/40A (A*STAR BMRC Strategic Positioning Fund, A*STAR-P&G Collaboration, R279-000-487-305) and R261-509-001-646 (3D Printing Initiatives), respectively. Pooya Davoodi greatly appreciates the National University of Singapore for a PhD graduate research scholarship. We thank Yu Kian Goh and You Kang Lim for technical help with experiments.References
- C. Frezza and C. P. Martins, Drug Resist. Updates, 2012, 15, 258–267 CrossRef CAS PubMed
.
- B. Hong, A. P. van den Heuvel, V. V. Prabhu, S. Zhang and W. S. El-Deiry, Curr. Drug Targets, 2014, 15, 80–89 CrossRef CAS PubMed
- M. H. Brodsky, W. Nordstrom, G. Tsang, E. Kwan, G. M. Rubin and J. M. Abrams, Cell, 2000, 101, 103–113 CrossRef CAS PubMed
- B. D. Lehmann and J. A. Pietenpol, J. Clin. Oncol., 2012, 30, 3648–3650 CrossRef CAS PubMed
- C. J. Brown, S. Lain, C. S. Verma, A. R. Fersht and D. P. Lane, Nat. Rev. Cancer, 2009, 9, 862–873 CrossRef CAS PubMed
- D. I. Zheleva, D. P. Lane and P. M. Fischer, Mini-Rev. Med. Chem., 2003, 3, 257–270 CrossRef CAS PubMed
- P. Chene, Mol. Cancer Res., 2004, 2, 20–28 CAS
- S. Shangary and S. Wang, Annu. Rev. Pharmacol. Toxicol., 2009, 49, 223–241 CrossRef CAS PubMed
- L. T. Vassilev, Trends Mol. Med., 2007, 13, 23–31 CrossRef CAS PubMed
- B. Vogelstein, D. Lane and A. J. Levine, Nature, 2000, 408, 307–310 CrossRef CAS PubMed
- K. H. Vousden and X. Lu, Nat. Rev. Cancer, 2002, 2, 594–604 CrossRef CAS PubMed
- A. G. Ostermeyer, E. Runko, B. Winkfield, B. Ahn and U. M. Moll, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 15190–15194 CrossRef CAS
- M. V. Poyurovsky, C. Katz, O. Laptenko, R. Beckerman, M. Lokshin, J. Ahn, I. J. Byeon, R. Gabizon, M. Mattia, A. Zupnick, L. M. Brown, A. Friedler and C. Prives, Nat. Struct. Mol. Biol., 2010, 17, 982–989 CAS
- D. A. Freedman, L. Wu and A. J. Levine, Cell. Mol. Life Sci., 1999, 55, 96–107 CrossRef CAS PubMed
- J. Momand, H. H. Wu and G. Dasgupta, Gene, 2000, 242, 15–29 CrossRef CAS PubMed
- X. Wu, J. H. Bayle, D. Olson and A. J. Levine, Genes Dev., 1993, 7, 1126–1132 CrossRef CAS PubMed
- S. V. Tokalov and N. D. Abolmaali, BMC Cancer, 2010, 10, 57 CrossRef PubMed
- G. Selivanova, FEBS Lett., 2014, 588, 2628–2638 CrossRef CAS PubMed
- Y. He, Y. Nie, L. Xie, H. Song and Z. Gu, Biomaterials, 2014, 35, 1657–1666 CrossRef CAS PubMed
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS PubMed
- R. Montes de Oca Luna, D. S. Wagner and G. Lozano, Nature, 1995, 378, 203–206 CrossRef CAS PubMed
- I. Ringshausen, C. C. O'Shea, A. J. Finch, L. B. Swigart and G. I. Evan, Cancer Cell, 2006, 10, 501–514 CrossRef CAS PubMed
- S. M. Mendrysa, M. K. McElwee, J. Michalowski, K. A. O'Leary, K. M. Young and M. E. Perry, Mol. Cell. Biol., 2003, 23, 462–473 CrossRef CAS PubMed
- A. V. Gudkov and E. A. Komarova, Nat. Rev. Cancer, 2003, 3, 117–129 CrossRef CAS PubMed
- W. H. Chappell, B. D. Lehmann, D. M. Terrian, S. L. Abrams, L. S. Steelman and J. A. McCubrey, Cell Cycle, 2012, 11, 4579–4588 CrossRef CAS PubMed
- Y. Peng, C. Li, L. Chen, S. Sebti and J. Chen, Oncogene, 2003, 22, 4478–4487 CrossRef CAS PubMed
- K. Kumar, G. Vulugundam, P. Kondaiah and S. Bhattachary, J. Mater. Chem. B, 2015, 3, 2318–2330 RSC
- X. Cai, Y. Li, D. Yue, Q. Yi, S. Li, D. Shi and Z. Gu, J. Mater. Chem. B, 2014, 2, 7210–7221 RSC
- Y. D. Kim, T. E. Park, B. Singh, K. S. Cho, J. N. Sangshetti, Y. J. Choi, R. B. Arote and C. S. Cho, J. Mater. Chem. B, 2016, 4, 2208–2218 RSC
- P. Davoodi, M. P. Srinivasan and C. H. Wang, Acta Biomater., 2016, 39, 79–93 CrossRef CAS PubMed
- W. Li, J. Shi, C. Zhang, M. Li, L. Gan, H. Xu and X. Yang, J. Mater. Chem. B, 2014, 2, 4901–4910 RSC
- A. Singh, M. Talekar, T. H. Tran, A. Samanta, R. Sundaram and M. Amiji, J. Mater. Chem. B, 2014, 2, 8069–8084 RSC
- H. Q. Mao, K. Roy, V. L. Troung-Le, K. A. Janes, K. Y. Lin, Y. Wang, J. T. August and K. W. Leong, J. Controlled Release, 2001, 70, 399–421 CrossRef CAS PubMed
- R. C. Petter, J. S. Salek, C. T. Sikorski, G. Kumaravel and F. T. Lin, J. Am. Chem. Soc., 1990, 112, 3860–3868 CrossRef CAS
- J. Xie, J. Jiang, P. Davoodi, M. P. Srinivasan and C.-H. Wang, Chem. Eng. Sci., 2015, 125, 32–57 CrossRef CAS PubMed
- P. Davoodi, F. Feng, Q. Xu, W. C. Yan, Y. W. Tong, M. P. Srinivasan, V. K. Sharma and C. H. Wang, J. Controlled Release, 2015, 205, 70–82 CrossRef CAS PubMed
- F. L. Mi, Y. Y. Wu, Y. L. Chiu, M. C. Chen, H. W. Sung, S. H. Yu, S. S. Shyu and M. F. Huang, Biomacromolecules, 2007, 8, 892–898 CrossRef CAS PubMed
- W. C. Yan, P. Davoodi, Y. W. Tong and C. H. Wang, AIChE J., 2016, 62, 4259–4276 CrossRef CAS
- P. Davoodi, W. C. Ng, W. C. Yan, M. P. Srinivasan and C. H. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 22785–22800 CAS
- R. J. Samuels, J. Polym. Sci., Part B: Polym. Phys., 1981, 19, 1081–1105 CrossRef CAS
- T. Ishii, Y. Okahata and T. Sato, Biochim. Biophys. Acta, 2001, 1514, 51–64 CrossRef CAS
- D. M. Drake, R. K. Keswani and D. W. Pack, Pharm. Res., 2010, 27, 2457–2465 CrossRef CAS PubMed
- Y. J. Kim, T. W. Kim, H. Chung, I. C. Kwon, H. C. Sung and S. Y. Jeong, Int. J. Pharm., 2003, 252, 241–252 CrossRef CAS PubMed
- P. Erbacher, S. Zou, T. Bettinger, A. M. Steffan and J. S. Remy, Pharm. Res., 1998, 15, 1332–1339 CrossRef CAS
- S. Nimesh, M. M. Thibault, M. Lavertu and M. D. Buschmann, Mol. Biotechnol., 2010, 46, 182–196 CrossRef CAS PubMed
- B. Halliwell, Plant Physiol., 2006, 141, 312–322 CrossRef CAS PubMed
- Y. I. Chirino and J. Pedraza-Chaverri, Exp. Toxicol. Pathol., 2009, 61, 223–242 CrossRef CAS PubMed
- Y. Kang, Breast Dis., 2006, 26, 129–138 CrossRef CAS PubMed
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed
- L. A. Liotta, Breast Cancer Res. Treat., 1988, 11, 113–124 CrossRef CAS PubMed
- D. M. Moran and C. G. Maki, Mol. Cancer Ther., 2010, 9, 895–905 CrossRef CAS PubMed
- A. J. Ridley, M. A. Schwartz, K. Burridge, R. A. Firtel, M. H. Ginsberg, G. Borisy, J. T. Parsons and A. R. Horwitz, Science, 2003, 302, 1704–1709 CrossRef CAS PubMed
- Q. Xu, S. E. Chin, C. H. Wang and D. W. Pack, Biomaterials, 2013, 34, 3902–3911 CrossRef CAS PubMed
- J. G. Teodoro, S. K. Evans and M. R. Green, J. Mol. Med., 2007, 85, 1175–1186 CrossRef CAS PubMed
- N. M. Martins, N. A. Santos, C. Curti, M. L. Bianchi and A. C. Santos, J. Appl. Toxicol., 2008, 28, 337–344 CrossRef CAS PubMed
- N. Andre, D. Braguer, G. Brasseur, A. Goncalves, D. Lemesle-Meunier, S. Guise, M. A. Jordan and C. Briand, Cancer Res., 2000, 60, 5349–5353 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb00481h |
| This journal is © The Royal Society of Chemistry 2017 |