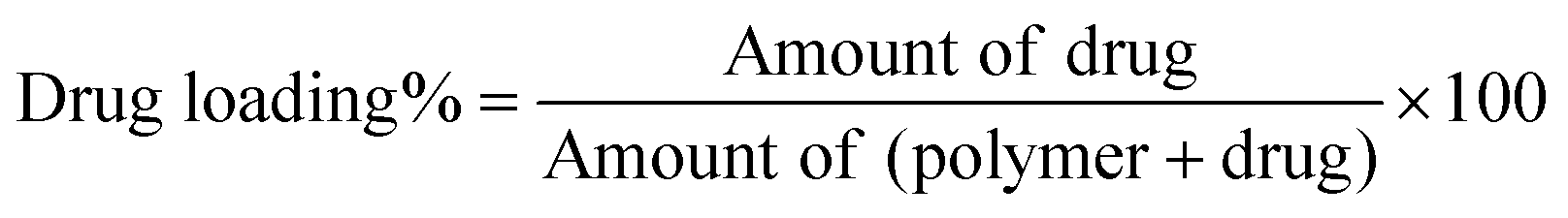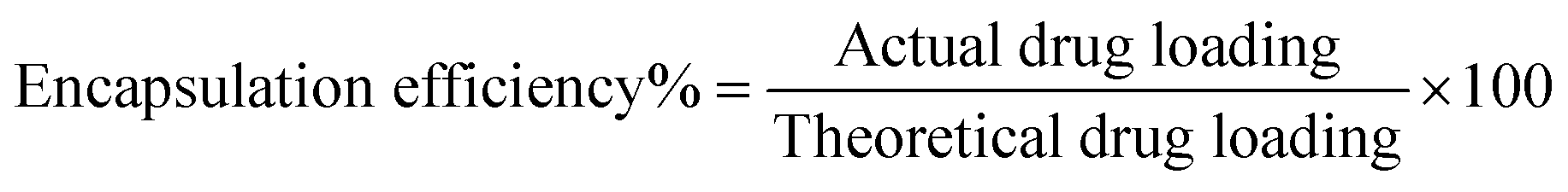Biodegradable nitric oxide precursor-loaded micro- and nanoparticles for the treatment of Staphylococcus aureus biofilms
Received
19th December 2016
, Accepted 3rd January 2017
First published on 9th January 2017
Abstract
Bacteria in biofilms are more difficult to eradicate than planktonic bacteria and result in treatment challenges for many chronic infectious diseases. Nitric oxide (NO) is an endogenous molecule that offers potential as an alternative to conventional antibiotics; however its sustained topical delivery to biofilms is not readily achieved. With this in mind, we report the development of biodegradable poly(lactide-co-glycolide) (PLGA) based microparticles (MP) and nanoparticles (NP) for encapsulation of the NO precursor isosorbide mononitrate (ISMN) and the controlled delivery to Staphylococcus aureus (S. aureus) biofilms. Firstly, water-in-oil-in-water (w/o/w) emulsification/solvent evaporation methods for PLGA NP and MP syntheses were experimentally optimised with respect to particle size and ISMN loading/encapsulation efficiency. The influence of various experiment parameters, such as the volume of inner aqueous phase, concentration of surfactants, mixing time on the particle size, drug loading and encapsulation efficiency were investigated systematically. Both PLGA MP and NP formulations enabled sustained ISMN release in physiological media over 3 to 5 days. PLGA MP with diameters of ∼3 μm and ISMN loading of 2.2% (w/w) were identified as the optimum delivery system and demonstrated significant antibacterial activity in S. aureus biofilms. This behaviour is considered to be due to targeted biofilm delivery through a combination of effective penetration and sustained release of ISMN.
Introduction
The increasing prevalence of Staphylococcus aureus (S. aureus) biofilm-associated infections such as skin and bone infections,1,2 endocarditis,3 sinus4 and device-related infections (catheters, implantable prosthetics)5,6 are placing significant burden on healthcare systems and society.7 Biofilms are complex, functional communities of one or more species of microorganisms that are encased in extracellular polymeric substance (EPS) and are attached to both a solid surface and to each other.8 The slow growth rates,9 low antibiotic penetration,10 high cell density,11 excessive extracellular matrices,12 pH alterations,13 mutations14 and altered nutrient requirements15 are well-known properties that give rise to greater antimicrobial resistance in microbial biofilms.11,16 The emergence of bacterial resistance and the innate tolerance of microbial biofilms to antibiotic therapy have intensified the problems in their eradication. Therefore, recent research has focused on the development of alternative antimicrobial therapies to treat bacterial biofilms.
Nitric oxide (NO) is a potential alternative for antimicrobial therapy to combat both free-floating (planktonic) bacteria and biofilm-associated bacteria as a result of its intrinsic antimicrobial properties.17 NO has been investigated for the treatment of chronic infectious diseases that are frequently associated with bacterial biofilms.15 NO is an endogenously produced gas and has been considered as a promising therapeutic agent in free radical biology investigations due to its remarkable vasorelaxation, antimicrobial and anti-tumour activity.18 The clinical application of NO is, however limited due to its short half-life, instability during storage, potential toxicity, lack of efficient localized and systemic delivery and poor dose control.19 To overcome these limitations, various NO donors with a range of stabilities have developed to deliver therapeutic levels of NO in biological systems. These NO donors are typically low molecular weight compounds including nitrates, nitrites, N-nitroso, C-nitroso, metal NO complexes and diazeniumdiolates.20 The most widely used NO donors are organic nitrates such as glycerol trinitrate (GTN) and isosorbide mononitrate (ISMN),21 which have been widely used in the treatment of cardiac diseases. However, the therapeutic potential of NO against biofilms is concentration dependent. Anti-biofilm effects of NO are achieved only at higher concentrations, whereas at suboptimal NO concentrations, S. aureus biofilm growth is enhanced.22 The dualistic properties of NO must therefore be taken into consideration when designing a topical delivery agent.23 To this end, obtaining therapeutic NO levels at an appropriate exposure time is the key challenge towards achieving potent anti-biofilm activity.
To date, there are few reports on the development of particle-based NO delivery systems in which NO donors either encapsulated into or covalently attached to macro- or micromolecular structures and their antibacterial activity in bacterial biofilms. Hetrick et al. synthesized two NONOate derived silica nanoparticle systems, N-methylaminopropyltrimethoxysilane/tetraethyl orthosilicate (MAP3/TEOS) and N-(6-aminohexyl) aminopropyltrimethoxysilane (AHAP3/TEOS) and reported increased MAP3/TEOS loading and faster NO release compared to AHAP3/TEOS particles.24 The MAP3/TEOS particles exhibited a 1000-fold increase in bactericidal efficacy against P. aeruginosa biofilms. Additionally, a significant reduction of planktonic bacteria was reported by using macromolecule NONOate modified silica nanoparticles compared to micromolecule N-diazeniumdiolate-modified proline (PROLI/NO).25 Friedman et al. introduced a new silane-based hydrogel nanoparticle platform. The core of this nanoparticle contained nitrite encapsulated within a composite matrix composed of tetramethyl orthosilicate, polyethylene glycol (PEG), chitosan and a glass forming disaccharide. During synthesis, nitrite is thermally reduced to NO.26 In a subsequent study, Martinez et al. evaluated the antimicrobial potential of silane based hydrogel nanoparticles.27 This study demonstrated that the NO releasing nanoparticles reduced 99.9% of bacterial growth for cutaneous methicillin-resistant Staphylococcus aureus (MRSA) in a murine infection model. However, the toxicity of silica may limit their potential as a future therapeutic agent.
There is increasing interest in the development of ISMN-based formulations for anti-biofilm applications, owing to its well- established efficacy and safety profile. Recently, our group reported on a novel ISMN liposomal formulation for the treatment of S. aureus biofilm-associated chronic rhinosinusitis.23 Following the exposure of S. aureus biofilm to various liposomal ISMN formulations, multilamellar vesicles (MLV) were found to have greater anti-biofilm effects compared to non-liposomal ISMN and unilamellar vesicle (ULV) liposomes. An increase in the lipid content of liposomal ISMN also improved the anti-biofilm efficacy of both MLV and ULV liposomes. A potential drawback of these systems is the presence of both free ISMN and encapsulated ISMN in the formulation that may cause uncontrolled NO donor delivery to biofilms and potential NO side effects such as hypotension.22 Furthermore, liposomal formulations suffer the drawback of poor stability, uncontrolled drug release and low encapsulation efficiency of water-soluble drugs due to rapid drug leakage. The potential use of a successful therapeutic depends on the bioavailability of the therapeutic agents at the site of action. Poly(lactide-co-glycolide) or PLGA micro/nanoparticles have been widely used for encapsulating therapeutic drugs in controlled release applications. Given that PLGA presents inherent advantages over other systems including controlled release rates, biocompatibility/biodegradability, low cytotoxicity and favourable degradation characteristics.28 Most importantly, PLGA are a family of FDA-approved biodegradable polymers that have been extensively studied as delivery vehicles for drugs, proteins and peptides.29,30 Therefore, PLGA-based formulations are a feasible approach for overcoming the challenges of NO delivery to bacterial biofilms.
Here, we explore the potential of PLGA particles to encapsulate and release ISMN in a controlled manner to kill microbial cells within in vitro S. aureus biofilms. We demonstrate that the rate of ISMN release can be tuned by the PLGA particle size and ISMN loading, and quantify anti-biofilm activities of the optimised ISMN-PLGA particulate formulations.
Materials and methods
Materials
The biodegradable polymer PLGA (LA/GA ratio 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, MW 30000–60000 Da), polyvinyl alcohol (PVA; 80% hydrolysed, MW 9000–10000 Da) and Tween 80 were purchased from Sigma Aldrich, Australia. Isosorbide mononitrate (ISMN) (purity >95%, MW 191.14) was procured from Bosche Scientific, USA. All other chemical reagents used including acetone, methanol, dichloromethane (Merck, Germany) were of analytical grade. High purity water was obtained from a Milli-Q Direct water purification system (Millipore, USA) and was used throughout the study.
:50, MW 30000–60000 Da), polyvinyl alcohol (PVA; 80% hydrolysed, MW 9000–10000 Da) and Tween 80 were purchased from Sigma Aldrich, Australia. Isosorbide mononitrate (ISMN) (purity >95%, MW 191.14) was procured from Bosche Scientific, USA. All other chemical reagents used including acetone, methanol, dichloromethane (Merck, Germany) were of analytical grade. High purity water was obtained from a Milli-Q Direct water purification system (Millipore, USA) and was used throughout the study.
ISMN-loaded PLGA particle preparation
ISMN-loaded PLGA microparticles (MPs).
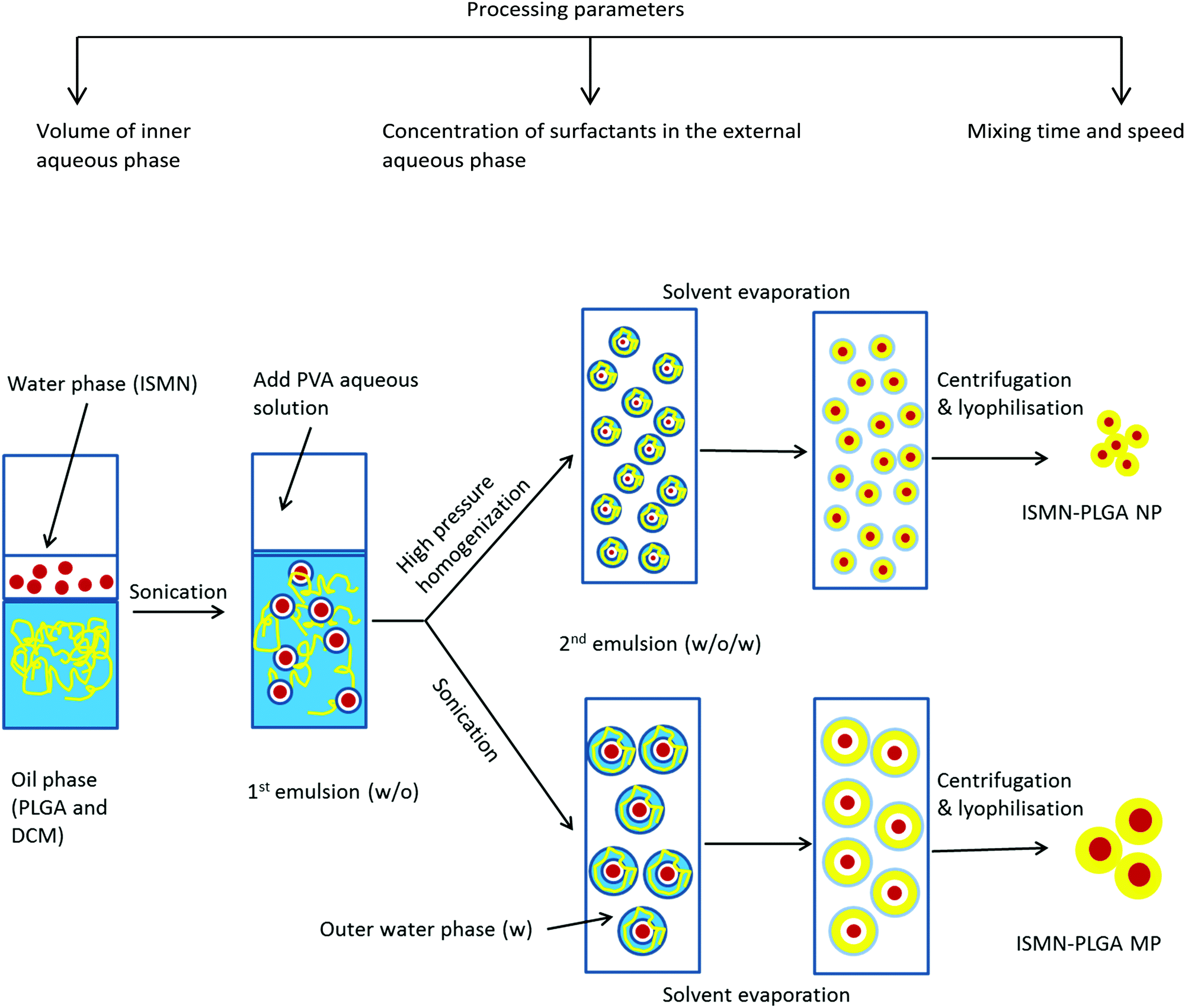
PLGA MPs containing various amount of ISMN were prepared using a w/o/w double emulsion solvent evaporation technique (Fig. 1).31 Briefly, 0.1, 0.5, and 1 mL of ISMN aqueous solutions (60 mg mL−1) or pure water (blank PLGA particles) as the internal aqueous phase were emulsified in 10 mL of dichloromethane containing PLGA (1%) using a Qsonica Q125 probe sonicator (Newton, CT) for the desired time (10, 30, 60, and 120 s) at 30 W to obtain a water-in-oil (w/o) emulsion. This primary w/o emulsion was rapidly added into 20 mL of a 1% (w/v) PVA aqueous solution (external aqueous phase) and emulsified again for the same times. The resultant w/o/w double emulsion was then diluted in 150 mL 0.3% (w/v) PVA aqueous solution in order to minimize droplet coalescence, and was stirred for 4 h at room temperature to allow evaporation of the organic solvent. The PLGA MPs were collected by centrifugation at 45000 × g (Haake Z36HK, Wehingen, Germany) for 20 min, washed with Milli-Q water three times, and freeze-dried overnight and stored at 4 °C until further use.
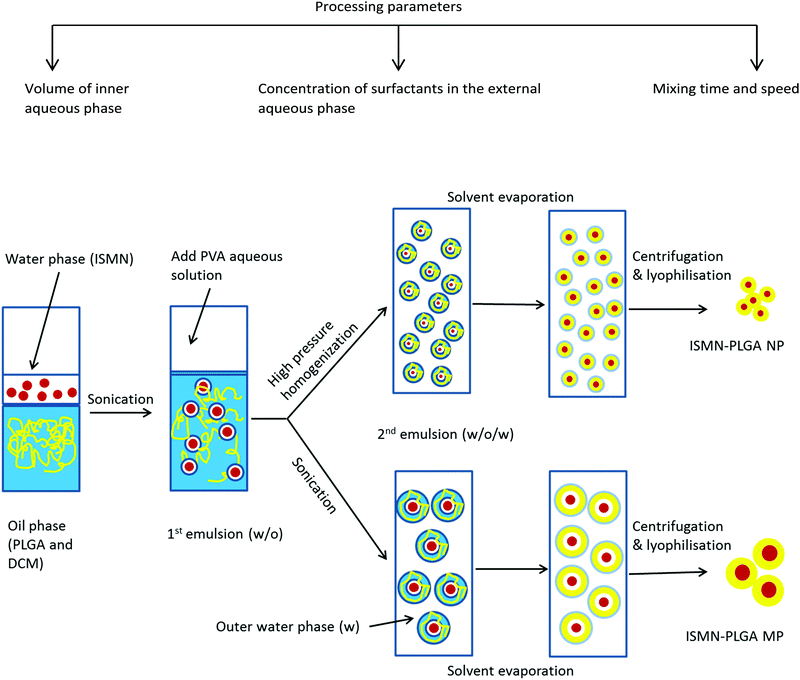 |
| | Fig. 1 Schematic of the preparation method of ISMN-loaded PLGA micro- and nanoparticles. | |
ISMN-loaded PLGA nanoparticles (NPs).
PLGA NPs were prepared by specific modification of the w/o/w emulsion technique,32i.e. using high pressure homogenization after the second emulsification step, thereby reducing the size of the dispersed droplets.33 Homogenisation was carried out for 3 cycles at 500 bar. The resulting homogenized emulsion was transferred gradually into 150 mL of a second outer phase containing 0.3% (w/v) of PVA stabilizer under continuous stirring to solidify the nanoparticles. The organic solvent was evaporated under magnetic stirring (700 rpm) for 4 hours at room temperature. The generated nanoparticles were collected by centrifugation at 45000 × g for 20 min and washed three times with Milli-Q water. Finally, the products were freeze-dried and stored at 4 °C.
Characterisation of PLGA particles
Determination of drug loading and encapsulation efficiency.
The freeze-dried PLGA MPs and NPs (10 mg) were dissolved in 2 mL of acetone to release the encapsulated ISMN. Then the mixture was diluted 10 times with HPLC mobile phase (80% Milli-Q water and 20% methanol), sonicated for 1 min, and centrifuged at 15000 rpm for 10 min. The supernatant was directly injected into a high performance liquid chromatography (HPLC) system (UFLC XR, Shimadzu, Japan). Chromatographic separation was performed on a LiChrospher RP C18 column (5 μm, 4.6 mm ID × 150 mm, Grace Davidson Discovery Science, Rowville, VIC) at a detection wavelength of 215 nm. The concentration of ISMN was calculated from a standard curve, prepared by measuring the peak areas of known concentrations of ISMN in methanol/water mixtures. Drug loading and encapsulation efficiency were calculated as follows:
Particle size and size distribution.
The mean particle diameter size and size distribution of the PLGA-NPs were measured by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern instruments, UK). The analysis was performed by using samples diluted with Milli-Q water. The particle size was expressed as the z-average diameter. Values reported are the mean diameter ± standard deviation for three independent replicate samples.
Scanning electron microscopy.
The morphology and particle sizes for both PLGA-MPs and PLGA-NPs were observed using scanning electron microscopy (SEM). Lyophilized particles were spread on carbon tape mounted on a SEM aluminum stub and imaged using a Quanta 450 field emission scanning microscope (FEI, Hillsboro, Oregon, USA) at an accelerating voltage of 1–2 kV. For size distribution analysis, the diameters of at least 50 particles were measured and analyzed with Image J software.34
Time-of-flight secondary ion mass spectrometry (ToF-SIMS).
ToF-SIMS has been used as a highly sensitive surface analytical techniques for characterisation of the ISMN-loaded PLGA microparticle drug delivery system. Specifically, spectral data from a freeze-dried PLGA microparticle powder were acquired to analyse the spatial distribution of the various identified mass fragments of interest. The sample powders were loaded by sprinkling onto adhesive paper, thereby obtaining a finely dispersed powder surface with a low background signal from the substrate. Surface chemical maps were acquired using a PHI TRIFT V nanoTOF instrument (Physical Electronics Inc. Chanhassen, MN, USA) operated with a 79+Au primary ion gun at 30 kV voltage under a vacuum of 5 × 10−6 Pa. ‘Unbunched’ instrumental settings were applied to optimize spatial resolution for obtaining images from areas roughly 100 × 100 μm, with an acquisition time of three minutes. The protonated molecular ion peaks were identified at m/z = 69.03, 85.03, 127.04 and 145.04 amu for ISMN and 69.07 amu for PLGA material respectively. Sample spectra and images were analysed with Wincadencen software (Physical Electronics Inc. Chanhassen, MN, USA).
In vitro release studies.
The ISMN release kinetics from PLGA MPs and NPs was evaluated in phosphate buffered saline (PBS) at pH 7.4. Briefly, 10 mg of dried ISMN-loaded PLGA MPs (corresponding to 220 μg ISMN) and NPs (corresponding to 40 μg ISMN) were suspended in 50 mL of 0.01 M PBS in a flask and incubated in a water bath at 37 °C with gentle magnetic stirring (150 rpm). At specified time intervals, aliquots (1 mL) were removed and centrifuged for 20 min at 25000 × g followed by dilution of the supernatants with mobile phase and ISMN quantification by HPLC as described above. Fresh PBS (1 mL) was added to the particle suspension after each sampling time point and agitation was continued. All release experiments were performed in triplicate.
In vitro bacteria studies
Bacterial strain and culture conditions.
A known biofilm-forming S. aureus strain, American Type Culture Collection (ATCC) 25923, was used. Briefly, a single loop of S. aureus was propagated from frozen stock, streaked on agar plates (Oxoid Australia) and allowed to grow at 37 °C for 18 to 24 hours. One or 2 bacterial colonies were suspended in 2 mL of sterile PBS and adjusted to 1 McFarland standard (equivalent to 3 × 108 colony forming units per mL). Then 400 μL of this solution was mixed with 5.6 mL of nutrient broth (1:15 dilution). This suspension served as the inoculum for the formation of biofilms.
Biofilm formation and treatment with ISMN solutions.
Sterile 96 well microtiter plates (Sarstedt, Nümbrecht, Germany) were inoculated with 150 μL per well of the inoculum and were incubated for 24 hours at 37 °C with continuously shaking at 70 rpm (Ratek Instruments, Australia) to allow adequate biofilm formation on the well's inner surfaces. After 24 h incubation the wells containing biofilms were washed twice with PBS to remove adhering planktonic cells. 150 μL of nutrient broth containing ISMN with three different concentrations (10, 30 and 60 mg mL−1) were added to the wells and incubated for 1, 3 and 24 hours at 37 °C. For quantification of biofilms after 1, 3 and 24 h ISMN exposure, the wells were washed twice with PBS, dried and then stained with 180 μL per well of 0.1% crystal violet (CV) solution. Then plates were incubated for 15–20 min at room temperature. After incubation, the wells containing CV solution were washed with water, dried and then 180 μL per well of acetic acid (30%) was added to solubilize CV for 30 min. 150 μL per well of the solubilized CV solution was then transferred into a new plate and the optical density at 550 nm (OD550) was measured using a plate reader (Model 680, Bio-Rad Laboratories, Hercules, CA, USA). All anti-biofilm assays were carried out in duplicate.
Minimum inhibitory concentration (MIC) studies.
The MIC was defined as the lowest concentration of ISMN at which no growth was observed after 24 hours incubation, indicated by an OD600 < 0.1 measured by a plate reader (Bio-Rad, CA, USA). The MIC value for encapsulated ISMN in PLGA-MPs was determined on planktonic S. aureus (ATCC 25923) using the broth dilution method.35 Briefly, 100 μL of PLGA-MP suspensions were prepared in nutrient broth using sterile tubes and later inoculated with an equal amount of bacterial suspension to the wells of a microtiter plate to achieve a series of final concentration (3.75–30 mg mL−1) of encapsulated ISMN. The plates were incubated for 24 hours at 37 °C with continuous shaking at 70 rpm. The MIC assays were performed in duplicate.
Minimum biofilm eradication concentration (MBEC) studies.
The MBEC was defined as the lowest concentration at which no growth of bacteria from the ISMN-exposed biofilms was observed as indicated by an OD < 0.1. The MBEC value for encapsulated ISMN in PLGA-MP was determined in S. aureus biofilm using the 96-well plate assay.36 Biofilm culture and formation were prepared as previously described. Briefly, the wells containing biofilm were washed twice with PBS to remove adhering planktonic cells after 24 h incubation, after which 150 μL of nutrient broth containing PLGA-MP at a range of 7.5–60 mg mL−1 ISMN were added to the wells and incubated for another 24h at 37 °C. The MBEC assays were performed in duplicate.
Anti-planktonic bacteria studies.
In vitro antibacterial studies on planktonic S. aureus where undertaken using the Alamar Blue assay. Briefly, 150 μL of bacterial suspension was added to tubes containing various amounts of ISMN-PLGA-MP. After 3 h incubation at 37 °C, a 100 μL aliquot was transferred into the wells of a new microtiter plate and added to premixed 100 μL nutrient broth and 20 μL Alamar Blue dye per well.37 The plates were then incubated on a rotating mixer at 10 rpm in the dark at room temperature for 1 hour. Fluorescence of each well was measured (λex: 560 nm and λem: 590 nm) after 1 h incubation at 37 °C by using a microtiter plate fluorometer (Bio-Rad, Australia).38 Assays were carried out in duplicate.
Anti-biofilm studies.
A biofilm forming S. aureus strains (ATCC 25923), was used to test ISMN either as free drug or loaded into PLGA MP using the Alamar Blue assay. S. aureus culture and biofilm formation were performed as describe in previous sections. Briefly, the wells containing biofilms were washed twice with PBS to remove adhering planktonic cells after 24 h incubation. 150 μL of nutrient broth containing PLGA-MP at two different ISMN concentration (500 mg mL−1 PLGA-MPs equivalent to 10 mg mL−1 and 2728 mg mL−1 PLGA-MPs equivalent to 60 mg mL−1 ISMN), free ISMN (10 mg mL−1 and 60 mg mL−1 ISMN) and blank MPs were added to the wells and incubated for another 24 h at 37 °C. For quantification of biofilms after 24 h treatments, the wells were washed twice with PBS and then stained with premixed 180 μL NB and 20 μL Alamar Blue dye per well. Plates were then incubated on a rotating mixer at 10 rpm in the dark at room temperature for 2 hours. Fluorescence (λex: 560 nm and λem: 590 nm) was measured after 2 h of incubation (37 °C). Anti-biofilm assays were carried out in duplicate.
Statistical analysis.
The statistical analyses of differences in the biofilm biomass resulting from the exposure to PLGA-MP and were tested by one-way analysis of variance (ANOVA) followed by multiple comparison with Bonferroni post hoc evaluation using Graph Pad Prism 5.03 (La Jolla, CA, USA). Differences were considered statistically significant at a level of p < 0.05.
Results and discussion
Synthesis and optimization of ISMN loaded PLGA particles
When considering PLGA particle preparation, the selection of a suitable encapsulation method is largely dependent on the physiochemical properties of the drug. For hydrophilic drug encapsulation, the double emulsion solvent evaporation technique is the most commonly utilized. However, the efficient encapsulation of a low molecular weight and water-soluble molecules such as ISMN into PLGA is a significant challenge. The common double emulsion technique39 suffers from poor encapsulation efficiency for water soluble drugs owing to their rapid migration and therefore loss into the aqueous phase. To improve the encapsulation of ISMN into PLGA particles we adjusted specific processing parameters including the volume of the internal aqueous phase, concentration of surfactants and mixing time and evaluated their effect on the particle size, ISMN loading and encapsulation efficiency.40,41
Effect of volume of inner aqueous phase.
The effect of volume of inner water phase on particle size, ISMN loading and encapsulation efficiency for both PLGA MPs and NPs is reported in Table 1. PLGA-MP and PLGA-NP sizes were significantly affected by the presence of ISMN. Moreover, when the volume of the inner aqueous phase was increased at the same initial ISMN concentration, a decrease in PLGA-MP particle size was observed from 9.8 μm to 2.8 μm. Since the particle size is related to the stability of the initial w/o emulsion,42 it is assumed that the optimal volume of the inner aqueous phase may help to stabilize the first emulsion and consequently hampers the fast coalescence of the emulsion droplets. For PLGA-NPs, the observed size increase from 172 nm to 556 nm with an increased volume of the inner aqueous phase (Table 1) is considered to have resulted from the less stable w/o emulsion and thus the coalescence of suspended droplets. Meanwhile, the ISMN loading and encapsulation efficiency increased upon increasing the internal aqueous phase volume. For PLGA-MPs, the ISMN loading and encapsulation efficiencies increased when the inner aqueous phase was increased up to 1 mL. The highest ISMN loading (2.2% w/w) and encapsulation efficiency (23.6%) for PLGA-MPs were obtained using an inner aqueous phase of 0.5 mL. This can be explained by the localization of ISMN at the interfaces. That is, a larger fraction of ISMN is adsorbed at the outer surface for the lower ISMN concentration and as the concentration is increased, the surface becomes saturated and more ISMN encapsulates in the aqueous core.32 If the ISMN amount is further increased, leakage from the inner into the external aqueous phase may occur32 hence reducing encapsulation efficiency. Therefore, the ISMN loading and encapsulation efficiency in PLGA-MPs showed a maximum at 0.5 mL of inner aqueous phase. Interestingly, PLGA-NPs showed no such maximum (Table 1). Considering the delicate balance between the particle size, ISMN loading and encapsulation efficiency, a 0.5 mL inner aqueous phase was used for all subsequent particle syntheses.
Table 1 Effect of volume of inner aqueous phase on ISMN-loaded PLGA particlesa
|
|
Microparticles |
Nanoparticles |
| Volume of internal phase (mL) |
Average sizeb (μm) |
DL (%) |
EE (%) |
Average size (nm) |
DL (%) |
EE (%) |
|
All batches were emulsified using an ultrasonicator (30 W) for 60 s, 10 mL PLGA (1%) dichloromethane solution was used as organic phase and 1% PVA (w/v) used as outer aqueous phase.
SEM, At least six representative PLGA MPs were used for the calculation, Data are given as mean ± SD (n = 3).
|
| 0.1 |
9.8 ± 1.8 |
0.8 ± 0.28 |
8.1 ± 5.0 |
172 ± 11 |
0.14 ± 0.03 |
1.6 ± 0.6 |
| 0.3 |
5.6 ± 2.2 |
1.9 ± 0.25 |
21.2 ± 1.5 |
255 ± 35 |
0.23 ± 0.03 |
2.7 ± 0.2 |
| 0.5 |
2.9 ± 1.4 |
2.2 ± 0.33 |
23.6 ± 1.4 |
264 ± 21 |
0.41 ± 0.02 |
5.8 ± 0.1 |
| 1 |
2.8 ± 1.7 |
1.3 ± 0.20 |
15.4 ± 0.7 |
556 ± 32 |
0.49 ± 0.07 |
6.3 ± 0.2 |
Effect of polyvinyl alcohol (PVA) concentration.
The influence of PVA concentration in the external water phase on the PLGA particles is shown in Table 2 (N.B. PLGA and ISMN concentrations and mixing times were kept constant). As the PVA concentration increased from 0.1% to 1%, the size of PLGA MPs decreased from 3.8 to 2.9 μm; the ISMN loading level (from 1.7% to 2.2%) and the encapsulation efficiency (from 18.1% to 23.6%) increased. A similar trend was observed for ISMN encapsulation into PLGA-NPs (but a less pronounced influence on size). This behaviour is likely due to enhanced stability of the emulsion droplets at a higher PVA concentration.
Table 2 Effect of PVA concentration in the external aqueous phase on ISMN-loaded PLGA particlesa
|
|
Microparticle |
Nanoparticle |
| PVA concentration (w/v) |
Average size (μm) |
DL (%) |
EE (%) |
Size (nm) |
DL (%) |
EE (%) |
|
All batches were emulsified using 0.5 mL ISMN aqueous solution (ISMN; 60 mg mL−1) was used as internal aqueous phase and 10 mL PLGA (1%) dichloromethane solution was used as organic phase.
|
| 0.1% |
3.8 ± 2.6 |
1.7 ± 0.51 |
18.1 ± 2.6 |
271 ± 17 |
0.33 ± 0.62 |
4.7 ± 0.3 |
| 0.5% |
3.4 ± 1.3 |
2.0 ± 0.63 |
21.1 ± 1.9 |
265 ± 9 |
0.36 ± 0.02 |
5.4 ± 0.1 |
| 1% |
2.9 ± 1.4 |
2.2 ± 0.33 |
23.6 ± 1.4 |
264 ± 21 |
0.41 ± 0.02 |
5.8 ± 0.1 |
Effect of ultrasonication time.
During the second emulsification step of the PLGA-MP preparation, the intensity and time of emulsification can significantly influence particle size.40 The particle size, ISMN loading and encapsulation efficiency as a function of ultra-sonication time are reported in Table 3. Increasing the ultrasonication time up to 60 seconds, decreased particle size, and increased ISMN loading and encapsulation efficiencies; this effect was not so pronounced for longer times. 60 seconds ultrasonication is considered suitable to form well dispersed emulsions, preventing droplet coalescence during the second emulsification step and subsequent ISMN leakage into the outer water phase.
Table 3 Effect of ultra-sonication time on ISMN-loaded PLGA-MPsa
| Ultra-sonication time (s) |
Average size (μm) |
DL (%) |
EE (%) |
|
All batches were emulsified using an ultrasonicator (30 W) for 60 s.
|
| 10 |
5.6 ± 1.1 |
0.8 ± 0.34 |
8.9 ± 6.1 |
| 30 |
4.7 ± 2.5 |
1.3 ± 0.29 |
14.1 ± 3.6 |
| 60 |
2.9 ± 1.4 |
2.2 ± 0.33 |
23.6 ± 1.4 |
| 90 |
3.1 ± 1.5 |
2.2 ± 0.17 |
24.8 ± 6 |
| 120 |
3.6 ± 2.6 |
2.3 ± 0.32 |
25.6 ± 6.6 |
Effect of homogenization pressure.
The particle size, ISMN loading and encapsulation efficiency of PLGA NPs were shown to depend on the intensity and extent of homogenization, see Table 4. The particle size decreased from 271 nm to 101 nm with increased homogenization pressure and number cycles. However, there was a corresponding reduction in ISMN loading (from 0.41% to 0.16%) and encapsulation efficiency (from 5.8% to 2.2%); this is likely due to ISMN leakage into the outer aqueous phase. 3 cycles at 500 bar pressure were selected to form well dispersed emulsion droplets, a narrow size distribution and to retain ISMN loading.
Table 4 Effect of homogenization pressure on ISMN-loaded PLGA nanoparticles
| Homogenization pressure (bar) |
Number of cycles |
Size (nm) |
DL (%) |
EE (%) |
| 500 |
1 |
271 ± 16 |
0.40 ± 0.29 |
5.6 ± 0.6 |
| 500 |
3 |
264 ± 21 |
0.41 ± 0.02 |
5.8 ± 0.3 |
| 500 |
5 |
243 ± 43 |
0.37 ± 0.21 |
4.8 ± 0.8 |
| 1000 |
1 |
139 ± 62 |
0.21 ± 0.02 |
2.7 ± 0.2 |
| 1000 |
3 |
121 ± 31 |
0.16 ± 0.04 |
2.2 ± 0.1 |
| 1000 |
5 |
101 ± 34 |
0.16 ± 0.08 |
2.2 ± 0.4 |
Based on the above data, the optimal processing parameters were defined as: 0.5 mL for the inner aqueous phase (ISMN, 60 mg mL−1), 1% PVA concentration (w/v), 1% PLGA in dichloromethane and either 60 seconds ultra-sonication for MP or 3 cycles of homogenization at 500 bars for NP.
Particle morphology.
SEM analysis images of the corresponding PLGA particles are shown in Fig. 2. PLGA-NPs showed evidence of aggregation and hollow structures (Fig. 2A). Conversely, PLGA-MPs displayed more spherical morphologies and homogenous size (Fig. 2B).
 |
| | Fig. 2 SEM images of the ISMN-loaded (A) PLGA nanoparticles (scale bar = 300 nm) and (B) PLGA microparticles (scale bar = 10 μm). | |
Particle surface analysis.
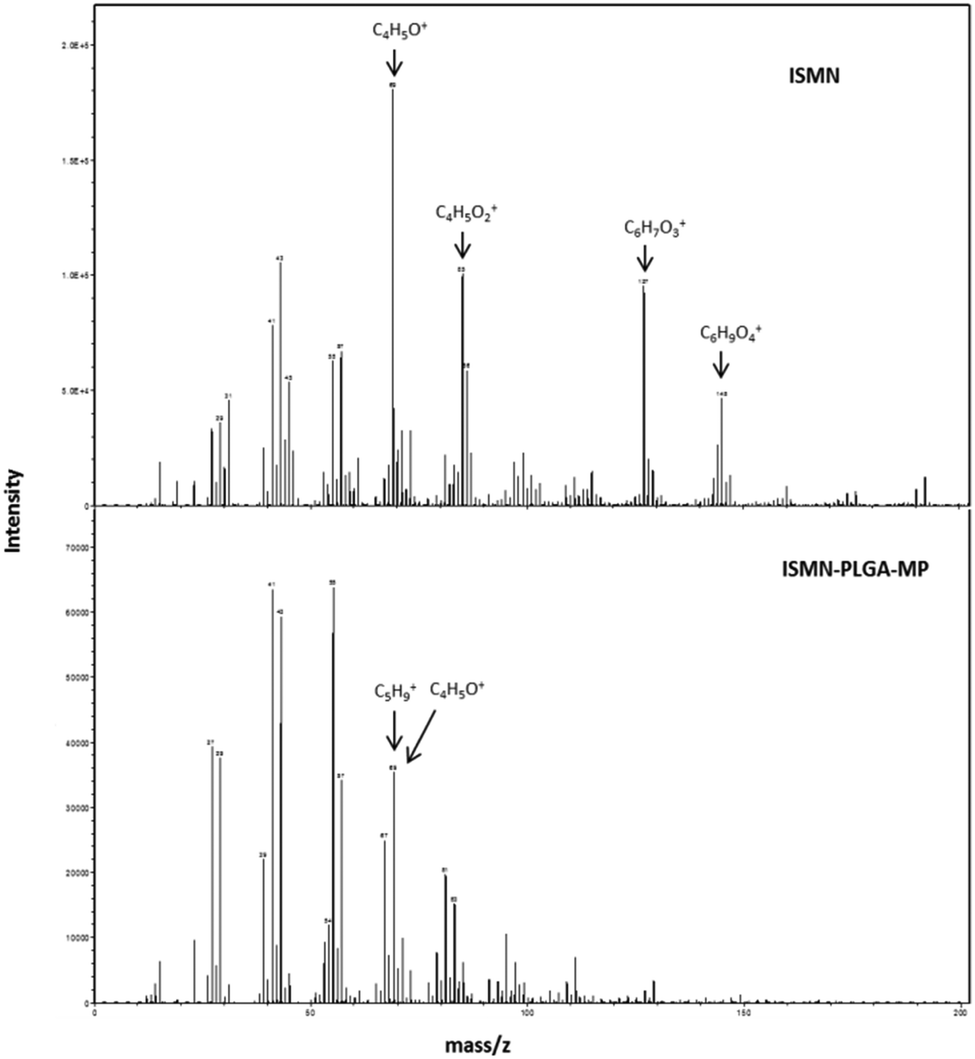
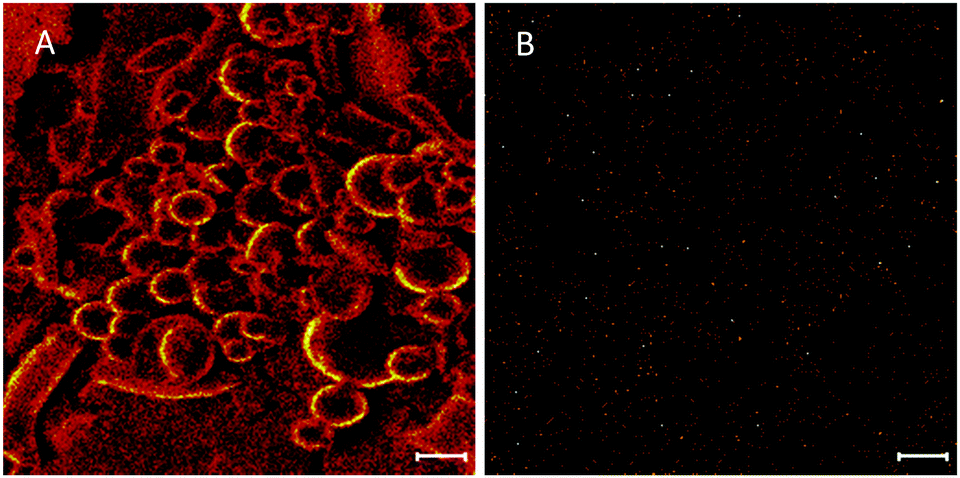
The technique of ToF-SIMS provides highly specific surface chemical information to elucidate surface chemistry and can determine the spatial distribution of molecules within a carrier material. ToFSIMS has been previously applied to pharmaceutical microparticle systems.43–45 In the current study, we probe the surface chemistry of ISMN containing PLGA microparticles to elucidate whether ISMN is distributed at the particle surfaces or not. Firstly, we determined ToFSIMS spectra of ISMN and PLGA particles to identify the specific mass fragments and the mass fragmentation patterns; these are shown in Fig. 3. No corresponding ISMN fragment peaks was observed except at mass to charge ratio (m/z) 69.03 amu correspond to ISMN fragment C4H5O+ and 69.07 amu to PLGA fragment C5H9+ would be overlapping in the spectra. Fig. 4A is a total ion image of the PLGA microparticles and Fig. 4B is an image associated with ISMN, which also confirm that the ISMN is not located at the particle surface, but fully encapsulated in the sub-surface/bulk of the PLGA particles.
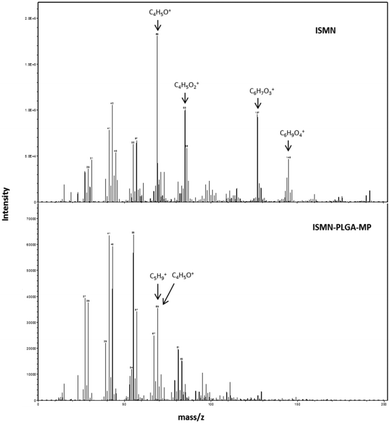 |
| | Fig. 3 ToF-SIMS spectra of the surface of ISMN and ISMN-loaded PLGA microparticles from a mass of 0 to 200, peaks used in the subsequent analysis are annotated (the unique ions are used to represent each constituent). | |
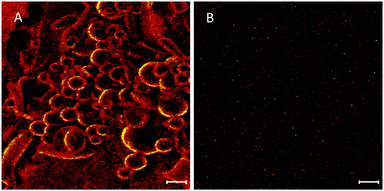 |
| | Fig. 4 (A) ToF-SIMS total ion image of PLGA particle and (B) image associated with ISMN (scale bar = 10 μm). | |
ISMN release in vitro.
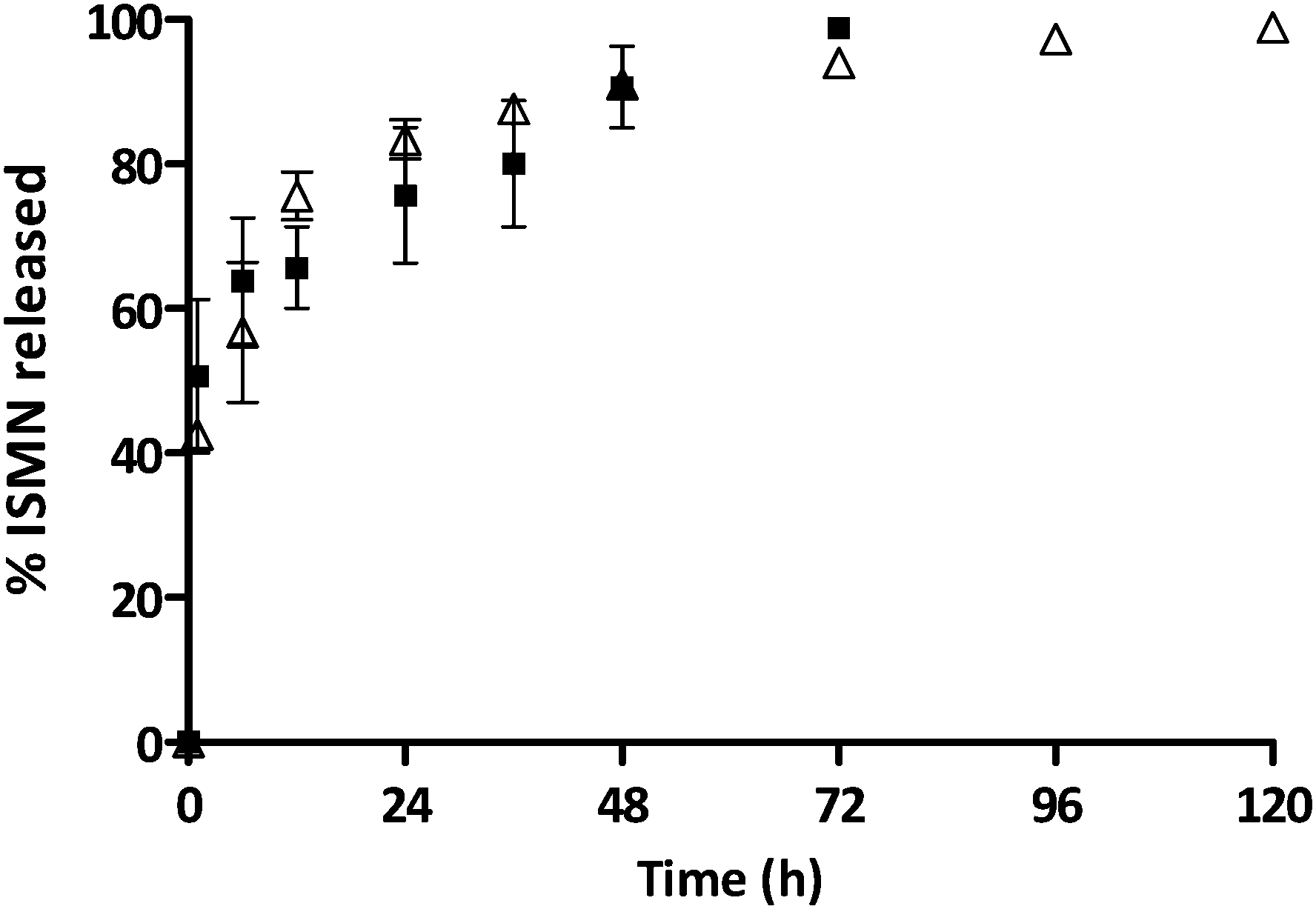
The release kinetics of ISMN from PLGA micro- and nanoparticles are shown in Fig. 5. It is likely that ISMN release from these carriers is controlled by some combination of diffusion and erosion mechanisms. For PLGA-NPs, ∼50% of ISMN was released within 1 hour (burst release) and further release was sustained for up to 3 days. For PLGA-MPs, ∼40% of the encapsulated ISMN displayed a burst release then, a further sustain release was observed for up to 5 days. The initial burst release is considered to be due to diffusion controlled mass transport processes,46,47i.e. the small and water soluble ISMN molecules that are encapsulated within the sub-surface of the PLGA particles can readily diffuse out of the polymeric particle matrix.
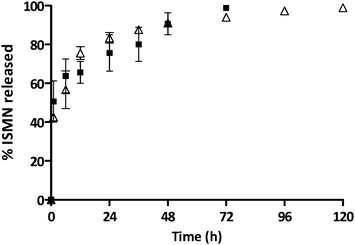 |
| | Fig. 5 The percentage of ISMN release from PLGA-MPs (Δ) and PLGA-NPs (■) at 37 °C in PBS (pH 7.4), data represent mean ± SD, n = 3. | |
In contrast, the sustained release phase results from several factors such as an increase in the diffusion pathways with time due to polymer swelling, degradation and erosion.47
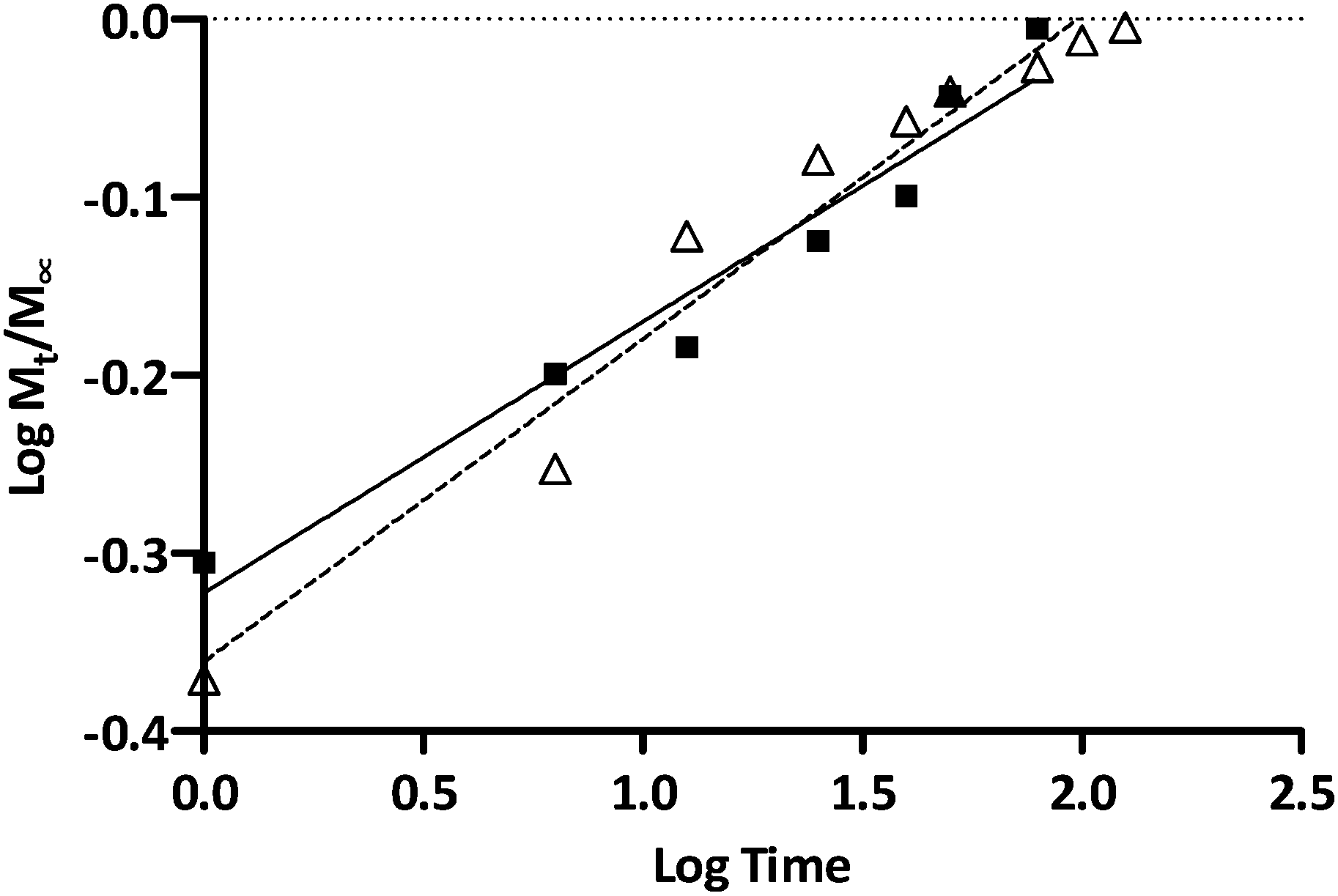
To further elucidate ISMN release mechanisms from these PLGA particles, ISMN release data were fitted to the Korsmeyer–Peppas equation as shown in Fig. 6,48i.e. Mt/M∞ = ktn, where Mt/M∞ is the fraction of ISMN released at time t, k is the rate constant and n is the drug release exponent. In this context, n values between 0.45 and 0.89 are indicative of non-Fickian release which refers to a combination of both diffusion and erosion drug release mechanisms.49 The observed n values for the release of ISMN from PLGA-MPs and PLGA-NPs of 0.74 and 0.66 respectively (R2 > 0.95) is confirmation of non-Fickian behaviour. Such release characteristics could be attributed to the increase in strong entanglement bonds between the polymer matrixes which resisted the erosion by the dissolution medium in the initial hours of ISMN release.47 The drug release from PLGA micro/nanoparticles is a complex process attributed to diffusion followed by degradation and is controlled by the molar ratio of the lactic and glycolic acid in the polymer chain, molecular weight of the polymer, the degree crystallinity and physico-chemical properties of the drug.30 Initial release of ISMN from both PLGA micro- and nanoparticles is considered a burst release of deposited or weakly bound ISMN molecules on the sub-surface of micro- and nanoparticles. Jameela et al.50 explained that when particles are prepared by the double emulsion method, most hydrophilic drugs exhibit higher tendency to migrate to the aqueous medium, thereby concentrating at the surface or sub-surface of the particles and rapidly released. A potential limiting factor in the preparation of ISMN-containing PLGA micro/nanoparticle is the hydrophobic nature of the PLGA polymer in contrast to the hydrophilic characteristics of ISMN. Generally, encapsulation of hydrophobic drug molecules in PLGA preparations is preferred due to strong hydrophobic interactions between the polymer and drug during formulation. Given the high aqueous solubility of ISMN (60 mg mL−1), it is not surprising that previous reports suggesting its presence on the outer surface or subsurface of particles, forming a molecular layer and thereby susceptible to be easily and rapidly released.51 Besides the polymer type, the drug loading also influenced the drug release rate. PLGA micro- and nanoparticle had different ISMN loadings (2.2% and 0.4% respectively) and these two ISMN release profiles have similar shape, but the PLGA-MPs with a higher ISMN loading leads to an increased initial rapid release.52 After the initial ISMN release from PLGA-MPs, there was a sustained ISMN release, indicating the constant degradation of the PLGA and the diffusion of encapsulated ISMN from the inner core of the PLGA-MPs.53 In contrast, faster release of ISMN from the PLGA-NPs could result from larger surface area and the shorter diffusion pathways compared to microparticles.
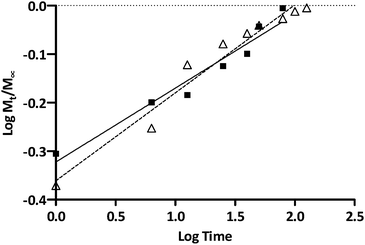 |
| | Fig. 6 Korsmeyer and Peppas kinetic plots of ISMN release from PLGA-MPs (Δ) and PLGA-NPs (■). | |
Anti-biofilm effect of ISMN in solution.
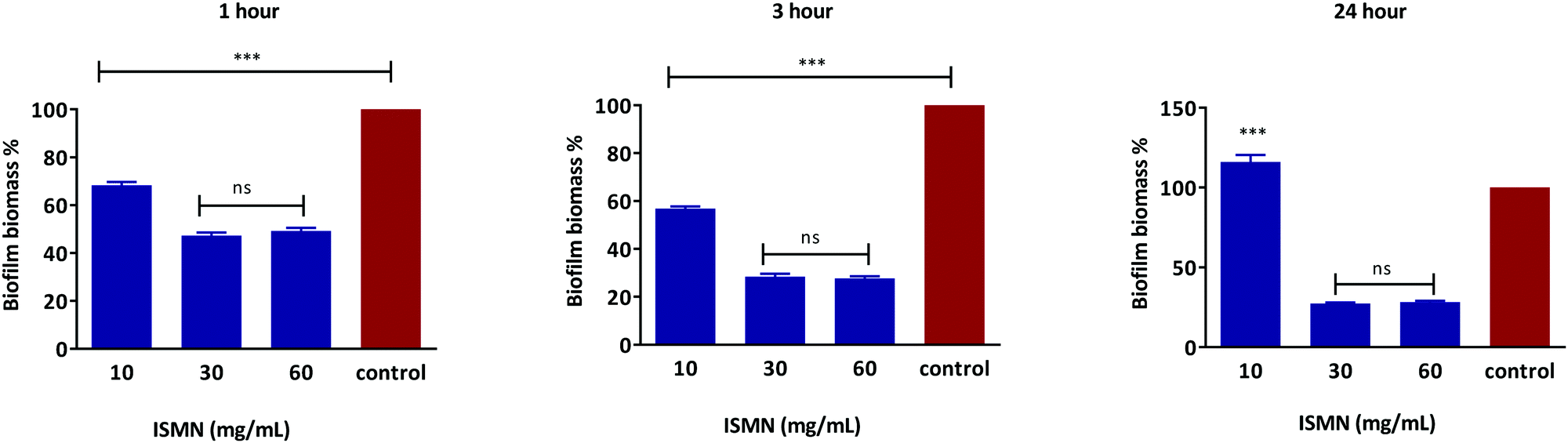
S. aureus biofilms grown for 1, 3 and 24 hours showed a concentration-dependent response to ISMN solutions, as shown in Fig. 7. ISMN solutions at concentration of 30 and 60 mg mL−1 showed a statistically significance anti-biofilm effect (P < 0.05) compared to the untreated control. This effect increased as the time of exposure increased from 1 to 3 to 24 hour. At an ISMN concentration of 10 mg mL−1 a less pronounced anti-biofilm effect was observed for up to 3 hours and then enhanced biofilm growth was observed at 24 hours of exposure. A stimulatory effect of biofilm growth by ISMN at low concentrations (i.e. 3.75 mg mL−1) has also been reported by Jardeleza et al.;23 this is considered due to a nutritional role of ISMN at sub-antimicrobial levels.
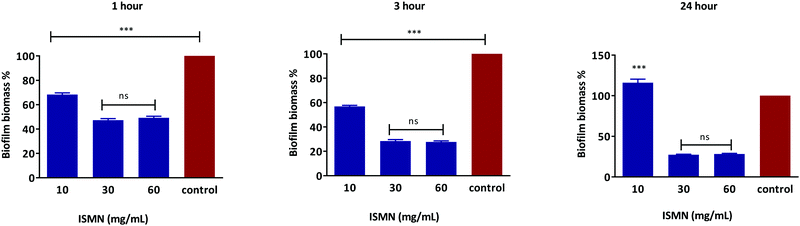 |
| | Fig. 7 Dose response of free ISMN on S. aureus biofilm biomass after 1, 3 and 24 hours treatment. Data represents as mean ± SD of a duplicate experiment. | |
MIC and MBEC assay of ISMN loaded PLGA-MP.
Owing to the requirement for high ISMN concentrations to exert anti-bacterial activity, PLGA nanoparticles were not considered for further antimicrobial evaluation due to their low drug loads. This section therefore focuses of ISMN containing PLGA microparticles (ISMN-PLGA-MP). The apparent MIC for ISMN encapsulated in PLGA-MPs was determined as 7.5 mg mL−1 (Table 5). The MIC observed for encapsulated ISMN showed a two-fold increase compared to that of the free ISMN (3.75 mg mL−1). The MBEC concentration for free ISMN was 15 mg mL−1, which is in accordance with previous studies,23 whereas the measured MBEC concentration for encapsulated ISMN in PLGA-MPs was determined as 30 mg mL−1 (Table 5). The higher MBEC values relative to MIC observed for both free and encapsulated ISMN are likely due to the incomplete drug release from PLGA-MPs within 24 h of incubation, effectively reducing the available drug concentration compared to treatment with a simple ISMN solution. An increase in the apparent MIC and MBEC of sustained release PLGA particles relative to drug solution has previously been observed for the antibiotic ciprofloxacin.39
Table 5 MIC and MBEC values for ISMN formulations
|
|
MIC (mg mL−1) |
MBEC (mg mL−1) |
| Free ISMN |
3.75 |
15 |
| MP-encapsulated ISMN |
7.5 |
30 |
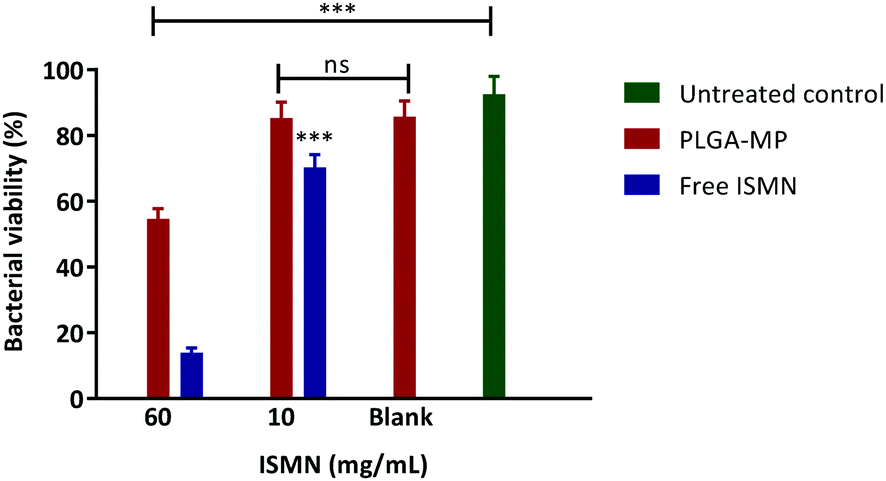
Anti-planktonic and biofilm effects of ISMN loaded PLGA-MP.
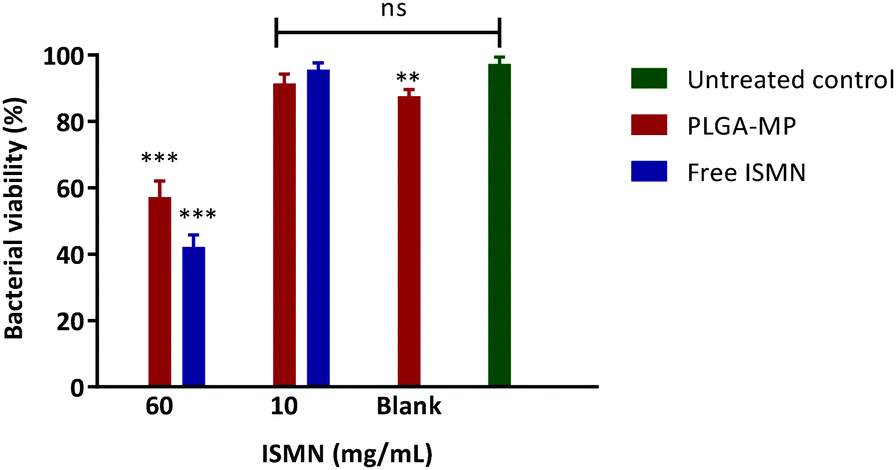
The influence of a 1 hour ISMN treatment (at both 10 and 60 mg mL−1) dosed as either ISMN solutions or PLGA MP's on S. aureus in planktonic cultures grown for 24 hours are shown in Fig. 8. Free ISMN at both 60 and 10 mg mL−1 significantly decreased (P < 0.05) the number of viable bacteria, i.e. a ∼86% and ∼30% reduction respectively. ISMN-PLGA-MP inhibited bacterial growth with less effectiveness than the free ISMN and this was only significant at 60 mg mL−1 ISMN. The blank PLGA-MPs exhibited no statistically significant inhibition of bacterial growth, highlighting that the antibacterial effects were elicited by ISMN itself. The anti-biofilm effects of ISMN were investigated in vitro using S. aureus biofilms grown in 96-well plates as previously described; these were challenged with both free ISMN and ISMN-loaded PLGA-MPs for 24 hours. As illustrated in Fig. 9, the percentage of bacterial cell viability following exposure to free ISMN (60 mg mL−1) was significantly reduced (57.8%, p > 0.05), whereas enhanced biofilm formation was observed for free ISMN at 10 mg mL−1 concentration. In agreement with previous observations, a low concentration of ISMN was associated with a stimulatory biofilm effect. PLGA-MPs containing 60 mg mL−1 ISMN exhibited a statistically significant anti-biofilm effect (42.2% bacterial viability reduction, p < 0.05), but with less potency than free ISMN (57.8% bacterial viability reduction). The anti-biofilm effect of blank microparticles and 10 mg mL−1 ISMN encapsulated in PLGA-MPs resulted in no statistically significant reduction of biofilm viability compared to the untreated control (p > 0.05). Subsequent analysis of ISMN release from PLGA-MPs demonstrated that the initial burst release followed by a constant sustained release enhanced the diffusion of ISMN from PLGA-MPs into biofilm. Therefore, continuous exposure of the biofilm to ISMN through sustained release from PLGA-MPs enhanced the efficacy compared to free ISMN despite incomplete ISMN release after 24 hours. Given the difficulty of biofilm penetration by PLGA-MPs, it may be possible to improve efficacy by modifying microparticle surfaces with a cationic polymer such as chitosan, potentially increasing the interaction with anionic mucopolysaccharide in the biofilm and maximizing biofilm exposure to ISMN. This is an area for future investigation.
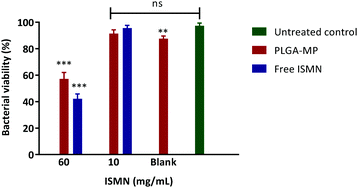 |
| | Fig. 8 Percentage of viable bacterial cells after 1 h treatments on planktonic S. aureus bacteria. PLGA MP-60 and 10: PLGA MP loaded with 60 and 10 mg mL−1 of ISMN and ISMN-60 and 10: free ISMN with 60 and 10 mg mL−1. Data represents as mean ± SD of a duplicate experiment. | |
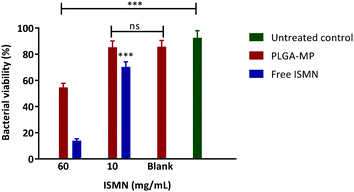 |
| | Fig. 9 Percentage of viable bacterial cells after 24 h treatments on S. aureus biofilm. Data represents as mean ± SD of a duplicate experiment. | |
Conclusion
A double emulsion solvent evaporation method has been successfully optimised to encapsulate the NO donor ISMN into PLGA nanoparticles and microparticles. Considerable sustained release of ISMN has been demonstrated. PLGA microparticles but not nanoparticles were able to deliver sufficient levels of ISMN to planktonic and biofilms of S. aureus to cause significant anti-bacterial effects. ISMN-PLGA-MP formulations have potential for the treatment of a number of biofilm-based infections.
Acknowledgements
This project has been supported by The National Health and Medical Research Council (NHMRC project grant GNT1047576 and GNT1090898). The University of South Australia and School of Pharmacy and Medical Sciences are acknowledged for providing a scholarship to SH. This work was performed in part at the South Australian node of the Australian National Fabrication Facility (ANFF) under the National Collaborative Research Infrastructure Strategy to provide nano- and microfabrication facilities for Australia's researchers.
References
- G. A. James, E. Swogger, R. Wolcott, E. D. Pulcini, P. Secor, J. Sestrich, J. W. Costerton and P. S. Stewart, Wound Repair Regen., 2008, 16, 37–44 CrossRef PubMed.
- R. A. Brady, J. G. Leid, J. H. Calhoun, J. W. Costerton and M. E. Shirtliff, FEMS Immunol. Med. Microbiol., 2008, 52, 13–22 CrossRef CAS PubMed.
- B. A. Cunha, M. V. Gill and J. M. Lazar, Infect. Dis. Clin. North Am., 1996, 10, 811–834 CrossRef CAS PubMed.
- A. Foreman and P.-J. Wormald, Laryngoscope, 2010, 120, 1701–1706 CrossRef PubMed.
- R. O. Darouiche, N. Engl. J. Med., 2004, 350, 1422–1429 CrossRef CAS PubMed.
- G. E. Thwaites, J. D. Edgeworth, E. Gkrania-Klotsas, A. Kirby, R. Tilley, M. E. Török, S. Walker, H. F. L. Wertheim, P. Wilson and M. J. Llewelyn, Lancet Infect. Dis., 2011, 11, 208–222 CrossRef PubMed.
- P. M. Hawkey, J. Antimicrob. Chemother., 2008, 62, i1–i9 CrossRef CAS PubMed.
- B. Vu, M. Chen, R. Crawford and E. Ivanova, Molecules, 2009, 14, 2535 CrossRef CAS PubMed.
- N. Høiby, T. Bjarnsholt, M. Givskov, S. Molin and O. Ciofu, Int. J. Antimicrob. Agents, 2010, 35, 322–332 CrossRef PubMed.
- I. d'Angelo, C. Conte, M. I. La Rotonda, A. Miro, F. Quaglia and F. Ungaro, Adv. Drug Delivery Rev., 2014, 75, 92–111 CrossRef PubMed.
- T.-F. C. Mah and G. A. O'Toole, Trends Microbiol., 2001, 9, 34–39 CrossRef CAS PubMed.
- C. A. Fux, J. W. Costerton, P. S. Stewart and P. Stoodley, Trends Microbiol., 2005, 13, 34–40 CrossRef CAS PubMed.
- P. S. Stewart, J. Bacteriol., 2003, 185, 1485–1491 CrossRef CAS PubMed.
- N. Høiby, O. Ciofu and T. Bjarnsholt, Future Microbiol., 2010, 5, 1663–1674 CrossRef PubMed.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS PubMed.
- C. A. Gordon, N. A. Hodges and C. Marriott, J. Antimicrob. Chemother., 1988, 22, 667–674 CrossRef CAS PubMed.
- G. N. Marcells, P. S. Phillips, N. A. Cohen, R. J. Harvey and R. Sacks, Otolaryngol.–Head Neck Surg., 2011, 144, 159–169 CrossRef.
- D. A. Sanchez, J. Nosanchuk and A. Friedman, Nanomedicine, 2012, 7, 933–936 CrossRef CAS PubMed.
- J. Saraiva, S. S. Marotta-Oliveira, S. A. Cicillini, J. D. O. Eloy and J. M. Marchetti, J. Drug Delivery, 2011, 936438 Search PubMed.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- A. Friedman and J. Friedman, Expert Opin. Drug Delivery, 2009, 6, 1113–1122 CrossRef CAS PubMed.
- C. Jardeleza, A. Foreman, L. Baker, S. Paramasivan, J. Field, L. W. Tan and P. J. Wormald, International Forum of Allergy and Rhinology, 2011, 1, 438–444 CrossRef PubMed.
- C. Jardeleza, S. Rao, B. Thierry, P. Gajjar, S. Vreugde, C. A. Prestidge and P. J. Wormald, PLoS One, 2014, 9, e92117 Search PubMed.
- E. M. Hetrick, J. H. Shin, H. S. Paul and M. H. Schoenfisch, Biomaterials, 2009, 30, 2782–2789 CrossRef CAS PubMed.
- E. M. Hetrick, J. H. Shin and M. H. Schoenfisch, 8th World Biomaterials Congress 2008, 2008, 4, 1845 Search PubMed.
- A. J. Friedman, G. Han, M. S. Navati, M. Chacko, L. Gunther, A. Alfieri and J. M. Friedman, Nitric Oxide, 2008, 19, 12–20 CrossRef CAS PubMed.
- L. R. Martinez, G. Han, M. Chacko, M. R. Mihu, M. Jacobson, P. Gialanella, A. J. Friedman, J. D. Nosanchuk and J. M. Friedman, J. Invest. Dermatol., 2009, 129, 2463–2469 CrossRef CAS PubMed.
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS PubMed.
- R. Jain, N. H. Shah, A. W. Malick and C. T. Rhodes, Drug Dev. Ind. Pharm., 1998, 24, 703–727 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- K. Dillen, J. Vandervoort, G. Van den Mooter, L. Verheyden and A. Ludwig, Int. J. Pharm., 2004, 275, 171–187 CrossRef CAS PubMed.
- J. Liu, Z. Qiu, S. Wang, L. Zhou and S. Zhang, Biomed. Mater., 2010, 5, 065002 CrossRef PubMed.
- J. O'Sullivan, M. Park and J. Beevers, J. Cereal Sci., 2016, 69, 77–84 CrossRef.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- I. Wiegand, K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
-
J. H. Merritt, D. E. Kadouri and G. A. O'Toole, Current Protocols in Microbiology, 2011 Search PubMed.
- E. Peeters, H. J. Nelis and T. Coenye, J. Microbiol. Methods, 2008, 72, 157–165 CrossRef CAS PubMed.
- I. Vandecandelaere, H. Van Acker and T. Coenye, Methods Mol. Biol., 2016, 1333, 53–66 Search PubMed.
- N. Thomas, C. Thorn, K. Richter, B. Thierry and C. Prestidge, J. Pharm. Sci., 2016, 105, 3115–3122 CrossRef CAS PubMed.
- U. Bilati, E. Allémann and E. Doelker, Pharm. Dev. Technol., 2003, 8, 1 CrossRef CAS PubMed.
- U. Bilati, E. Allémann and E. Doelker, J. Microencapsulation, 2005, 22, 205–214 CrossRef CAS PubMed.
- T. Verrecchia, G. Spenlehauer, D. V. Bazile, A. Murry-Brelier, Y. Archimbaud and M. Veillard, J. Controlled Release, 1995, 36, 49–61 CrossRef CAS.
- A. Rafati, A. Boussahel, K. M. Shakesheff, A. G. Shard, C. J. Roberts, X. Chen, D. J. Scurr, S. Rigby-Singleton, P. Whiteside, M. R. Alexander and M. C. Davies, J. Controlled Release, 2012, 162, 321–329 CrossRef CAS PubMed.
- J. G. Y. Chan, H. K. Chan, C. A. Prestidge, J. A. Denman, P. M. Young and D. Traini, Eur. J. Pharm. Biopharm., 2013, 83, 285–292 CrossRef CAS PubMed.
- A. M. Belu, M. C. Davies, J. M. Newton and N. Patel, Anal. Chem., 2000, 72, 5625–5638 CrossRef CAS PubMed.
- N. Faisant, J. Siepmann, J. Richard and J. P. Benoit, Eur. J. Pharm. Biopharm., 2003, 56, 271–279 CrossRef CAS PubMed.
- A. N. Ford Versypt, D. W. Pack and R. D. Braatz, J. Controlled Release, 2013, 165, 29–37 CrossRef CAS PubMed.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- J. Vysloužil, P. Doležel, M. Kejdušová, V. Košál, L. Beneš and K. Dvoráčková, Pharm. Dev. Technol., 2016, 21, 214–221 CrossRef PubMed.
- S. R. Jameela, N. Suma and A. Jayakrishnan, J. Biomater. Sci., Polym. Ed., 1997, 8, 457–466 CrossRef CAS PubMed.
- C. Ahlneck and G. Zografi, Int. J. Pharm., 1990, 62, 87–95 CrossRef CAS.
- F. Cui, D. Cun, A. Tao, M. Yang, K. Shi, M. Zhao and Y. Guan, J. Controlled Release, 2005, 107, 310–319 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Benjamin
Thierry
b and
Clive A.
Prestidge
*a
a,
Benjamin
Thierry
b and
Clive A.
Prestidge
*a