
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the local solvent composition at a drug carrier surface: the effect of dimethyl sulfoxide/water mixture on the photofunctional properties of hypericin–β-lactoglobulin complexes
P.
Delcanale
a,
B.
Rodríguez-Amigo
b,
J.
Juárez-Jiménez
 c,
F. J.
Luque
c,
S.
Abbruzzetti
ad,
M.
Agut
b,
S.
Nonell
*b and
C.
Viappiani
*ad
c,
F. J.
Luque
c,
S.
Abbruzzetti
ad,
M.
Agut
b,
S.
Nonell
*b and
C.
Viappiani
*ad
aDipartimento di Scienze Matematiche, Fisiche e Informatiche, Università di Parma, Parco Area delle Scienze 7A, 43124 Parma, Italy. E-mail: cristiano.viappiani@unipr.it
bInstitut Quimic de Sarrià, Universitat Ramon Llull, Via Augusta 390, 08017 Barcelona, Spain. E-mail: santi.nonell@iqs.url.edu
cDepartment of Nutrition, Food Science, and Gastronomy, and Institute of Biomedicine, University of Barcelona, Av. Prat de la Riba 171, 08921 Santa Coloma de Gramenet, Spain
dNEST, Istituto Nanoscienze, Consiglio Nazionale delle Ricerche, Piazza San Silvestro 12, 56127 Pisa, Italy
First published on 20th January 2017
Abstract
Aggregation is a major problem for the anti-microbial photodynamic applications of hydrophobic photosensitizers since it strongly reduces the amount of singlet oxygen generated in aqueous solutions. Binding of hypericin (Hyp) to the milk whey protein β-lactoglobulin (βLG), occurring at the two hydrophobic cavities located at the interface of the protein homodimer, can be exploited to confer water-solubility and biocompatibility to the photosensitizer. The introduction of a small amount of the organic cosolvent dimethyl sulfoxide (DMSO) leads to a remarkable improvement of the photophysical properties of the complex Hyp–βLG by increasing its fluorescence emission and singlet oxygen photosensitization quantum yields. Surprisingly, the ability of the complex to photo-inactivate bacteria of the strain Staphylococcus aureus is strongly reduced in the presence of DMSO, despite the higher yield of photosensitization. The reasons for this apparently contradictory behavior are investigated, providing new insights into the use of carrier systems for hydrophobic photosensitizers.
Introduction
The use of photosensitization for antimicrobial purposes is gaining interest in medical applications for the treatment of localized infections,1,2 and for the decontamination and disinfection processes in the food industry.3Photosensitization is a photo-reaction involving an otherwise non-toxic molecule, called a photosensitizer (PS), visible light and molecular oxygen. The absorption of a photon by the PS results in the formation of a triplet state (with yield ΦT), showing a relatively long lifetime (usually in the μs time scale). The triplet state is efficiently quenched by interaction with molecular oxygen leading to the production of cytotoxic reactive oxygen species (ROS), in most cases the non-radical electronically-excited singlet oxygen (1O2).4 The high reactivity of 1O2 towards multiple cellular components (e.g. membrane lipids, proteins and nucleic acids) precludes the selection of photo-resistant species.5,6
Hypericin (Hyp) is a naturally occurring PS, found in the plants belonging to the genus Hypericum (e.g. St. John's wort).7 Hyp has proved to be efficient as an antibacterial,8–10 antiviral11 and antifungal19 photodynamic agent. When dissolved in organic solvents, Hyp is endowed with a good quantum yield of fluorescence emission (ΦF = 0.35 ± 0.02 in dimethyl sulfoxide (DMSO)12) and singlet oxygen sensitization (ΦΔ = 0.32 in ethanol,13 0.39 ± 0.01 in methanol14) upon excitation with visible light. One of the main problems to overcome in the delivery of Hyp to targeted cells is the very low water-solubility that leads to the aggregation of the compound and almost complete loss of fluorescence emission and singlet oxygen sensitization.15,16 Hyp is thus usually delivered by means of suitable carriers like liposomes17 or nanoparticles,10,18 in some cases endowed with targeting capabilities.19 Among other carriers, proteins are highly biocompatible systems that warrant bioavailability of the PS. We recently demonstrated that the complex between Hyp and apomyoglobin (apoMb) is an effective photosensitizing agent for Gram positive bacteria,20 with fluorescence properties that allow us to localize the PS with sub-diffraction resolution on bacterial cells, using STED (Stimulated Emission Depletion) microscopy.21
We also demonstrated the use of β-lactoglobulin (βLG) as a carrier for Hyp in aqueous solutions such as phosphate-buffered saline (PBS), with the advantage, relative to apoMb, that it could carry more Hyp molecules per protein unit.22 Since βLG is one of the most abundant proteins of the bovine milk, its use is of special interest for antimicrobial applications in the dairy industry. However, although the complex Hyp–βLG proved effective as a photosensitizing agent, its spectral properties and the low fluorescence and singlet oxygen quantum yields suggest that Hyp is not fully monomeric when bound to the protein, which prevents the full realization of its photosensitizing potential.22
Since βLG can be coated with DMSO23 we hypothesize that the DMSO layer around βLG could provide a better environment for the bound Hyp molecules. In this work, we seek to study whether the use of DMSO as a cosolvent improves the photophysical properties of Hyp–βLG, thereby assessing its potential as a photodisinfection agent for materials contaminated with bacteria. Indeed, the results confirm that DMSO as a cosolvent leads to an improvement in the fluorescence and singlet oxygen yields of bound Hyp. Surprisingly, the photo-inactivating action of the complex against bacteria was decreased. This apparently contradictory finding demonstrates that a high efficiency of 1O2 photosensitization may not necessarily translate into higher photodynamic efficiency against bacteria.
Results and discussion
Hyp and βLG form a stable complex in a DMSO:PBS mixture
βLG spontaneously forms homodimers (2βLG) under physiological conditions and under the conditions encountered in milk processing.24,25 Dimerization involves the antiparallel alignment of the strand βI (residues 163–167), not involved in the eight-stranded β-barrel, assisted by the formation of hydrogen bonds between the interacting strands, and the intersubunit interface is also shaped by the loop AB, where residues Asp49 and Arg66 participate in salt bridges. As previously shown by molecular modelling, Hyp can a priori be accommodated in the two clefts formed at the interface of 2βLG. In the narrower cleft, which is shaped by the α-helix that precedes the strand βI, Hyp can be inserted only as a monomer. The other cleft, formed by loops AB and CD, which contains Trp77, is wider and might accommodate the binding of dimeric Hyp, and possibly higher order aggregates.22Experimental proof for the formation of a stable complex between Hyp and 2βLG is provided by a combination of spectroscopic techniques. Hyp is readily soluble in polar organic solvents, like DMSO, where it shows a structured absorption spectrum, with two sharp bands at 555 nm and 599 nm, and intense fluorescence emission with two bands centered at 604 nm and 653 nm (Fig. 1A and B, red). In PBS buffer, absorption bands are lowered in intensity and less structured, and the fluorescence emission is almost completely quenched (Fig. 1A and B, black), as a consequence of the aggregation of the molecule. In the presence of 2βLG in the PBS buffered solution, the structured absorption spectrum is partially recovered, with bands centered around 560 nm and 598 nm, along with fluorescence emission (Fig. 1A and B, blue). As previously observed for apoMb20 and bovine serum albumin,26 these facts are a consequence of the binding of Hyp to the protein, which can accommodate the molecule in a less-polar environment than water, thus preventing the aggregation. Time-resolved fluorescence emission measurements for Hyp in solution in the presence of 2βLG afford a biexponential decay with lifetimes (τF) of 3.9 ns (35%) and 6.7 ns (65%), consistent with the values observed for Hyp bound to apoMb, where the high concentration of the long-lived species was attributed to Hyp shielded from the aqueous solvent.20 As can be observed in Fig. 1C, Hyp in solution with 2βLG shows a non-zero anisotropy fluorescence excitation spectrum, indicating that the rotational diffusion of the molecule is limited because of the interaction with the protein scaffold. In comparison, Hyp dissolved in pure DMSO gives a zero anisotropy spectrum (Fig. 1C, red), due to the faster rotational diffusion.
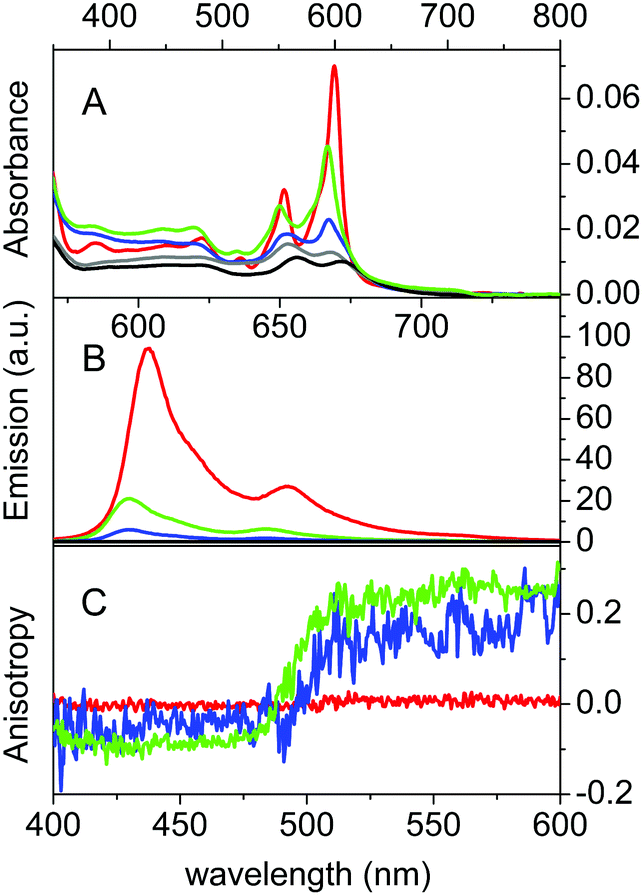 | ||
| Fig. 1 Absorption (A), fluorescence emission (B) and fluorescence anisotropy (C) spectra of Hyp in DMSO (red), Hyp in PBS (black), Hyp in 20% PBS–DMSO (grey), Hyp2βLG in PBS (blue) and Hyp2βLG in 20% PBS–DMSO (green). The Hyp concentration is 4 μM and that of βLG, when present, is 200 μM. Emission spectra are collected after excitation at 554 nm, T = 20 °C. Fluorescence anisotropy spectra are obtained from excitation spectra (λem = 620 nm, T = 20 °C). | ||
Fluorescence Correlation Spectroscopy (FCS) provided additional proof for the formation of a stable complex between 2βLG and Hyp. FCS is useful to determine the diffusion coefficient of molecules taking advantage of the spontaneous thermal fluctuation in the fluorescence emission intensity of the freely diffusing fluorophores in solution.27,28Fig. 2A shows the cross-correlation function determined for 1 nM Hyp in the presence of 30 μM βLG. The cross-correlation signal is well described by a model comprising diffusional motion, on the 10 μs – 1 ms time scale, and triplet decay, occurring on the 1–10 μs time scale. The diffusion coefficient of the dominant diffusing species is D = 70 μm2 s−1, consistent with the value expected for Hyp bound to 2βLG.
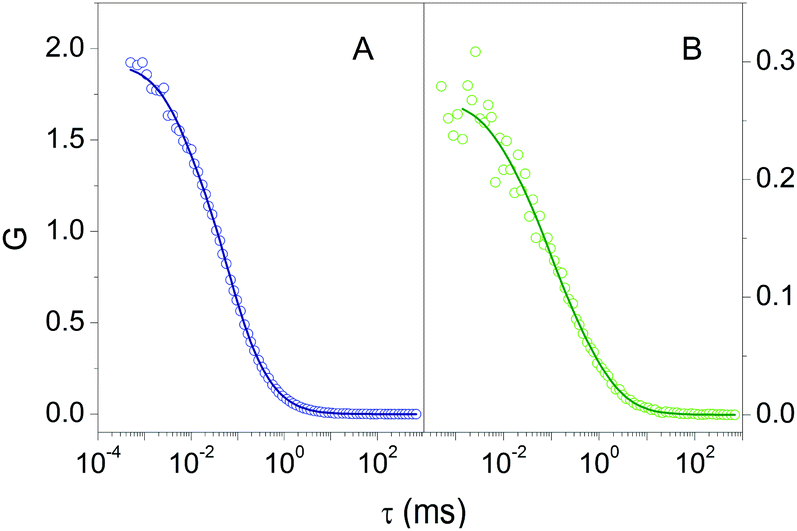 | ||
| Fig. 2 Cross correlation functions for 1 nM Hyp in PBS buffer in the presence of 30 μM βLG (A – open circles) and 1 nM Hyp in the mixture PBS–DMSO in the presence of 36 μM βLG (B – open circles). The solid lines are the results of the fit using a model comprising triplet state decay and diffusion. Excitation 475 nm, detection at 670 nm. | ||
Coating βLG with DMSO improves the photofunctional properties of bound Hyp
A key issue for potential applications in photosensitization-based antimicrobial treatments is that the PS retains the ability to populate the triplet state upon photo-excitation and photosensitize the production of 1O2. As shown in Table 1, the complex Hyp2βLG in PBS buffer has a quantum yield for the triplet state formation ΦT = 0.050 ± 0.002. The triplet state shows a lifetime decay (τT) of around 10 μs, considerably longer than the value obtained for Hyp in DMSO (1.4 μs) and similar to the one reported for Hyp bound to apoMb (11.1 μs), where this relatively long lifetime was attributed to the shielding of Hyp from the solvent by the protein scaffold.20 Hyp2βLG photosensitizes 1O2 with a yield ΦΔ = 0.065 ± 0.010. The similar ΦΔ and ΦT values measured for Hyp2βLG in PBS buffer indicate that the efficiency of the energy transfer between the triplet state of Hyp and 3O2, leading to the production of 1O2, is close to unity. However, the value retrieved for ΦΔ is quite low in comparison to the one observed for Hyp in pure DMSO (0.28 ± 0.05) or Hyp bound to apoMb (0.14 ± 0.03).31 According to molecular modeling studies, the reduced fraction of active PS may be due to the preferential binding of Hyp, possibly as an aggregate, to the wide cleft in 2βLG, where it is stacked against Trp77. Moreover, when bound to the wide cleft, one face of Hyp is exposed to the aqueous solvent, an interaction which may be detrimental to the photophysics of the compound.| Sample | Solvent | τ F (ns) | Φ F | τ T (μs) | Φ T | τ Δ (μs) | Φ Δ |
|---|---|---|---|---|---|---|---|
| a Laser flash Photolysis. b Time-resolved NIR phosphorescence detection. c Fluorescence correlation spectroscopy. | |||||||
| Hyp | DMSO | 5.5 ± 0.1 (100%) | 0.35 ± 0.0212 | 1.4 ± 0.1a | 0.3513 | 5.5 ± 0.1 | 0.28 ± 0.0529 |
| Hyp | PBS + S. aureus | 0.2 (48%) | |||||
| 4.2 (52%) | |||||||
| Hyp | PBS–DMSO 20% | 3.5 (100%) | |||||
| +S. aureus | 0.5 (48%) | ||||||
| 3.6 (52%) | |||||||
| Hyp2βLG | PBS | 3.9 (35%) | 0.03 ± 0.01 | 10 ± 2a | 0.050 ± 0.002 | 2.3 ± 0.1 | 0.065 ± 0.010 |
| 6.7 (65%) | 8.6b | ||||||
| 9.6c | |||||||
| +S. aureus | 4.6 (45%) | 8 ± 1a | |||||
| 7.0 (55%) | |||||||
| Hyp2βLG | PBS–DMSO 20% | 0.2 (20%) | 0.06 ± 0.01 | 8.2 ± 0.4a | 0.170 ± 0.002 | 2.5 ± 0.1 | 0.12 ± 0.05 |
| 5.6 (80%) | 8.4b | ||||||
| +S. aureus | 5.5 (100%) | 7.5c | |||||
| 9 ± 1a | |||||||
To overcome these limitations we have taken advantage of the preferential interactions between DMSO and 2βLG. Since DMSO (up to 50%) is known to coat 2βLG without affecting its structure to any appreciable extent,23 we have introduced a small amount of DMSO as a cosolvent, with the aim of providing a better environment to bound Hyp, thus increasing the fraction of photo-active bound molecules. We chose a DMSO concentration of 20% (VDMSO/VPBS), corresponding to ∼17% (VDMSO/Vtot), well below the limit at which denaturation is observed. Molecular dynamics (MD) simulations performed for dimeric βLG in a DMSO/water mixture at this concentration reveal that DMSO molecules tend to fill specific pockets in hydrophobic patches of the protein surface, including the areas corresponding to the narrow and wide clefts formed at the interface of the interacting monomers (Fig. 3). This agrees with the experimental finding that at low DMSO concentrations there is a negative preferential binding of DMSO molecules,23 reflecting the favorable interactions of DMSO with aliphatic and aromatic residues. Furthermore, mapping of the highly populated binding sites for organic probes is valuable to identify ‘hot spots’ for the binding of small molecules.30–33 The distribution of DMSO molecules on the protein surface is consistent with the observation, based on THz spectroscopy, that βLG is a relatively hydrophobic protein not displaying an extended long-range hydration dynamics,34 as is observed for other hydrophilic proteins.35,36
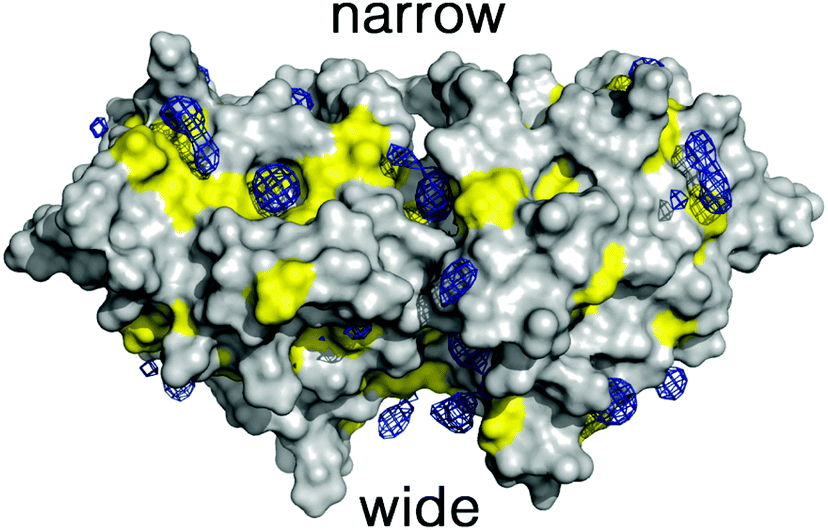 | ||
| Fig. 3 Preferential distribution of DMSO molecules around the protein surface of dimeric βLG. Blue isocontours denote a DMSO density of 0.3 g cm−3. Yellow areas denote the surface contribution arising from amino acids with hydrophobic side chains. The location of the narrow and wide clefts at the protein–protein interface is also shown. | ||
Hence, these results suggest that DMSO might stabilize the attachment of monomeric Hyp to the clefts located at the interface of 2βLG.
The formation of a stable Hyp2βLG complex in the binary DMSO–PBS mixture was confirmed by fluorescence anisotropy and FCS measurements. The anisotropy spectrum (Fig. 1C, green) shows non-zero values, consistent with the ones observed for Hyp2βLG in pure PBS buffer. The cross-correlation function measured using FCS for Hyp2βLG (10 nM Hyp with 36 μM βLG) in the mixture PBS–DMSO (Fig. 2B), is well-described by a model considering molecular diffusion and triplet decay. The diffusion coefficient of the dominant species is D = 40 μm2 s−1. The lower value of D observed in PBS–DMSO with respect to the pure PBS buffer arises from the higher viscosity of the mixture and the different hydrodynamic radius of the DMSO-coated protein.37,38 The spectroscopic properties of the complex Hyp2βLG considerably change in the DMSO:PBS mixture: the absorption spectrum (Fig. 1A, green) is more structured than that in pure PBS, with absorption bands similar to those observed for Hyp dissolved in pure DMSO, and the intensity of the fluorescence emission is increased (Fig. 1B, green). As can be observed in Table 1, the presence of DMSO induces a nearly two-fold increase in the values of ΦF, ΦT, and ΦΔ, which become comparable to those observed when Hyp is bound to apoMb.21Table 1 also shows that the fluorescence decay of Hyp2βLG in DMSO:PBS occurs with a biexponential relaxation, where the main component has a 5.6 ns lifetime, identical to the one of Hyp in DMSO. This is in keeping with the picture derived from MD simulations, where Hyp bound to 2βLG is coated by DMSO. The minor fast component (lifetime 0.3 ns) may arise from scattering.
The effect of local solvent composition on protein bound Hyp has been discussed in the case of human and bovine serum albumins, where the network of hydrogen bonds has been suggested to be responsible for the observed solvatochromism.42 While the presence of DMSO in close vicinity of the bound Hyp may affect the excited state proton transfer of the compound, and thus modulate its photophysics,39–41 the above results suggest that disruption of Hyp aggregates by specific DMSO solvation may be even more relevant.
Given the similar concentration of dissolved molecular oxygen in PBS buffer and in the 20% DMSO–PBS mixture,43 the Hyp2βLG complex can be considered more efficient in the photosensitization of 1O2 when assembled in a PBS–DMSO mixture.
The increment observed in the values of ΦF, ΦT and ΦΔ for Hyp2βLG in DMSO–PBS cannot be attributed to a larger fraction of monomeric free Hyp molecules in the mixture, since the spectra recorded for Hyp alone (Fig. 1; grey curves) show the typical broadened and weaker absorption bands, a barely detectable fluorescence emission (ΦF < 0.001), and a negligible formation of triplet state. Thus, the increased quantum yields observed in the presence of DMSO indicate a larger fraction of better solvated Hyp molecules bound to the protein. In fact, molecular modelling studies support the structural integrity of the Hyp2βLG complex. As noted in Fig. 4, which shows the last snapshot collected at the end of a MD (250 ns) simulation, the presence of DMSO neither alters the binding of monomeric Hyp to the narrow cleft nor impedes the binding of Hyp, either as a monomer or a dimer, to the wide cleft, where it is stacked against the indole ring of Trp77 (the average distance between the Hyp and indole rings is close to 4.8 Å). Although the present results do not allow us to discern the relative stability of the binding to the two clefts, it is worth noting the large population of DMSO molecules around bound Hyp, especially for the monomeric species in the narrow and wide clefts (blue contour in Fig. 4). Moreover, monomeric Hyp is deeply inserted into the wide cleft, while the larger size of dimeric Hyp leads to a more superficial interaction with the protein. The preferential solvation by DMSO molecules is expected to result in a more effective shielding of bound Hyp from surrounding water molecules, which would stabilize the binding of monomeric Hyp to the protein.
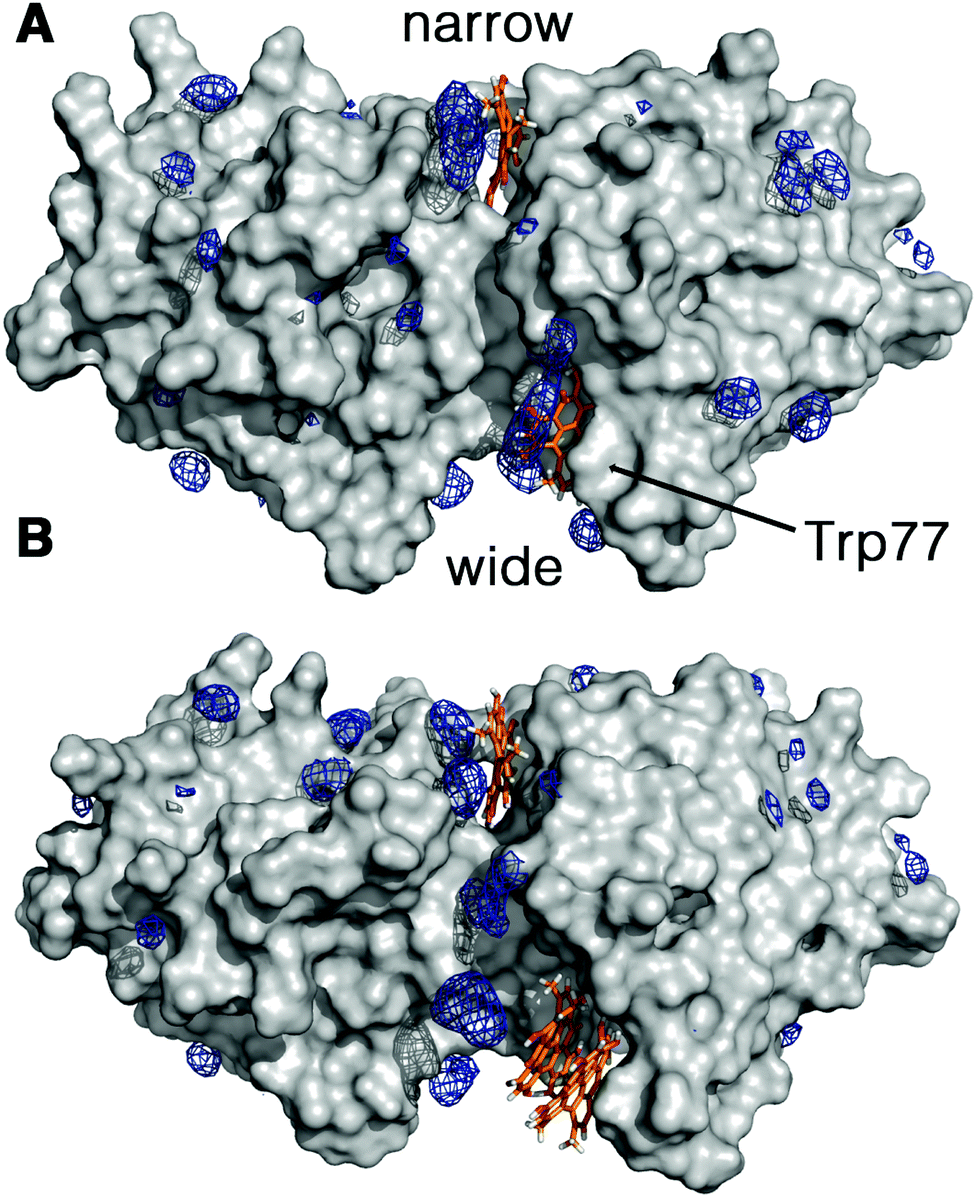 | ||
| Fig. 4 Preferential distribution of DMSO molecules around the protein surface of dimeric βLG bound to Hyp molecules as monomers in the narrow cleft, and either (A) monomers or (B) dimers in the wide cleft. Blue isocontours denote a DMSO density of 0.3 g cm−3. Hyp molecules are shown as orange-based sticks. | ||
Antimicrobial activity of the complex between βLG and Hyp
The enhanced photophysical properties in the presence of DMSO suggest that Hyp2βLG may have improved antimicrobial activity. We then compared the photoinactivation of S. aureus suspensions incubated with Hyp in DMSO, Hyp2βLG in PBS and Hyp2βLG in the mixed solvent PBS–DMSO. In addition, after the incubation period, suspensions were centrifuged and the pellet containing the cells was re-suspended in an equal volume of solvent, while the supernatant was removed by suction. This washing procedure allows for the removal of the species that are not tightly combined to the cells after the incubation, and thus can be separated by centrifugation.In pure PBS buffer, Hyp2βLG induces a relevant inactivation of S. aureus, with a decrease of ∼6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log units in the number of bacterial CFU upon irradiation (Fig. 5A, green). This effect is comparable to the one observed when free Hyp in pure DMSO is added to the bacteria suspension (Fig. 5A, red), whose antimicrobial activity against Gram-positive bacteria is known.8,9,44,45 The results obtained for the washed suspensions are very similar: a decrease of ∼6log units in the number of CFU for free Hyp (Fig. 5A, magenta) and 5 to 6log units for Hyp2βLG, with a slight reduction of the dark toxicity (Fig. 5A, dark green). Fig. 5B shows the results of the experiments carried out using the 20% PBS–DMSO mixture as a solvent. Control experiments demonstrated that the presence of DMSO at this concentration is not toxic for these bacteria (Fig. 5B, orange). As observed in pure PBS buffer, when free Hyp in pure DMSO is added, excellent photoinactivation is obtained, with a reduction of 6–7log units in the number of CFU (Fig. 5B, red). No substantial change occurs after washing by centrifugation (Fig. 5B, magenta). Surprisingly, the complex Hyp2BLG considerably reduces its photodynamic action against S. aureus in the mixture PBS–DMSO, inducing a decrease in the number of CFU of ∼3log units upon irradiation (Fig. 5B, green). This result is in apparent contradiction with the measured value of ΦΔ of the complex, which is about two-fold higher in the mixed solvent PBS–DMSO than in pure PBS.
log units in the number of bacterial CFU upon irradiation (Fig. 5A, green). This effect is comparable to the one observed when free Hyp in pure DMSO is added to the bacteria suspension (Fig. 5A, red), whose antimicrobial activity against Gram-positive bacteria is known.8,9,44,45 The results obtained for the washed suspensions are very similar: a decrease of ∼6log units in the number of CFU for free Hyp (Fig. 5A, magenta) and 5 to 6log units for Hyp2βLG, with a slight reduction of the dark toxicity (Fig. 5A, dark green). Fig. 5B shows the results of the experiments carried out using the 20% PBS–DMSO mixture as a solvent. Control experiments demonstrated that the presence of DMSO at this concentration is not toxic for these bacteria (Fig. 5B, orange). As observed in pure PBS buffer, when free Hyp in pure DMSO is added, excellent photoinactivation is obtained, with a reduction of 6–7log units in the number of CFU (Fig. 5B, red). No substantial change occurs after washing by centrifugation (Fig. 5B, magenta). Surprisingly, the complex Hyp2BLG considerably reduces its photodynamic action against S. aureus in the mixture PBS–DMSO, inducing a decrease in the number of CFU of ∼3log units upon irradiation (Fig. 5B, green). This result is in apparent contradiction with the measured value of ΦΔ of the complex, which is about two-fold higher in the mixed solvent PBS–DMSO than in pure PBS.
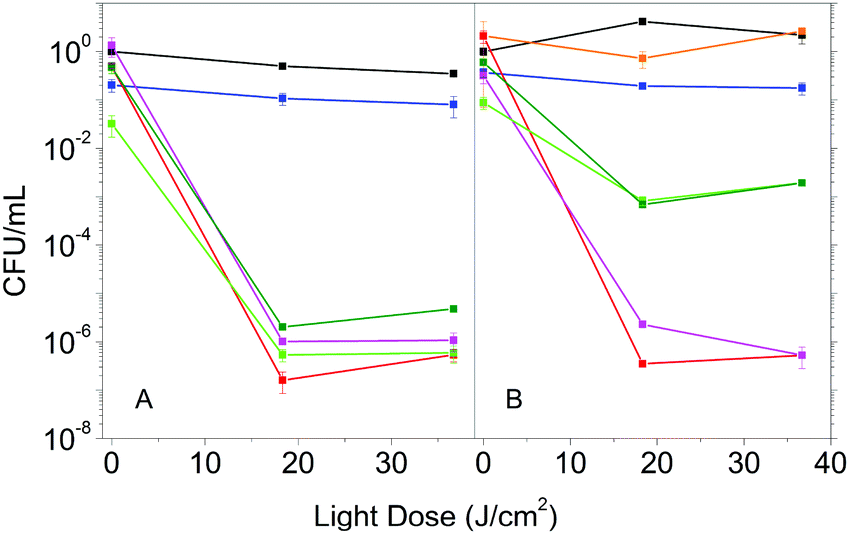 | ||
| Fig. 5 Photoinactivation results for S. aureus cells in suspension incubated with: 4 μM Hyp (red), 4 μM Hyp and washed by centrifugation (magenta), Hyp 4 μM complexed with βLG 200 μM (green), Hyp 4 μM complexed with βLG 200 μM and washed by centrifugation (dark green). The same experiments are performed in a PBS buffered solution (A) and in a 20% PBS–DMSO mixture (B). Controls are made for S. aureus suspension in PBS (black), in 20% PBS–DMSO (orange), and incubated with βLG 200 μM (blue). | ||
The different photodynamic activity towards S. aureus in the mixed solvent PBS–DMSO does not arise from a reduced photostability of the complex in the mixed solvent as compared to PBS. Exposing Hyp2βLG to the same light dose as used in the photoinactivation experiments leads to the same level of bleaching of the compound in PBS and in PBS–DMSO (data not shown).
Spectroscopic analysis in the presence of bacteria
The comparison of the fluorescence emission spectra in Fig. 6 can provide some indications about the distribution of Hyp in the presence of S. aureus cells. As previously discussed, free Hyp is largely aggregated in PBS buffer and no fluorescence can be detected at the concentration used (Fig. 6A, red). When free Hyp is placed in the mixture PBS–DMSO, the fluorescence emission is detectable with a maximum at 593 nm, but still weak (Fig. 6B, red). After incubation with bacterial cells, the emission spectra of free Hyp undergo minor but important changes: a slight increase in the emission is observed in pure PBS (Fig. 6A, black, peak at 598 nm), while an increase of the emission and a band-shift are observed in the mixture PBS–DMSO (Fig. 6B, black, peak at 598 nm). Fluorescence decay is very similar for bacteria bound Hyp, either with or without DMSO (Table 1). This indicates re-distribution of Hyp, where Hyp molecules are placed in a different environment than the bulk solvent, provided by the cell.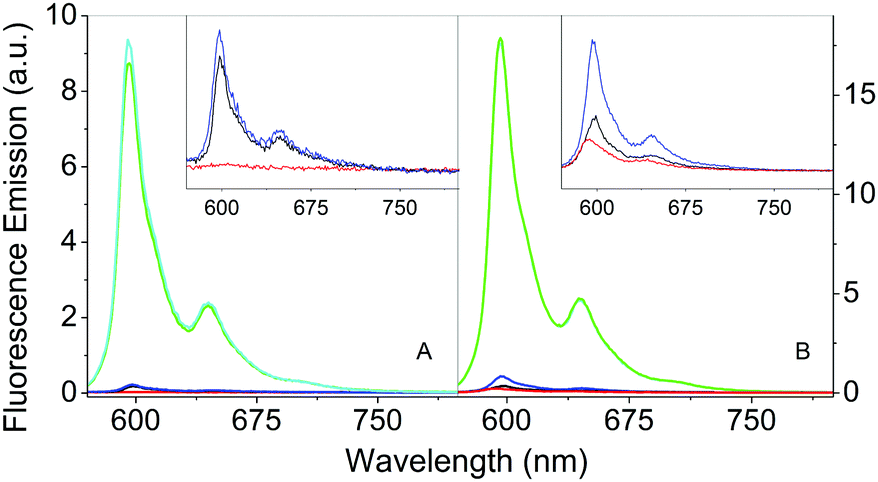 | ||
| Fig. 6 Fluorescence emission spectra in PBS buffer (A) and in the 20% PBS–DMSO mixture (B) for Hyp 4 μM (red), Hyp 4 μM incubated with S. aureus (black), Hyp 4 μM complexed with βLG 200 μM (green), Hyp 4 μM complexed with βLG 200 μM incubated with S. aureus (cyan), Hyp 4 μM complexed with βLG 200 μM, incubated with S. aureus and washed by centrifugation (blue). The insets represent a zoom of the less intense signals. Excitation at 554 nm. | ||
The interaction between Hyp or Hyp2βLG and S. aureus cells is crucial for the photodynamic inactivation induced by the photosensitizing species.
As previously discussed, the complex Hyp2βLG in pure PBS is considerably more fluorescent than free Hyp (Fig. 6A, green), and its emission becomes even more intense in PBS–DMSO (Fig. 6B, green). No significant changes in the emission spectra of Hyp2βLG are detected after incubation with S. aureus cells, either in pure PBS (Fig. 6A, cyan) or PBS–DMSO (Fig. 6B, cyan). When solutions are washed by means of centrifugation after incubation with bacteria, the fluorescence emission is largely reduced (Fig. 6A and B, blue), becoming comparable with the one observed for free Hyp incubated with bacteria. The small but clearly detectable emission spectra of the washed bacteria indicate that an appreciable fraction of Hyp2βLG is tightly bound to the bacteria. The position of the emission peak for this fraction is very similar in the absence (598 nm) and in the presence of DMSO (597 nm). All of the above results are unchanged whether bacteria are incubated with Hyp2βLG at room temperature, or at 37 °C.
When S. aureus cells are incubated with Hyp2βLG in PBS, time resolved fluorescence emission from the PS occurs through a biexponential relaxation with similar weights as, and slightly longer lifetimes than, those observed in the absence of bacteria (Table 1). This fact indicates that the fluorophore is kept in the same type of environment with or without bacteria, provided by the protein scaffold. Similarly, the fluorescence decay observed for S. aureus incubated with Hyp2βLG in PBS–DMSO is essentially the same as that observed for Hyp2βLG in PBS–DMSO and for Hyp in DMSO (Table 1). This suggests that the patches of DMSO, coating the hydrophobic areas of the protein surface, persist also when Hyp2βLG is bound to the bacteria.
Importantly, triplet decay of Hyp2βLG bound to S. aureus is very similar in PBS and PBS–DMSO, with a lifetime of approx. 9 μs, identical to the value measured in the absence of bacteria (Table 1). The long lifetime of the triplet state indicates that Hyp remains bound to the protein carrier, since transfer to the bacterial membrane would lead to a shorter triplet state lifetime (approx. 1–2 μs).
Finally, the interaction between Hyp2βLG and S. aureus cells is further confirmed by FCS experiments, which demonstrate for all these samples the presence of fluorescent species with a diffusion coefficient consistent with that of large objects such as S. aureus cells (data not shown).
Since experimental evidence suggests that the complex between Hyp and 2βLG remains intact even after binding to S. aureus, the different type of interaction of the whole complex Hyp2βLG coated by DMSO with the cells is most likely the reason for the reduced efficiency in the microbial photoinactivation. In spite of the higher amount of photosensitized 1O2 by Hyp2βLG, the DMSO coating thus appears to decrease the bioavailability of the photoactive compound for yet to be understood reasons.
Conclusions
The complex between the dimeric milk whey protein βLG and the natural photosensitizer Hyp has been fruitfully used as photoactive system in antibacterial photosensitization-based treatments.22 However, the presence of the wide cleft, capable of accommodating more than one PS molecule, and the partial exposure of bound Hyp to the aqueous environment lowers both triplet and fluorescence quantum yields, in comparison with cases where bound Hyp is strictly monomeric and the shielding from solvent is more efficient.20Taking advantage of the known peculiar solvation properties of βLG, we show that it is possible to coat the dimeric protein with the cosolvent DMSO, which provides a better environment for the bound Hyp molecules. While this increases fluorescence, triplet, and singlet oxygen yields, it turns out that the presence of DMSO is detrimental to the antibacterial treatment. This most likely arises from a change in the interaction between Hyp2βLG and the cellular constituents, which prevents the DMSO coated nanostructure from reaching more photosensitive regions in the bacterial wall.
The present work demonstrates that the improved photophysical properties of the delivery system are not necessarily translated into more effective photoinactivation of bacterial cells. The overall efficiency is the result of a combination of suitable photosensitization yield and interaction with the target environment.
Experimental section
Hypericin was obtained from HWI Analytik GmbH (Ruelzheim, Germany). β-Lactoglobulin (isoform B) from bovine milk was obtained from Sigma Aldrich (St. Louis, MO). All samples were used as received.For the spectroscopic studies on the complex Hyp2βLG, we choose experimental conditions comprising an excess of protein, so that the fraction of free Hyp molecules is negligible. Typical experimental values are [βLG] = 200 μM, corresponding to [2βLG] ≈ 86 μM, and [Hyp] = 4 μM. Under these conditions, ∼86% of βLG is found as a homodimer (dimerization constant kD = 8.6 μM24) and ∼97% of Hyp is bound to the protein (dissociation constant kd = 0.71 ± 0.03 μM22). For FCS experiments, Hyp was kept at 1 nM in the presence of 30 μM βLG.
General spectroscopic instrumentation
Absorption spectra were recorded using a Jasco V-650 (Jasco Europe). Fluorescence spectra were recorded using a Spex-Fluoromax 4 (Horiba Jobin Yvon, Edison, NJ) or a Perkin Elmer LS50 spectrofluorometer (PerkinElmer, Waltham, MA). For steady state anisotropy studies, two Glan Taylor polarizers were placed before sample excitation and before fluorescence detection. Fluorescence decays were recorded by using a Fluotime 200 time-correlated single photon counting system (PicoQuant GmbH, Berlin, Germany) with pulsed LED excitation at 457 or 375 nm and detection via a PicoQuant PMA 182-M single photon detector.Singlet oxygen measurements
The time-resolved 1O2 phosphorescence signals and ΦΔ values were determined by direct detection of 1O2 phosphorescence at 1275 nm using a modified PicoQuant Fluotime 200 system. A diode-pumped pulsed Nd:YAG laser (FTSS355-Q, Crystal Laser, Berlin, Germany) working at a 10 kHz repetition rate (λexc = 532 nm, 1.2 μJ per pulse) was used for excitation; the NIR luminescence exiting from the sample was detected at 90° by using a H9170-45 NIR-PMT module (Hamamatsu) working in photon counting mode and a NanoHarp 250 multichannel scaler (PicoQuant, Germany). The time-resolved phosphorescence signals, showing the typical rise and decay, were fitted using the equation: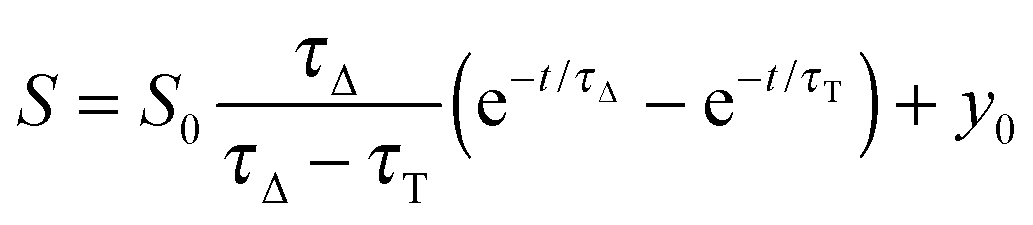 | (1) |
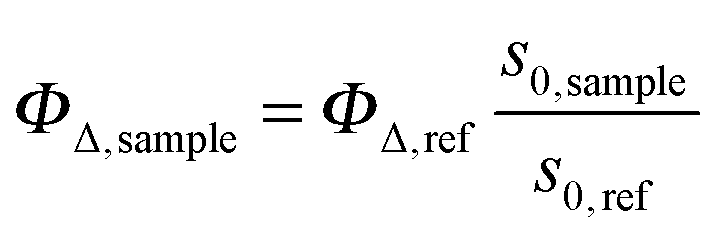 | (2) |
Fluorescence correlation spectroscopy (FCS)
FCS experiments were performed using a Microtime 200 system from PicoQuant, based on an inverted confocal microscope (Olympus IX70) and equipped with two SPADs (single photon avalanche diodes) used in the cross-correlation mode. Excitation was achieved by using a 475 nm picosecond diode laser. Fluorescence emission by Hyp was collected through a bandpass filter and split with a 50/50 splitter between the two detection channels. The Hyp concentration was kept in the nM range, so that only a few molecules (always below 10) were detected in the confocal volume.Laser flash photolysis
Triplet state decay of Hyp was monitored at 520 nm after photoexcitation with the second harmonic (532 nm) of a nanosecond Nd:YAG laser (Spectron Laser) using a previously described setup.47Molecular modeling
000 atoms, including the protein–ligand complex, 900 DMSO molecules, 14000 water molecules and 9 Na+ counterions in a simulation box of 625000 Å3.
The geometry of the system was minimized in five cycles that combined 3500 steps of steepest descent algorithm followed by 6500 of conjugate gradient. Thermalization was performed in 3 steps of 125 ps, where the temperature was gradually increased from 50 K to 298 K, while the protein and ligands were restrained with a force constant that was concurrently reduced from 1 kcal mol−1 Å−2 to 0.1 kcal mol−1 Å−2. Prior to the production runs, each system was subjected to a 0.5 ns simulation on the NPT ensemble to equilibrate the density of the system. During the thermalization and equilibration stages a timestep of 1 fs was employed using SHAKE to constraint bonds involving hydrogen atoms. The production runs consisted of 250 ns using SHAKE for bonds involving hydrogen atoms, a time step of 2 fs, periodic boundary conditions at constant volume and temperature (298 K; Langevin thermostat with a collision frequency of 3 ps−1), particle mesh Ewald to handle long-range electrostatic interactions, and applying a cutoff of 10 Å to all nonbonded interactions. The sander module was employed for the minimization stage, while the CUDA accelerated version of PMEMD62 was used thorough the heating, equilibration and production stages, with both modules being available in the standard distribution of AMBER15.
Microbial strain growth and photoinactivation
The bacterial strain used was Staphylococcus aureus CECT 239 obtained from the Spanish Type Culture Collection (CECT). Vegetative bacterial cells were grown in sterile Tryptic soy broth at 37 °C until an optical density at 600 nm corresponding to 0.4. The cells suspensions were then washed three times in sterile PBS by means of centrifugation and re-suspension, diluted 1:2 with a double concentrated solution containing the photosensitizer and incubated in the dark for 30 min at room temperature. For the photoinactivation experiments, the suspensions were placed in 96-wells plates, irradiated with green light for 15 or 30 min (18 and 37 J cm−2) and serially diluted until 10−6 times the original concentration. Colony forming units (CFUs) were counted after 24 h incubation in the dark at 37 °C to calculate the survival fraction. Experiments were carried out in duplicate.
Acknowledgements
CV acknowledges S. E. Braslavsky for the kind donation of the Spectron Laser. This work has been supported by the Spanish MINECO, grant No. CTQ2013-48767-C3-1-R, and Generalitat de Catalunya, SGR2014-1189. FJL acknowledges the ICREA foundation for financial support.Notes and references
- T. Dai, Y.-Y. Huang and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2009, 6, 170–188 CrossRef CAS PubMed.
- G. B. Kharkwal, S. K. Sharma, Y. Y. Huang, T. Dai and M. R. Hamblin, Lasers Surg. Med., 2011, 43, 755–767 CrossRef PubMed.
- Z. Luksiene and L. Brovko, Food Eng. Rev., 2013, 5, 185–199 CrossRef CAS.
- Singlet Oxygen. Applications in Biosciences and Nanosciences, ed. S. Nonell and C. Flors, London, 2016 Search PubMed.
- G. Jori, M. Camerin, M. Soncin, L. Guidolin and O. Coppellotti, in Photodynamic Inactivation of Microbial Pathogens: Medical and Environmental Applications, ed. M. R. Hamblin and G. Jori, The Royal Society of Chemistry, 2011, pp. 1–18 Search PubMed.
- E. Alves, M. A. F. Faustino, M. G. P. M. S. Neves, A. Cunha, J. Tome and A. Almeida, Future Med. Chem., 2014, 6, 141–164 CrossRef CAS PubMed.
- N. Duràn and P. S. Song, Photochem. Photobiol., 1986, 43, 677–680 CrossRef.
- K. Kairyte, S. Lapinskas, V. Gudelis and Z. Luksiene, J. Appl. Microbiol., 2012, 112, 1144–1151 CrossRef CAS PubMed.
- C. M. N. Yow, H. M. Tang, E. S. M. Chu and Z. Huang, Photochem. Photobiol., 2012, 88, 626–632 CrossRef CAS PubMed.
- N. Nafee, A. Youssef, H. El-Gowelli, H. Asem and S. Kandil, Int. J. Pharm., 2013, 454, 249–258 CrossRef CAS PubMed.
- J. M. Jacobson, L. Feinman, L. Liebes, N. Ostrow, V. Koslowski, A. Tobia, B. E. Cabana, D. H. Lee, J. Spritzler and A. M. Prince, Antimicrob. Agents Chemother., 2001, 45, 517–524 CrossRef CAS PubMed.
- D. S. English, K. Das, J. M. Zenner, W. Zhang, G. A. Kraus, R. C. Larock and J. W. Petrich, J. Phys. Chem. A, 1997, 101, 3235–3240 CrossRef CAS.
- A. Darmanyan, L. Burel, D. Eloy and P. Jardon, J. Chim. Phys., 1994, 91, 1774–1785 CAS.
- M. Roslaniec, H. Weitman, D. Freeman, Y. Mazur and B. Ehrenberg, J. Photochem. Photobiol., B, 2000, 57, 149–158 CrossRef CAS.
- T. Yamazaki, N. Ohta, I. Yamazaki and P. S. Song, J. Phys. Chem., 1993, 97, 7870–7875 CrossRef CAS.
- J. L. Wynn and T. M. Coton, J. Phys. Chem., 1995, 99, 4317–4323 CrossRef CAS.
- M. Fadel, K. Kassab and T. Youssef, Lasers in Medical Science, 2010, 25, 675–683 CrossRef PubMed.
- M. Zeisser-Labouèbe, N. Lange, R. Gurny and F. Delie, Int. J. Pharm., 2006, 326, 174–181 CrossRef PubMed.
- O. Planas, E. Boix-Garriga, B. Rodriguez-Amigo, J. Torra, R. Bresolí-Obach, C. Flors, C. Viappiani, M. Agut, R. Ruiz-González and S. Nonell, in Photochemistry, ed. A. Albini and E. Fasani, The Royal Society of Chemistry, London, 2014 Search PubMed.
- J. Comas-Barceló, B. Rodríguez-Amigo, S. Abbruzzetti, P. D. Rey-Puech, M. Agut, S. Nonell and C. Viappiani, RSC Adv., 2013, 3, 17874–17879 RSC.
- P. Delcanale, F. Pennacchietti, G. Maestrini, B. Rodríguez-Amigo, P. Bianchini, A. Diaspro, A. Iagatti, B. Patrizi, P. Foggi, M. Agut, S. Nonell, S. Abbruzzetti and C. Viappiani, Sci. Rep., 2015, 5, 15564 CrossRef CAS PubMed.
- B. Rodríguez-Amigo, P. Delcanale, G. Rotger, J. Juárez-Jiménez, S. Abbruzzetti, A. Summer, M. Agut, F. J. Luque, S. Nonell and C. Viappiani, J. Dairy Sci., 2015, 98, 89–94 CrossRef PubMed.
- T. Arakawa, Y. Kita and S. N. Timasheff, Biophys. Chem., 2007, 131, 62–70 CrossRef CAS PubMed.
- D. Mercadante, L. D. Melton, G. E. Norris, T. S. Loo, M. A. K. Williams, R. C. J. Dobson and G. B. Jameson, Biophys. J., 2012, 103, 303–312 CrossRef CAS PubMed.
- E. Dufour, C. Bertrand-Harb and T. Haertlé, Biopolymers, 1993, 33, 589–598 CrossRef CAS PubMed.
- P. Miskovsky, D. Jancura, S. Sanchez-Cortez, E. Kocisova and L. Chinsky, J. Am. Chem. Soc., 1998, 120, 6374–6379 CrossRef CAS.
- D. Magde, E. Elson and W. W. Webb, Phys. Rev. Lett., 1972, 29, 705–708 CrossRef CAS.
- E. Haustein and P. Schwille, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 151–169 CrossRef CAS PubMed.
- A. Losi, Photochem. Photobiol., 1997, 65, 791–801 CrossRef CAS.
- C. Mattos and D. Ringe, Nat. Biotechnol., 1996, 14, 595–599 CrossRef CAS PubMed.
- E. Liepinsh and G. Otting, Nat. Biotechnol., 1997, 15, 264–268 CrossRef CAS PubMed.
- A. C. English, C. R. Groom and R. E. Hubbard, Protein Eng., 2001, 14, 47–59 CrossRef CAS PubMed.
- D. Alvarez-Garcia and X. Barril, J. Med. Chem., 2014, 57, 8530–8539 CrossRef CAS PubMed.
- H. Vondracek, J. Dielmann-Gessner, W. Lubitz, M. Knipp and M. Havenith, J. Chem. Phys., 2014, 141, 22D534 CrossRef PubMed.
- S. Ebbinghaus, S. J. Kim, M. Heyden, X. Yu, U. Heugen, M. Gruebele, D. M. Leitner and M. Havenith, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20749–20752 CrossRef CAS PubMed.
- S. Ebbinghaus, K. Meister, B. Born, A. L. DeVries, M. Gruebele and M. Havenith, J. Am. Chem. Soc., 2010, 132, 12210–12211 CrossRef CAS PubMed.
- J. M. G. Cowie and P. M. Toporowski, Can. J. Chem., 1961, 39, 2240–2243 CrossRef CAS.
- R. G. LeBel and D. A. I. Goring, J. Chem. Eng. Data, 1962, 7, 100–101 CrossRef CAS.
- F. Gai, M. J. Fehr and J. W. Petrich, J. Phys. Chem., 1994, 98, 5784–5795 CrossRef CAS.
- F. Gai, M. J. Fehr and J. W. Petrich, J. Phys. Chem., 1994, 98, 8352–8358 CrossRef CAS.
- K. Das, A. V. Smirnov, M. D. Snyder and J. W. Petrich, J. Phys. Chem. B, 1998, 102, 6098–6106 CrossRef CAS.
- E. I. Kapinus, Biophysics, 2010, 55, 188–193 CrossRef.
- H. Lawrence Clever, R. Battino, H. Miyamoto, Y. Yampolski and C. L. Young, J. Phys. Chem. Ref. Data, 2014, 43, 033102 CrossRef.
- C. Cecchini, A. Cresci, M. M. Coman, M. Ricciutelli, G. Sagratini, S. Vittori, D. Lucarini and F. Maggi, Planta Med., 2007, 73, 564–566 CrossRef CAS PubMed.
- K. Aponiene, E. Paskeviciute, I. Reklaitis and Z. Luksiene, J. Food Eng., 2015, 144, 29–35 CrossRef CAS.
- S. Nonell and S. E. Braslavsky, in Singlet oxygen, UV-A, and Ozone, ed. L. P. A. H. Sies, Academic Press, San Diego, 2000, pp. 37–49 Search PubMed.
- S. Abbruzzetti, S. Bruno, S. Faggiano, E. Grandi, A. Mozzarelli and C. Viappiani, Photochem. Photobiol. Sci., 2006, 5, 1109–1120 CAS.
- D. Alvarez-Garcia and X. Barril, J. Chem. Theory Comput., 2014, 10, 2608–2614 CrossRef CAS PubMed.
- C. C. G. Inc., ed. S. 1010 Sherbooke St. West, Montreal, QC, Canada, H3A 2R7, 2016.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Z. J. L. Sonnenberg, M. Hada, M. Ehara, K. T. R. Fukuda, J. Hasegawa, M. Ishida, T. N. Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. B. C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Y. A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Z. G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Fox and P. A. Kollman, J. Phys. Chem. B, 1998, 102, 8070–8079 CrossRef CAS.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2005, 26, 114 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- D. J. Price and C. L. Brooks, J. Chem. Phys., 2004, 121, 10096–10103 CrossRef CAS PubMed.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- D. A. Case, T. A. Darden, I. T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. P. J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. B. T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov and A. K. P. A. Kollman, AMBER12, San Francisco, 2012 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- I. S. Joung and T. E. Cheatham, J. Phys. Chem. B, 2008, 112, 9020–9041 CrossRef CAS PubMed.
- I. S. Joung and T. E. Cheatham, J. Phys. Chem. B, 2009, 113, 13279–13290 CrossRef CAS PubMed.
- R. Salomon-Ferrer, A. W. Goetz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |