
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface modification strategies for combating catheter-related complications: recent advances and challenges
Koon Gee
Neoh
 *a,
Min
Li
a,
En-Tang
Kang
a,
Edmund
Chiong
b and
Paul Anantharajah
Tambyah
c
*a,
Min
Li
a,
En-Tang
Kang
a,
Edmund
Chiong
b and
Paul Anantharajah
Tambyah
c
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 21 Lower Kent Ridge Road, Singapore 119077. E-mail: chenkg@nus.edu.sg; Fax: +65-67791936; Tel: +65-6516-2176
bDepartment of Surgery, National University of Singapore, 21 Lower Kent Ridge Road, Singapore 119077
cDepartment of Medicine, National University of Singapore, 21 Lower Kent Ridge Road, Singapore 119077
First published on 22nd February 2017
Abstract
Indwelling medical devices such as catheters are a ubiquitous and indispensable component in modern medical practice for improving therapeutic outcomes for patients. Yet at the same time, they can be a cause of healthcare-associated infections contributing to patient morbidity and mortality, and healthcare costs. Other surface-related complications can also arise from interactions of the catheter with biological components in the in vivo environment. This review summarizes the progress made in the development of antimicrobial surfaces, and the application of surface modification strategies to three important classes of catheters: urinary catheters, intravascular catheters and peritoneal dialysis catheters. The review also provides a perspective on the challenges in translating favorable developments from in vitro studies into similar clinical outcomes.
 Koon Gee Neoh | Koon Gee Neoh is currently a Professor in the Department of Chemical & Biomolecular Engineering at the National University of Singapore (NUS). She received her Bachelor's and ScD degrees in Chemical Engineering from MIT. Her main research focus is on surface/molecular engineering of materials to control their interactions with biomolecules, microorganisms and cells, and application of the technology to address biomedical issues such as preventing healthcare-associated infections and targeted drug delivery. |
 Min Li | Dr Min Li received his BEng and MSc degrees in Chemical Engineering from the Tianjin University, China, and obtained his PhD from the National University of Singapore (NUS). He is working as a Research Fellow in NUS. He has research interests in anti-fouling and anti-infective surfaces, and polymers in nanoscience and biomolecular engineering. |
 En-Tang Kang | E. T. Kang obtained his PhD from the State University of New York at Buffalo in 1983. He is currently a professor in the Department of Chemical & Biomolecular Engineering at the National University of Singapore. His research interests include surface functionalization via molecular design, molecular/polymer electronics, polymers in nanoscience and biomolecular engineering and polymers for environmentally-benign applications. He received the National Young Scientist and Engineer Award in 1989 and the National Science Award of Singapore in 1996. He is an Associate Editor for the journal ACS Applied Materials & Interfaces and serves on the Editorial Advisory Board of several international journals. |
 Edmund Chiong | A/Prof. Edmund Chiong received his MBBS and PhD from the National University of Singapore (NUS), and obtained his post-graduate medical qualifications from the UK and Singapore. He is presently an Associate Professor at NUS, a Senior Consultant Urologist, Director of Urologic Oncology and Director of Research, in the Department of Urology, National University Health System, Singapore, and an Associate Chairman (Research) of the Medical Board at the National University Hospital. He is also currently the President of the Singapore Urological Association. He has research interests in investigating new diagnostic and therapeutic modalities for urologic cancers, urinary tract infection prevention, and medical device innovation. |
 Paul Anantharajah Tambyah | Paul Anantharajah Tambyah completed medical school at the National University of Singapore and after national service in the Singapore Armed Forces Medical Corps, went on to train in Infectious Diseases at the University of Wisconsin under Dr Dennis Maki. He is currently a Professor of Medicine and a senior consultant infectious diseases physician. He is also a board member of the Asian Association of UTI and STD and the Secretary General of the Asia Pacific Society of Clinical Microbiology and Infection. His main research interests are device-associated infections in particular catheter-associated urinary tract infections and emerging infectious diseases including antimicrobial resistant pathogens. |
1. Introduction
Implantable medical devices such as catheters, endotracheal tubes, orthopedic devices, and pacemakers have become an integral part of modern medical practice for saving lives or improving therapeutic outcomes. However, such devices are also risk factors for healthcare-associated infections (HAIs), which are defined as infections that patients develop while they are receiving care in a healthcare setting for another condition.1 Infections associated with implantable medical devices constitute three of the five most common HAIs, which are surgical site infections, diarrhea caused by Clostridium difficile (C. difficile), catheter-associated urinary tract infections (CAUTIs), central line-associated blood stream infections (CLABSIs) and ventilator-associated pneumonia (VAP).2 The United States Centers for Disease Control and Prevention (CDC) reported that there were over 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases of HAIs in US acute care hospitals in 2011, and more than 10% of the patients with HAIs died during their hospitalization.3 The situation in Europe is just as serious, with over 4 million patients estimated by the European Center for Disease Control and Prevention (ECDC) to acquire a HAI in the European Union each year.4 Comparable data from Asia are scarce but there are indications that rates are higher than in either the US or Europe.5 HAIs, besides a major cause of morbidity and mortality, also impose a heavy financial burden resulting from prolonged hospital stay and treatment. It has been estimated that the annual costs attributable to the HAIs mentioned for US adult inpatients at acute care hospitals amounted to $9.8 billion (adjusted to 2012 dollars), with CLABSI being the most costly HAI on a per-case basis at about $46000.6
000 cases of HAIs in US acute care hospitals in 2011, and more than 10% of the patients with HAIs died during their hospitalization.3 The situation in Europe is just as serious, with over 4 million patients estimated by the European Center for Disease Control and Prevention (ECDC) to acquire a HAI in the European Union each year.4 Comparable data from Asia are scarce but there are indications that rates are higher than in either the US or Europe.5 HAIs, besides a major cause of morbidity and mortality, also impose a heavy financial burden resulting from prolonged hospital stay and treatment. It has been estimated that the annual costs attributable to the HAIs mentioned for US adult inpatients at acute care hospitals amounted to $9.8 billion (adjusted to 2012 dollars), with CLABSI being the most costly HAI on a per-case basis at about $46000.6
The high human and economic costs of HAIs have spurred international agencies such as the CDC and ECDC to increase surveillance and issue guidelines for prevention and control of HAIs. Among the findings of the 2016 CDC’s Annual National and State Healthcare-Associated Infections Progress Report (2014 data) were a 50% decrease in CLABSI between 2008 and 2014 but no change in the overall CAUTI between 2009 and 2014.3,7 The National Healthcare Safety Network report of 2013 showed that urinary catheter utilization (defined as the ratio of device-days to patient-days) ranged from 0.48 to 0.75 in adult critical care units while the corresponding range for central line (also known as central venous catheters, CVCs) utilization is from 0.37 to 0.66.8 In the US, about 300 million peripheral intravenous catheters (PIVCs),9,10 5 million CVCs and 30 million urinary catheters11–13 are used annually. In view of the prevalence of catheters in healthcare institutions, prevention of infections attributable to these devices is an important goal to reduce HAIs. Depending on the function of the catheter and the needs of the patients, periods of catheterization may range from a few days to weeks and may even be on a permanent basis. It is well recognized that an important risk factor for catheter-associated infections is the duration of catheterization.14–19 Microorganisms can adhere to the surfaces of an indwelling catheter and subsequently colonize the device. These microorganisms may be either the patient’s endogenous flora (e.g. from skin or mucous membranes) or from exogenous sources such as other patients, healthcare personnel, inanimate environmental surfaces or objects that have become contaminated. A review of 1022 outbreaks of nosocomial infection indicated that the most frequent source was cross transmission from individual patients followed by medical equipment or devices, the hospital environment, healthcare personnel, contaminated drugs, contaminated food, and contaminated care equipment.20
Since infection is a problem associated with all indwelling devices, this review will first highlight the process of microbial colonization of medical devices. Next, the complications specific to three important classes of catheters: urinary catheters, intravascular catheters and peritoneal dialysis catheters will then be presented (Table 1). Urinary catheters and intravascular catheters are selected for this review due to their prevalence, and peritoneal dialysis catheters are selected because of the extended duration of use of this class of catheters. Lastly, strategies to modify the surfaces of the three classes of catheters to inhibit microbial colonization and other complications specific to the particular class of catheter will be discussed.
| Indwelling catheter | Major complications | Microorganisms | References |
|---|---|---|---|
| Urinary catheter | Infection and encrustation | Staphylococcus epidermidis, Escherichia coli, Klebsiella pneumoniae, Enterococcus faecalis, Proteus mirabilis | 24, 51, 56 and 57 |
| Central venous catheter | Infection and catheter occlusion | Coagulase-negative staphylococci, Staphylococcus aureus, Enterococcus faecalis, Klebsiella pneumoniae, Pseudomonas aeruginosa, Candida species | 24, 69, 74 and 75 |
| Peritoneal dialysis catheter | Infection and omental wrapping | Coagulase-negative staphylococci, Staphylococcus aureus, Pseudomonas species, Enterobacter species, Candida species | 39, 103 and 104 |
2. Bacterial adhesion and biofilm formation on medical devices
When a medical device is placed in the body, host molecules such as ions and proteins will rapidly interact with its surface to form a conditioning film.21–23 Planktonic bacterial cells migrate to the surface via Brownian motion, van der Waals attraction, gravitational forces, surface electrostatic charges and hydrophobic interactions, and adhere to the conditioning film.21 In general, rougher and more hydrophobic surfaces are more prone to bacterial adhesion but the presence of the conditioning film may change the surface characteristics of the substrate.24 The initial adhesion process is reversible, but with time as the adherent cells multiply, form microbial aggregates with the same or different species, and secrete extracellular polymeric substances (EPS), the process becomes irreversible. EPS comprise mainly polysaccharides, extracellular DNA and proteins.25 The term “biofilm” is used to describe these aggregates of cells encased in the matrix of EPS on the device surface.26 In most biofilms, the microorganisms account for less than 10% of the dry mass, whereas the matrix can account for over 90%.26 Single cells or aggregates of cells may detach from the biofilm due to fluid shear, or as a result of internal biofilm processes, such as endogenous enzymatic degradation, or by the release of EPS or surface-binding proteins, and these released cells may proceed to colonize other sites.26,27 A schematic representation of the process of biofilm formation is shown in Fig. 1.28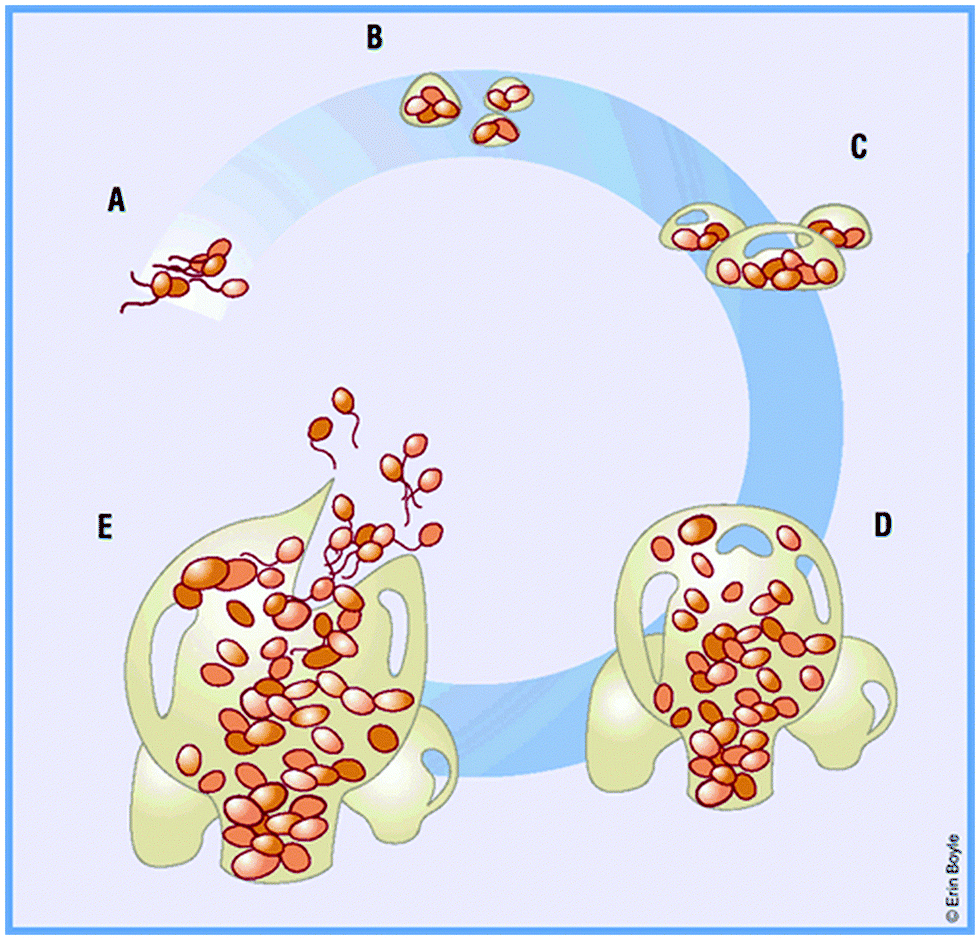 | ||
| Fig. 1 (A) Bacteria reversibly attach to a solid support. (B) Bacteria become irreversibly attached, and aggregate to form a matrix. (C) Maturation phase: cells become layered and the effects of quorum sensing begin. (D) Clusters reach maximum thickness. (E) Escape of planktonic bacteria from matrix dispersion. Reproduced from ref. 28 with permission from Nature Publishing Group. | ||
The cells embedded within the EPS of the biofilm are in an environment that is different from the planktonic state.26 The biofilm-associated cells grow more slowly than their planktonic counterparts probably because the former are limited by nutrient supply and/or oxygen depletion.27 The biofilm provides protection to the associated cells against a wide range of environmental challenges including host defenses and antimicrobial agents. Biofilm bacteria can be orders of magnitude less susceptible to antibiotics29–31 as well as to commonly used hospital biocides like sodium hypochlorite, benzalkonium chloride, chlorhexidine and triclosan.32,33 Three mechanisms have been proposed to explain the generally higher resistance of biofilm bacteria to biocidal agents as compared to their planktonic counterparts.26–28 In the first mechanism, the EPS matrix may serve as a barrier by physically impeding the transport of the antimicrobial agent or by binding and neutralizing the agent. Since it has been demonstrated that some antibiotics can penetrate biofilms and yet the bacteria in the biofilm are able to resist the effects of the antibiotics,34 this mechanism cannot fully account for the observed resistance. The second mechanism may be related to the slow growth state of the bacteria in the biofilm which reduces their uptake of the antimicrobial agent which primarily targets dividing cells. It has been demonstrated that biofilm bacteria exhibit increased susceptibility to antibiotics with an increase in the growth rate.34,35 The last mechanism may be the existence of subpopulations of resistant phenotypes in the biofilm known as “persisters”. These persister cells usually comprise about 1% in biofilms, and are likely to arise due to a state of dormancy, i.e. being metabolically inactive.36
The association of biofilms with medical devices was established in the 1980s when investigators used electron microscopy techniques to examine the surfaces of intravascular catheters37 and endocardial pacemaker leads.38 Biofilm formation on indwelling medical devices and the associated decrease in susceptibility to antimicrobial agents have significant implications for public health. Microorganisms that are commonly involved in infections associated with selected indwelling catheters are given in Table 1.24,39 These microorganisms may be bacteria or fungi, and may form monomicrobial or polymicrobial biofilms. Microorganisms growing in mixed species biofilms can have higher resistance and virulence. For example, a mixed species biofilm of Candida albicans (C. albicans) and Staphylococcus epidermidis (S. epidermidis) enhances the growth of the latter and increases its resistance to vancomycin.40 An in vivo model of systemic intraperitoneal infection showed that infection with sub-lethal doses of C. albicans and Staphylococcus aureus (S. aureus) resulted in no mortality, but with the same doses, co-infection resulted in 100% mortality.41 Device-related infections are notoriously difficult to treat due to the resistance of the biofilm microorganisms. While planktonic cells in the bloodstream during a device-related infection can be killed by antibiotics, biofilm bacteria frequently survive the treatment and the biofilm may not be eliminated from the infected device and reinfection can result.42 In many cases, removal of the colonized device is necessary to eradicate a biofilm-related infection.43,44 In view of the difficulty in treating device-related infection, alternative approaches of preventing biofilm formation warrant serious consideration. Research on modification of the surfaces of implanted devices to inhibit microbial adhesion and colonization is actively being pursued, and some advances made in this approach for intravascular catheters, urinary catheters and peritoneal dialysis catheters and the challenges encountered are described in Section 4.
3. Catheter-related complications
In this section, the complications associated with urinary catheters, intravascular catheters and peritoneal dialysis catheters will be presented. While infections can arise with the use of all three classes of catheters, there are other complications that are specific to each class of catheter since they are used in different biological environments.3.1 Urinary catheter
Indwelling urinary catheters are common medical devices utilized in hospitals and nursing homes to relieve urinary retention and treat incontinence. It has been reported that between 15% and 25% of hospitalized patients have a urinary catheter placed some time during their hospital stay.45 However, the use of urinary catheters has been associated with urinary tract infections, and the greatest risk factor for catheter-associated urinary tract infections (CAUTIs) is prolonged catheterization.46 The duration of catheterization varies from ≤4 days to >20 days, depending on the hospital and type of ward.45–48 In addition to hospitals, urinary catheters are frequently used in nursing homes. Catheter prevalence in nursing homes is reported to range from 5% to 22% of all residents, and on any given day, 80000 to 352000 of the approximately 1.5 million US post-acute care patients and long-stay nursing home residents have a urinary catheter.49 Among catheterized patients, the incidence of CAUTIs is ∼5% per day.50 Patients with CAUTIs suffer increased morbidity and possibly mortality, and such infections are collectively the most common cause of secondary bloodstream infections.51 Due to this high incidence of CAUTIs, the overall cost for medical intervention is very high, accounting for an annual direct medical cost of $340–370 million in the US alone.45 The American Urological Association recently published a study which showed that hospitalization due to catheter-associated complications has increased over a 10 year period from 2001 to 2010, and urinary tract infections were the primary diagnosis (87% in 2010) for admission.52 As of 2008, Medicare (the US government’s health insurance program for senior citizens and persons with certain disabilities) has stopped reimbursing hospitals in the US for CAUTIs that develop during hospitalization.53 This has provided a new impetus for the development of strategies to combat CAUTIs.
Urinary tract infections are clinically categorized as uncomplicated if the affected individuals are otherwise healthy and have no structural or neurological urinary tract abnormalities (these primarily refer to bladder infections in young women).51 On the other hand, complicated urinary tract infections are defined as those associated with factors that compromise the urinary tract or host defense or occur in men, and it is estimated that 70–80% of complicated urinary tract infections in the US are attributable to indwelling catheters.51 The indwelling catheter not only provides a surface for microbial attachment, but the insertion of the catheter may also damage the protective uroepithelial mucosa, leading to the exposure of new binding sites for bacteria. Furthermore, the presence of the indwelling catheter in the urinary tract disrupts normal host mechanical defenses, resulting in unimpeded entry of microorganisms into the bladder.54 The microorganisms causing CAUTIs may be endogenous such as perineal organisms or exogenous from contaminated equipment or hands of healthcare workers. The microorganisms can enter the urinary tract during catheter insertion or by migrating from the perineum in the thin mucous sheath on the catheter surface (extraluminal route). Alternatively, intraluminal contamination can occur when microorganisms gain access to the catheter lumen from a contaminated collection bag or catheter–drainage tube junction (Fig. 2).55,56 The extraluminal route has been reported to be dominant, accounting for 66% of identified CAUTIs.56 Upon insertion of a urinary catheter, a host of urinary components, including proteins, electrolytes, and other organic molecules, deposit on the surface of the catheter resulting in the formation of a conditioning film. Planktonic bacteria adhere to the conditioned catheter surface, and over time bacterial multiplication and secretion of extracellular polysaccharide result in the formation of a biofilm. The biofilm protects the bacteria against antimicrobial agents and host immune responses, and its formation is crucial for the development and progression of CAUTI.56 The common causative agents of CAUTIs are Escherichia coli (E. coli) (most prevalent), Enterococcus spp., Klebsiella pneumoniae (K. pneumoniae), Candida spp., coagulase-negative Staphylococci, S. aureus, Proteus mirabilis (P. mirabilis), and Pseudomonas aeruginosa (P. aeruginosa).51,56,57
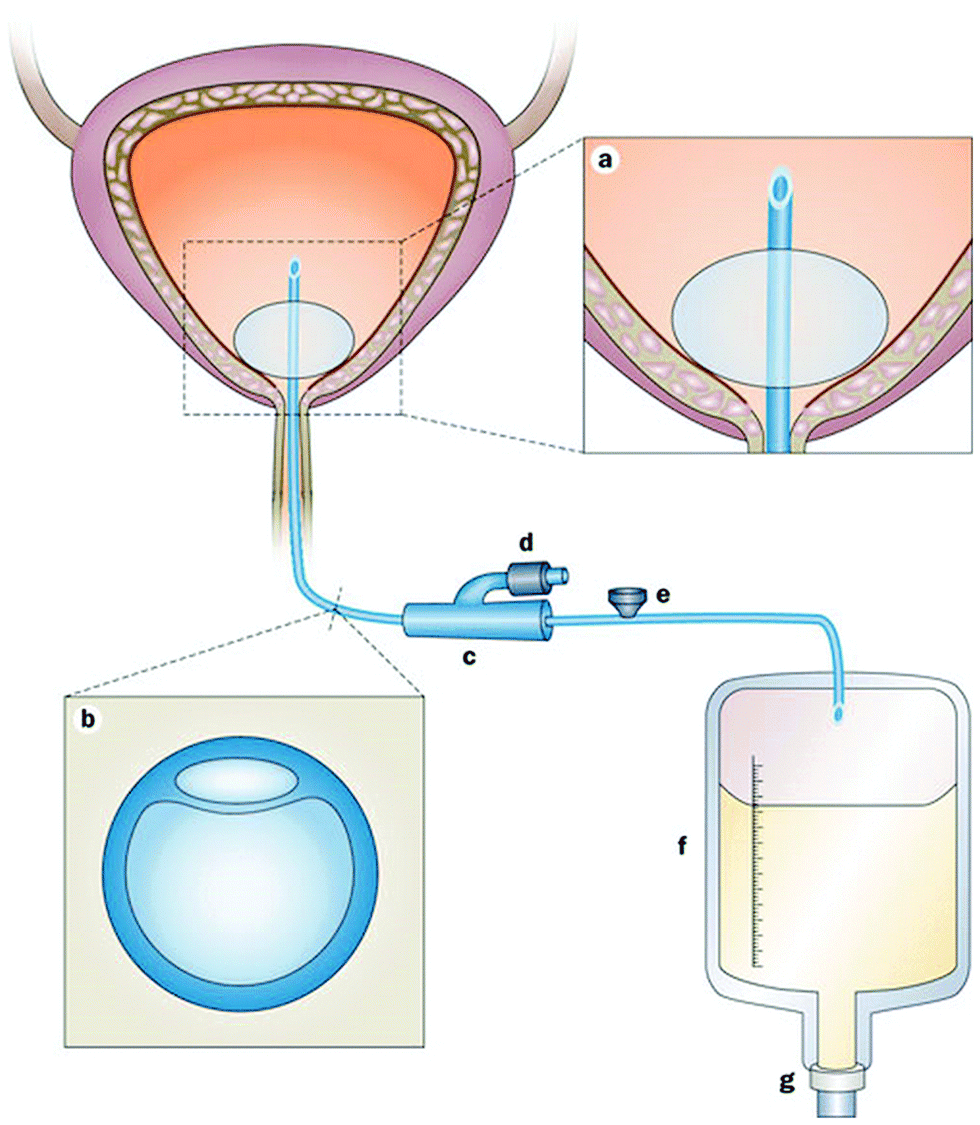 | ||
| Fig. 2 Urinary bladder with an indwelling urinary catheter. (a) An eyelet at the proximal end of the catheter allows drainage of urine from the bladder. The reservoir balloon is routinely filled with sterile saline. (b) Cross-section of the commonly used double-lumen indwelling urinary catheter. The large lumen is for urinary drainage; the small lumen is connected to the port. (c) The preconnected, presealed junction of the indwelling catheter and the drainage system should not be broken. (d) This port is connected to the small lumen of the catheter and is used to inflate the reservoir balloon with sterile saline. (e) This port is a one-way sampling port through which urine specimens for microbiological testing are collected. (f) Draining urine accumulates in the collection bag. (g) Urine is ultimately emptied into the external environment via the outlet valve. Reproduced from ref. 56 with permission from Nature Publishing Group. | ||
A major complication in patients undergoing long-term indwelling bladder catheterization is the formation of encrustations (usually composed of calcium and magnesium phosphates) on the surfaces of the catheter due to urease-producing uropathogens.58 Urease catalyzes the hydrolysis of urea in urine to carbon dioxide and ammonia, resulting in an increase in pH. As urine turns alkaline, precipitation of hydroxylated calcium phosphate (apatite) and magnesium ammonium phosphate (struvite) is promoted in urine as well as on the catheter surface.59 Colonization of these crystalline deposits by bacteria and entrapment of the crystals within the bacterial polysaccharide matrix result in the formation of a crystalline biofilm (Fig. 3). These deposits can block the catheter lumen leading to bladder distension, reflux of urine to the kidneys or leakage around the catheter. If the blocked catheter is not changed, further progression to serious complications such as pyelonephritis, septicemia and septic shock may result.51 The hard crystalline deposits can also cause trauma to the bladder mucosa and to the urethra on withdrawal of the catheter.60P. mirabilis is the bacterium mainly responsible for the formation of crystalline biofilms on urinary catheters. The urease produced by P. mirabilis is able to hydrolyze urea several times faster than those produced by other species.58 The presence of fine hair-like fimbriae (adhesins) on the cell wall enables P. mirabilis to attach to conditioning films on the catheter as well as directly to catheter surfaces. Irregularities on the catheter surface also entrap bacteria. P. mirabilis has the ability to transform from 2 μm long cells into elongated cells of up to 80 μm in length which organize into raft-like groups moving rapidly in a coordinated manner (swarming) over surfaces. Evidence suggests that swarming may well have played a role in the initiation of CAUTI by facilitating the movement of P. mirabilis from the skin at the catheter-insertion site, along the outside of the catheter into the bladder.58
 | ||
| Fig. 3 (a) Encrustation around the eyelet of a catheter removed from a patient after just 5 days, and (b) bacilli, cocci and microcrystalline aggregates of calcium phosphate in the crystalline biofilm around the eyelet of the catheter. Reproduced from ref. 59 with permission from Springer. | ||
3.2 Intravascular catheters
Intravascular catheters, such as CVCs, peripherally inserted venous catheters, peripherally inserted central catheters (PICCs), intravenous catheters, arterial catheters, and umbilical vein catheters, are used to sample blood, monitor blood pressure or administer medicines, fluids, nutrients, or blood products. The major complications associated with intravascular catheters include thrombosis and infections.61 Upon insertion into the bloodstream, the surface of the catheter is rapidly coated with host-derived matrix proteins such as fibrin, fibrinogen, fibronectin, collagen, elastin and laminin.62,63 A fibrin sheath, initially composed of fibrinogen, albumin, lipoproteins, and coagulation factors, forms within 24 h of insertion of the catheter.64 It attracts platelets and coagulation factors and promotes leukocyte adherence as it continues to evolve over time.64–66 The fibrin sheath (Fig. 4) is one of the most common causes of thrombotic occlusion as discussed below.67,68 Microorganisms can also bind to the adsorbed proteins and start to colonize the medical device, forming a biofilm, as early as 24 h after catheter placement.69 The microorganisms can infect intravascular catheters via an extraluminal route arising from the skin at the catheter insertion site or intraluminally from the catheter hub or less commonly from contaminated intravenous fluids.69–71 The extraluminal route of infection predominates soon after insertion, whereas the intraluminal route predominates with prolonged dwelling times or in outbreaks.70 A catheter-related bloodstream infection (CRBSI) is clinically defined as a systemic blood infection where the catheter has been identified as the source of the infection.72,73 The term “CRBSI” has often been used interchangeably with CLABSI, which is a term used by CDC’s National Healthcare Safety Network to denote a primary bloodstream infection (BSI) in a patient who had a central line within the 48 hour period before the development of the BSI and that is not a bloodstream infection related to an infection at another site.73 Since it may not be possible to precisely establish if a BSI is catheter-related due to the clinical needs of the patient or limited resources, CLABSI is simpler to use for surveillance purposes but it may overestimate the true incidence of CRBSI.73 The incidence of CRBSIs varies with the catheter type as shown in Table 2.74 Among the intravascular catheters, the incidence of infection (defined as infections per catheter placed) of long-term CVCs is the highest due to the long period they remain in place. On the other hand, the incidence densities (defined as the number of infections that occur over the number of days that the line is in place) of these infections are lower indicating a reduced incidence over time.74 The predominant microorganisms accounting for CRBSIs are the Gram-positive Staphylococci (S. aureus and coagulase-negative Staphylococci) and Enterococci, followed by Gram-negative bacilli E. coli, K. pneumoniae, Pseudomonas species, Enterobacter species, and Candida species.69,74,75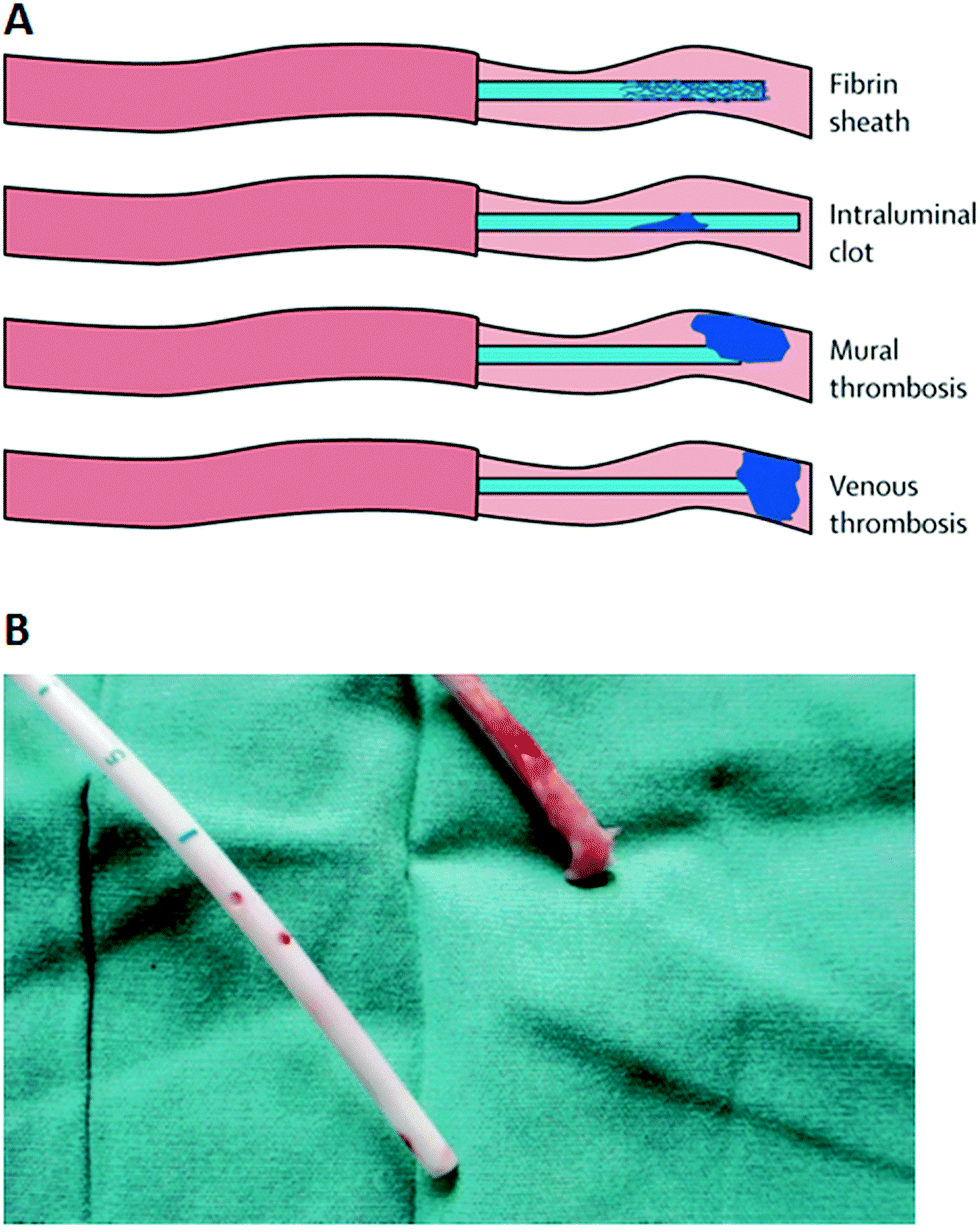 | ||
| Fig. 4 (A) Types of thrombotic occlusions that may be associated with intravascular catheters. (B) Photograph of the Tesio line (tunneled hemodialysis catheter) removed from a patient showing a clean venous line (blue lower line), and a red upper line with an extensive fibrin sheath attached. (A) Reproduced from ref. 67 with permission from Elsevier. (B) Reproduced from ref. 68 with permission from Oxford University Press. | ||
| Catheter | Catheter-related bloodstream infection | |
|---|---|---|
| Incidence (%) | Incidence density (number/1000 line days) | |
| Reproduced from ref. 74 with permission from SAGE Publications. | ||
| Peripheral venous catheter | 0.1 | 0.5 |
| Arterial catheter | 0.4 | 0.2 |
| Peripherally inserted central catheter (PICC) | 2.4 | 2.1 |
| Short-term central venous catheter | 4.4 | 2.7 |
| Percutaneous, tunneled long-term central venous catheter | 22.5 | 1.6 |
| Fully implanted, tunneled long-term central venous catheter | 3.6 | 0.1 |
Catheter occlusion can be due to mechanical causes (such as kinking), precipitation of medications or parenteral nutrition, or thrombotic events.65,67 It is estimated that catheter-related thrombosis occurs in up to 50% of children and 66% of adults with a long-term CVC, and this can lead to further complications such as increased risk of subsequent catheter infections, pulmonary embolism, post-thrombotic syndrome, and persistent vascular compromise.67 Thrombotic events that may be associated with intravascular catheters are shown schematically in Fig. 4A. The fibrin sheath may cause a partial obstruction by creating a one-way valve over the catheter tip that allows the infusion of fluid but inhibits blood withdrawal. It may also lead to mural thrombosis.76 A mural thrombus forms when fibrin from a blood vessel wall injury binds to the fibrin-covered catheter.64 It can occlude the tip of the catheter, but does not completely occlude the vein in which the catheter is positioned, unlike venous thrombosis which occludes the vein. On the other hand, intraluminal clots, which account for 5–25% of all catheter occlusions, may cause complete catheter obstruction.67 Since the blood–catheter interface triggers a complex series of events including protein adsorption, adhesion and activation of platelets and leukocytes, complement activation and coagulation, the physicochemical properties of the catheter material such as surface wettability and roughness may influence the propensity of thrombosis.77 In general, a hydrophobic surface is more receptive to platelet adhesion, and rough material surfaces have microscopic crevices that may adsorb fibrinogen and permit platelet adhesion and activation of the intrinsic coagulation pathway.64,77 Earlier studies comparing polyurethane and other types of intravascular catheters indicate that the polyurethane catheter is more prone to thrombosis, and this is coincident with the rougher and more irregular surface of this type of catheter.78–80 A detailed description of the interfacial processes occurring at the surface of blood-contacting medical devices, and associated thrombus formation, is given in a review by Jaffer et al.81 Nevertheless, catheter-related thrombosis is a multicausal problem, and besides the catheter itself, the position of the catheter in the vascular system as well as the presence of malignancy in the patient are major risk factors.82
The proteins within the thrombus, such as fibrinogen and fibronectin, can also attract bacterial species and enhance their adherence to the catheter surface, thereby increasing the risk of catheter infection.62,83 It is known that several specific adhesins expressed on the surface of S. aureus interact with a number of host proteins, such as fibronectin, fibrinogen, collagen, vitronectin and laminin.84,85 Bacteria can also interact with platelets, resulting in platelet activation/aggregation which subsequently leads to blood coagulation and thrombosis.86–88 Thus, there is strong evidence that catheter-related thrombosis and infection are interrelated and should therefore not be seen as separate entities.76,89
3.3 Peritoneal dialysis catheters
Peritoneal dialysis (PD) is a home-based dialysis treatment modality that has been established as an effective alternative to hemodialysis for patients with end-stage kidney failure. Its primary advantage compared to hemodialysis is that it offers the patient more flexibility in scheduling and self-autonomy. In PD, a catheter is inserted permanently into the peritoneal cavity for the introduction of dialysate solution and its subsequent drainage. The peritoneum acts as a filter allowing excess water and waste products from the blood to pass into the dialysate solution and be removed from the body. The prevalence of PD among the end-stage renal disease population varies widely among different countries. In the United States, PD accounts for only 7–8%,90 and possible reasons include concerns regarding the patient well-being, infectious complications, level of physician comfort in dealing with a particular treatment modality, and subtle financial incentives.91 On the other hand, Hong Kong has adopted a “PD-first” policy since the mid-1980s, and 70–80% of its end-stage renal disease patients are on PD.92 The costs associated with PD were deemed to be lower than hemodialysis especially in developed countries.93,94 Two studies comparing the mortality of PD and hemodialysis patients indicated that PD is associated with lower mortality in the first 2 years of dialysis,95,96 although over time, the risk of death with PD equals or becomes greater than with in-center hemodialysis.95The success of PD is highly dependent on a well-functioning catheter providing long-term access to the peritoneal cavity. Thus, the PD catheter is considered the lifeline of patients but it also serves as a nidus for infection. PD catheter-related infection remains an important cause of morbidity, technique failure, and mortality.97 Bacteria can gain access to the peritoneal cavity via the catheter lumen or infections at the catheter exit-site can lead to tunnel tract infection and peritonitis as the infectious process progresses from the skin towards the peritoneum.98,99 Peritonitis is a major complication of PD, being a leading cause of hospitalization and technique failure100 and it has been deemed to be a contributing factor to 16% of deaths of patients on PD.101 The average incidence of peritonitis among PD patients shows considerable variation depending on factors such as the location, PD technique employed, and patient category, ranging from >2 episodes per patient per year to <1 every 4th year.102 Gram positive cocci such as S. epidermidis, other coagulase-negative Staphylococci, and S. aureus are the most frequent causative agents of PD-associated peritonitis.39,103 Fungal peritonitis is relatively uncommon but is associated with high mortality rates, in the range of 15 to 50% partly due to patient factors.104 A history of previous bacterial peritonitis and prolonged treatment with antibiotics increase the risk of fungal infections.104
Another complication that is commonly encountered in PD is catheter malfunction. It is estimated that catheter malfunction develops in 3.9–13.3% of patients.105 Catheter malfunction can result from catheter migration or kinking, fibrin deposition, omental wrapping and intraperitoneal adhesion. Omental wrapping (Fig. 5) is a leading cause of outflow failure,105–109 and surgical intervention is often required for catheter salvage or replacement. Studies on the laparoscopic salvage of peritoneal catheters have found an incidence of omental wrapping ranging from 57 to 92%.109 The omentum, a fold of the peritoneum, plays a central role in peritoneal defense, absorbing bacteria and contaminants, and has the ability to adhere to areas of inflammation or foreign bodies in the abdomen such as a catheter.110 Omental wrapping usually develops early (<30 days) after catheter placement.105,111 As a result of technique failure in PD arising commonly from peritonitis and catheter-related problems, or from patient choice, it is estimated that 10–20% of PD patients permanently transfer to hemodialysis each year.112,113 However, such transfer may also result in unfavorable outcomes and those transfer patients who commence hemodialysis with a CVC have a higher risk of mortality.113
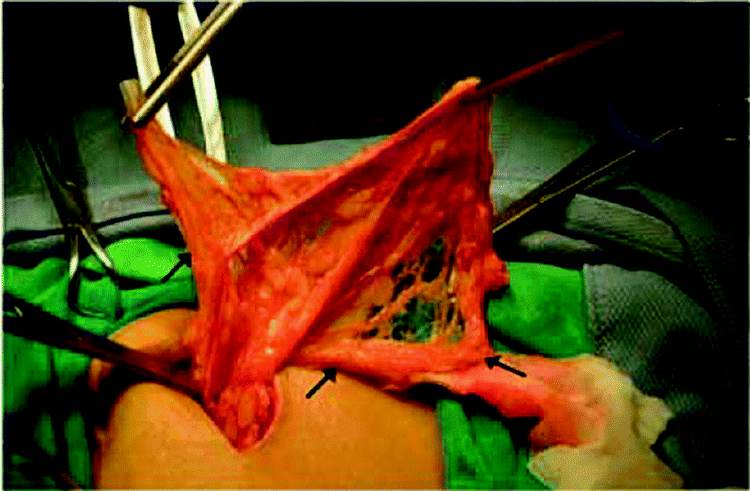 | ||
| Fig. 5 Intra-abdominal portion of a peritoneal catheter completely wrapped by omentum (arrow). Reproduced from ref. 108 with permission from Elsevier. | ||
4. Surface modification strategies to mitigate catheter-related complications
Since the insertion of a catheter into the body provides a surface for bacterial colonization and constitutes a risk factor for infection, there has been much research into the development of catheter surfaces that can potentially inhibit bacterial colonization. While the choice of the catheter material is often application dependent, polymeric materials such as silicone, polyurethane or latex are commonly used. Bacteria adhere readily to such polymers, and hence to prevent bacterial colonization, the surface of the catheter has to be modified to be anti-adhesive or bactericidal or both anti-adhesive and bactericidal. In this section, some strategies which can be applied for imparting polymeric surfaces with anti-adhesive and bactericidal properties will be first discussed followed by specific developments in urinary, intravascular and peritoneal dialysis catheters.4.1 Anti-adhesive coatings
Bacterial adhesion to surfaces is a very complex phenomenon that is affected by the physicochemical properties of the surface and bacteria (e.g. hydrophobicity and surface charge) as well as the environment where the adhesion takes place (e.g. pH, the presence of proteins or electrolytes).114 Anti-adhesive properties can be conferred on surfaces via modification of the chemistry or topography of the surface. This review will focus on chemical modification strategies. Since the adsorption of proteins on indwelling devices facilitates bacterial attachment and subsequent biofilm formation, anti-adhesive coatings are usually designed to resist protein adsorption. Two major classes of polymer coatings based on polyhydrophilic and polyzwitterionic materials (Fig. 6A) have been used for resisting protein adsorption and bacterial adhesion.115,116 It is believed that the anti-adhesive properties of these coatings are tightly correlated with the formation of a hydration layer near the surface, and this layer acts as a physical and energetic barrier preventing protein adsorption.117,118 The “surface-bound” water is associated via hydrogen bonding with the polyhydrophilic materials while zwitterionic materials bind water more strongly via ionic solvation.115 When protein molecules approach a surface with tethered anti-adhesive polymer chains, the compression of these chains results in repulsive forces, and the expulsion of water molecules from the hydrated polymer chains creates osmotic stresses.119,120 Thus, the physicochemical properties of polymer chains, their packing density, chain length and conformation play important roles in determining the anti-adhesive property of the coating.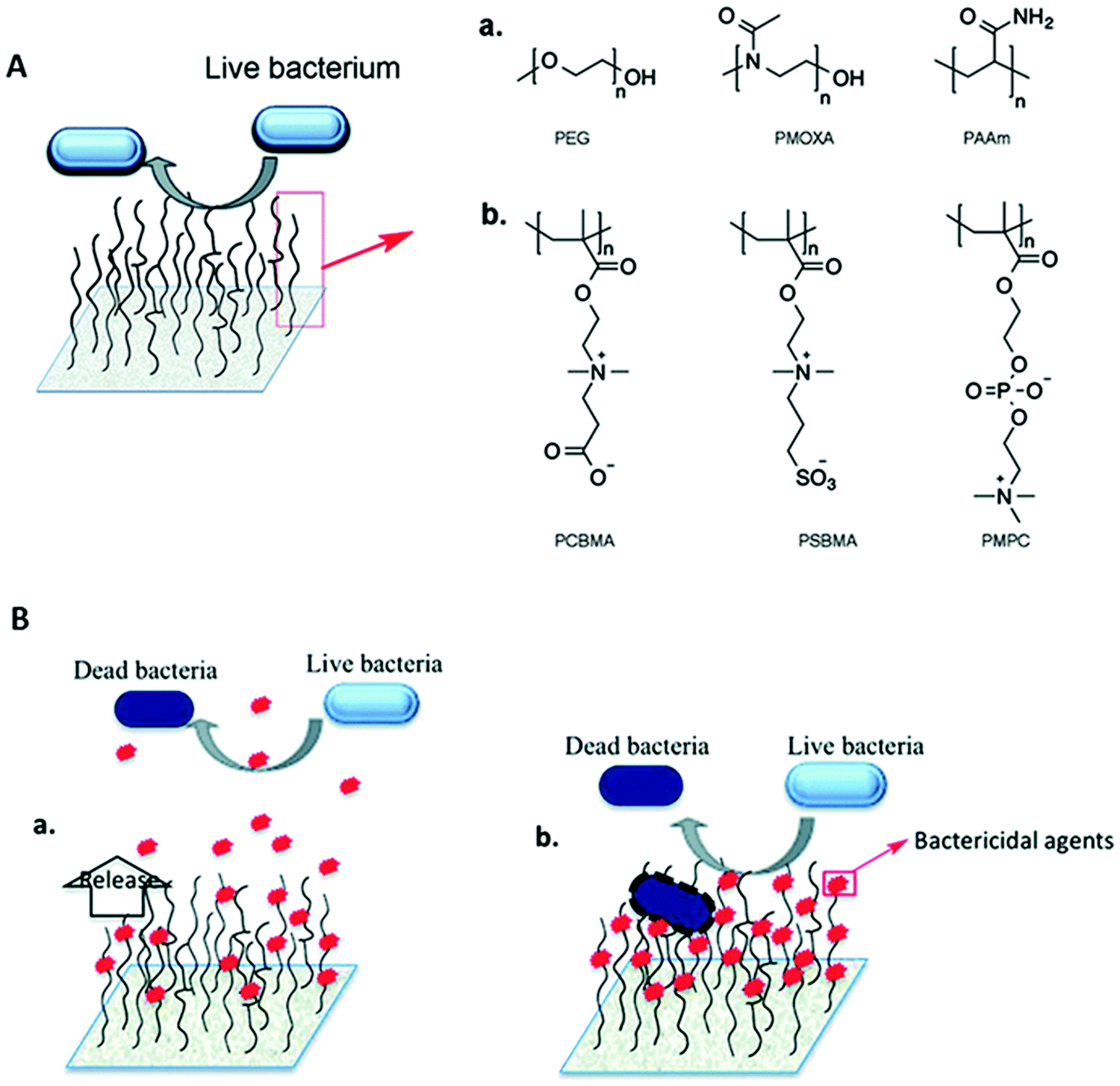 | ||
| Fig. 6 (A) Hydrophilic polymer brushes as an anti-adhesive coating. Representative chemical structures of (a) neutral polymers, polyethylene glycol (PEG), poly(2-methyl-2-oxazoline) (PMOXA) and polyacrylamide (PAAm), and (b) zwitterionic polymers, poly(carboxybetaine methacrylate) (PCBMA), poly[N-(3-sulfopropyl)-N-methacryloxyethyl-N,N-dimethyl ammonium betaine] (PSBMA) and poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC), used for the development of hydrophilic polymer brush systems. (B) Representation of polymer brush-based (a) bactericide-releasing coatings and (b) contact killing coatings where bactericidal agents are conjugated covalently to the polymer chains. Reproduced from ref. 116 with permission from the Royal Society of Chemistry. | ||
Since bacteria are negatively charged in neutral media, positively charged surfaces are expected to increase bacterial adhesion over negatively charged surfaces. This was indeed the finding from a number of studies121–123 although a lack of influence of surface charge on bacterial adhesion has also been reported.124 The neutral hydrophilic polymer, poly(ethylene glycol) (PEG), and its derivatives are widely investigated as anti-adhesive coatings.115,125,126 However, it has been reported that PEG chains exhibit weak hydrophobic interactions with proteins in aqueous solution,127 and under in vivo conditions, the terminal hydroxyl group of PEG may be oxidized to aldehyde.128 As a result, PEG may not be suitable as a long term anti-adhesive surface for in vivo applications, and other polymers are being investigated as possible alternatives. Zwitterionic polymers have attracted great interest since coatings prepared with these polymers exhibit stronger resistance to protein adsorption127 and higher efficacy in inhibiting bacterial adhesion and biofilm formation than PEG coatings.129 Investigators using a high-throughput material discovery methodology have reported that weakly amphiphilic poly(meth)acrylates also show resistance to bacterial attachment, and a pendant group with both a nonpolar nature and increased rigidity correlates with decreased bacterial attachment.130,131 Besides synthetic polymers, natural or modified polysaccharides such as ulvan, hyaluronic acid and agarose have also shown promise in inhibiting bacterial adhesion.132–135 Since an anti-adhesive coating does not kill bacteria and it is not likely that it can completely prevent bacteria from adhering, a few adhering bacterial cells can eventually form a biofilm. Thus, a combination of anti-adhesive and bactericidal properties may be necessary especially when high bacterial loading is expected.
4.2 Bactericidal coatings
Bactericidal coatings which depend solely on one or multiple eluting agents have a number of limitations since the amount of agent(s) that can be loaded is finite. It is difficult to ensure that the concentration of the released agents can be maintained at an effective level throughout the period of application. With time, as the amount of agent in the coating diminishes, its release rate will slow down and the concentration of the released agent will eventually drop below the effective concentration. Once the surviving planktonic bacteria form a biofilm, the concentration of agent required to eliminate the bacteria may be orders of magnitude higher than that needed to kill the planktonic bacteria. Recent evidence suggests that frequent exposure of bacteria to sub-inhibitory concentrations of antimicrobial agents is likely to have an important role in the evolution of resistance.146,147 It is also possible that the sub-inhibitory level of antimicrobial agents can enhance bacterial attachment and biofilm formation by some bacterial strains.148 Thus, it is a significant challenge to design coatings with release kinetics that can maintain the released antimicrobial agent within the concentration range sufficient to kill bacteria over the period of application but low enough to limit cytotoxicity for in vivo applications.
Antimicrobial peptides (AMPs) or host defense peptides are produced by organisms such as plants, insects, and vertebrates as components of their innate immune system.162,163 AMPs are considered promising bactericidal agents due to their broad activity spectrum, efficacy at lower concentrations compared to QACs and antibiotics, and limited ability to promote bacterial resistance.164–166 AMPs vary widely in composition but they generally have two common physical features: a cationic charge and a significant proportion of hydrophobic residues.167,168 With a cationic charge, the AMPs preferentially target negatively charged microbial cytoplasmic membranes over zwitterionic mammalian membranes and the hydrophobic residues facilitate interactions with the fatty acyl chains in the bacterial membrane.167,168 These interactions promote the disruption of the bacterial membrane, and additionally, some AMPs may have intracellular targets resulting in the disruption of the cell wall, and protein and nucleic acid synthesis.167
AMPs have been immobilized on surfaces to provide an antibacterial coating.164,169–171 However, some investigations on the antibacterial efficacy of AMPs covalently conjugated to surfaces highlighted the importance of providing the conjugated peptide with lateral mobility and appropriate orientation for its mode of action on the bacterial membrane.169,170 Direct conjugation of an AMP, cathelin LL37, to a surface without a spacer resulted in no antibacterial activity.169 Another study showed that immobilization via a longer spacer may result in less reduction in bactericidal activity than a shorter one.170 Besides the spacer length, the manner in which the AMP is conjugated to the spacer also affects the bactericidal activity, as shown by LL37 randomly conjugated to a PEG spacer having no antibacterial activity in contrast to the peptide that is conjugated to the PEG spacer via its N-terminus.169 The use of a hydrophilic spacer to conjugate the AMP to the surface also confers an anti-adhesive property to the surface. This is an important consideration since the positively charged nature of AMPs like that of QACs is likely to promote protein adsorption onto the coated surface with subsequent loss of bactericidal activity in vivo. Thus, for antibacterial coatings in in vivo applications, a combination of anti-adhesive moieties and AMPs will be necessary and the proportions of each must be tuned for the respective in vivo environment. There are some potential limitations associated with the use of AMPs clinically such as enzymatic degradation in physiological fluids, the possible toxicity of AMPs toward host tissue cells and the high cost of production.171,172
4.3 Coatings for urinary catheters
A number of trials have been carried out to test the efficacy of commercial antimicrobial urinary catheters in reducing the risk of CAUTI compared with standard indwelling catheters. A multicenter randomized controlled trial involving over 6000 participants requiring short-term (≤14 days) catheterization compared commercially available silver alloy-coated latex catheters and nitrofurazone-impregnated silicone catheters with standard polytetrafluoroethylene-coated latex catheters.173 The study concluded that the silver alloy-coated catheters were not effective in reducing the incidence of symptomatic CAUTI, and the reduction in CAUTI associated with nitrofurazone-impregnated catheters was less than that regarded as clinically important. As such, the trial did not support the routine use of antimicrobial-impregnated catheters.173 Another review of 26 trials involving short-term catheterization in >40000 hospitalized adults also indicated that silver alloy-coated catheters were not associated with a statistically significant reduction in symptomatic CAUTI, and are considerably more expensive. While the nitrofurazone-impregnated catheters were shown to reduce the risk of symptomatic CAUTI and bacteriuria, the magnitude of reduction was low and not sustained, and hence may not be clinically important.174 The nitrofurazone-impregnated catheters are more expensive than standard catheters, and also more likely to cause discomfort in the patients.174
In view of the lack of success of commercial antimicrobial catheters, a number of groups are investigating different strategies for endowing the surfaces of urinary catheters with antimicrobial properties. A comprehensive summary of various organic and inorganic coatings developed for urinary catheters is given in a recent review by Mandakhalikar et al.175 The vast majority of these coatings have not been tested in in vivo models. Ciprofloxacin-eluting coatings have been prepared by immersing acrylic acid-grafted or poly(ethylene glycol) methacrylate-grafted poly(vinyl chloride) urinary catheters in ciprofloxacin solution.176 However, the release of ciprofloxacin can only be sustained for a few hours.176 In another study, ciprofloxacin was loaded into liposomes entrapped in a PEG gelatin hydrogel coated on silicone catheters.177 The release of ciprofloxacin was sustained for ∼7 days, and the median time for the development of bacteriuria in rabbits inserted with ciprofloxacin-containing hydrogel coated catheters was 6.25 days compared to 3.5 days for hydrogel coated and 3.25 days for untreated catheters.177 Besides antibiotics, coatings containing antiseptics were also fabricated for urinary catheters. Chlorhexidine mixed with ethyl cellulose and polyethylene glycol to a final concentration of 1% was applied as a coating on urinary catheters, which were then inserted into dogs and left in place for 1 to 7 days.178 Over this period, the coated catheters exhibited reduction in biofilm formation.178 In another study, urinary catheters were impregnated with a mixture of two antibiotics, rifampicin and sparfloxacin, and an antiseptic, triclosan.179 The loading was carried out by swelling the catheter segments in a chloroform solution of the antimicrobial agents. Bacterial challenge was carried out by inoculating the catheter with a dose of bacteria each week under flow conditions. While the control catheters were colonized within 1 day, the impregnated catheters withstood the challenge for 2 to >12 weeks depending on the bacterium.179 However, the safety of triclosan has come under scrutiny over concerns that it may be associated with antibiotic resistance,180 disruption of thyroid hormone homeostasis181 and promotion of liver tumor growth.182
Silver is known to be a potent antimicrobial agent although commercial silver alloy-coated urinary catheters have not been shown to be effective in reducing symptomatic CAUTI. As such, research has focused on the development of other means of coating silver on urinary catheters. Urinary catheters coated with 8–21 nm silver nanoparticles (AgNPs) by immersing the catheter in an AgNP suspension for 24 h showed a 80–90% reduction in biofilm formation by different bacteria in vitro as compared with the uncoated catheter over 24 h.183 Ag/TiO2 nanocomposite coated silicone catheters were fabricated by immersing TiO2-coated catheters in AgNO3 solution under UV irradiation.184 These catheters were reported to achieve a killing efficacy of 99% of bacteria in contact with the surface within 20 to 90 min depending on the bacterium.184 Since these two studies were conducted over short periods of time, the antibacterial efficacy of these AgNP coatings over the duration of application expected for urinary catheters is unknown. Another study attempted to provide a sustained release of silver over weeks by means of a micron-scale coating comprising AgNPs–polydopamine multilayers and an outer anti-adhesive poly(sulfobetaine methacrylate-co-acrylamide) (poly(SBMA-co-AAm)) layer (Fig. 7A).185 The outer poly(SBMA-co-AAm) layer prevents the deposition of a conditioning film and ensures free diffusion of the silver from the coating. While the poly(SBMA-co-AAm) coating alone can reduce bacterial adhesion and biofilm formation by >92% for P. mirabilis, >94% for P. aeruginosa and >96% for E. coli compared with that on the pristine silicone catheter, with added AgNPs, (P2 and P3 catheters in Fig. 7A) colonization of the urinary catheter by these uropathogens can be further reduced (Fig. 7B). The coated catheter with one and two AgNP–polydopamine bilayers (P2 and P3 in Fig. 7A, respectively) resisted encrustation in artificial urine spiked with P. mirabilis for 12 and 45 days, respectively, compared with ∼6 days for the commercial Dover™ silver-coated catheter. After 7 days, the surface of the Dover™ catheter was covered by a crystalline mass, while those of the P2 and P3 catheters remained relatively free of deposits (Fig. 7C). Only ∼14% of the silver in the Dover™ catheter was released before encrustation occurred, while the corresponding amount released from the P3 catheter was 50%.185 Thus, unless the rate of silver release can be sustained at a high enough level to kill P. mirabilis in urine, the presence of silver on a urinary catheter is insufficient to ensure that encrustation can be inhibited since the surface can be rapidly covered by crystalline deposits.
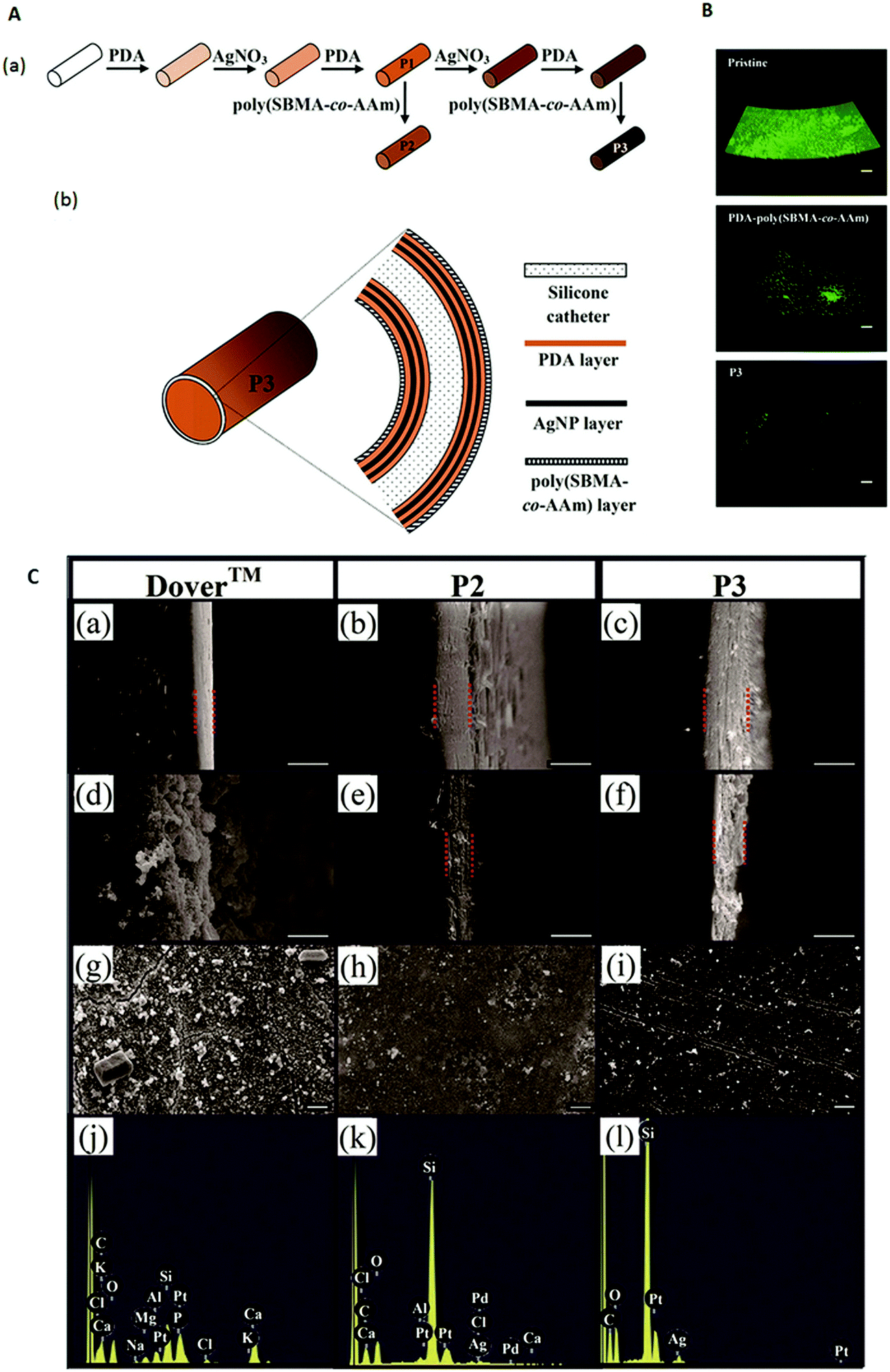 | ||
| Fig. 7 (A) Schematic diagram illustrating the (a) steps for modifying a silicone catheter surface and (b) structural layers of a P3-coated catheter. (B) Confocal laser scanning microscopy images (volume view) of a P. mirabilis biofilm on the intraluminal surface of pristine, PDA–poly(SBMA-co-AAm)-, and P3-coated catheter segments after incubation in culture medium containing 105 cells per mL for 24 h. Scale bars represent 100 μm. (C) Scanning electron microscopy images of the (a–f) cross-section of the intraluminal coating and (g–i) intraluminal surface; (a–c) before the encrustation test, (d, e and g, h) after 7 days of encrustation test, and (f and i) after 40 days of encrustation test; (j–l) energy dispersive X-ray spectra of the surfaces shown in (g–i), respectively. (a, d, g and j): Dover™ silver-coated catheter; (b, e, h and k): P2-coated catheter; and (c, f, i and l): P3-coated catheter. The Si signal from the silicone surface in the P2-coated and P3-coated catheters remained prominent unlike the Dover™ silver-coated catheter which showed strong Ca and P signals. Scale bars represent 10 μm. Adapted from ref. 185 with permission from John Wiley and Sons. | ||
Although NO is a well-known antimicrobial agent that causes nitrosative and oxidative damage to bacteria,186,187 it would be difficult for catheters impregnated with NO to achieve long term antibacterial efficacy under dynamic urine flow.188,189 Since gaseous NO is highly reactive and short-lived under physiological conditions, NO donor molecules capable of efficiently storing and delivering NO locally, such as S-nitrosothiols (RSNOs), have been investigated as an active agent for incorporation into biomaterials.189,190 Commercial silicone urinary catheters have been impregnated via a solvent swelling method with S-nitroso-N-acetylpenicillamine (SNAP), a synthetic RSNO, and the impregnated catheters generate NO surface-fluxes >0.7 × 10−10 mol min−1 cm−2 for over one month.190 The leaching loss of SNAP during the first week in PBS at 37 °C was reported to be 11%. Biofilm formation by S. epidermidis and P. mirabilis on the SNAP-impregnated catheter was reduced by ∼4 and 6logs compared to the unmodified catheter after 14 days in a CDC biofilm reactor. Another RSNO, S-nitroso-tertdodecyl mercaptan (SNTDM), was found to have a lower leaching rate than SNAP after impregnation in silicone tubing due to its highly lipophilic character.189 The impregnated tubing reduced the surface levels of S. aureus by 4logs during the first two weeks, and by 3logs after the third week. However, light and ethylene oxide sterilization reduced the SNTDM levels, and less than 10% of the loaded SNTDM remained after storage in a cupboard for 80 days compared to 75% when stored in a freezer. Thus, more work is needed to increase the stability and shelf life of RSNO-impregnated NO eluting catheters.
Recently, bacteriophages (or phages) have been used to pretreat catheters as a strategy to inhibit biofilm formation.191–193 Phages can replicate inside bacterial hosts, causing cell lysis and release of phage progeny, which in turn infect neighboring bacterial cells.191 Some phages also produce enzymes that degrade the EPS of biofilms.194 Phage treatment of urinary catheters can be easily carried out by exposing the catheter lumen to a phage cocktail for a few hours. Phage pretreatment can achieve 4log and 2log reductions in biofilm formation by P. aeruginosa and P. mirabilis, respectively, over 48 h in in vitro experiments.192 Phages from different families have a different adhesion strength on surfaces and this affects their release rate from the surface and long term efficacy against biofilm formation.193 Crystalline deposits and the biofilm matrix itself may also inhibit the phages from infecting the target cells.193 Efficacy may also be affected by other phage-dependent factors, such as the production of depolymerases, the ability to penetrate the EPS matrix,195 and environmental factors such as pH and ions.196
Urinary catheters with surface-immobilized bactericidal moieties have been prepared with QACs, antimicrobial enzymes or peptides. Poly(2-methacryloyloxyethyl)trimethylammonium chloride was grafted onto silicone catheters via a 2-step process: (i) initial grafting of polydopamine-2-bromoisobutyryl bromide onto the catheter surface to serve as an ATRP activator, followed by (ii) the ATRP of [2-(methacryloyloxy)ethyl]trimethylammonium chloride under N2 using a CuCl2 catalyst and a tris(2-pyridylmethyl)amine ligand.197 The coated catheter reduced biofilm formation by ∼50% and ∼75% for E. coli and P. aeruginosa, respectively, over 48 h. Quaternary ammonium and zwitterionic side chains have been incorporated into a copolymer and assembled on 3-aminopropyl triethoxysilane-pretreated silicone via electrostatically driven layer-by-layer assembly with poly(styrenesulfonate) as the polyanion to form anti-adhesive and antibacterial coatings.198 Different configurations involving different fractions of zwitterionic groups in the copolymer, and the incorporation of a hydrogen peroxide-producing enzyme, cellobiose dehydrogenase, as a polyanion within the multilayers to supplement the bactericidal activity were tested. The coatings were stable for up to 10 days in water and in artificial urine, and the highest extent of reduction in adherent bacteria achievable was 53%. Cellobiose dehydrogenase has also been covalently grafted onto plasma-activated silicone urinary catheters using aminopropyl triethoxysilane and glutaraldehyde as linkers.199 The immobilized enzyme was relatively stable in artificial urine over 16 days, retaining 20% of its initial activity. Over 7 days, the enzyme-coated catheter reduced S. aureus biofilm formation in an artificial bladder model by 70%.
AMPs have been immobilized on catheter surfaces via different conjugation methods.200–202 Lasioglossin-III, a salt tolerant AMP, was chemically modified at the N-terminal with a cysteine residue, and immobilized via sulfhydryl chemistry to a PEG spacer coupled to an allyl glycidyl ether (AGE) brush-grafted silicone catheter.200 The AMP-coated catheter reduced biofilm formation by E. faecalis and E. coli by 60% and 40% over 24 h, respectively. Two short arginine, lysine and tryptophan rich AMPs engineered from the C-terminus of a salt-resistant human beta defensin 28 variant template were conjugated to an AGE brush-grafted silicone catheter, and reduction in biofilm formation by E. coli, S. aureus and C. albicans was also demonstrated.201 The same group also showed that an AMP-coated urinary catheter can be obtained via a simple method of dip-coating the catheter pre-coated with polydopamine in a solution of an in-house-designed synthetic AMP.202 The AMP-coated catheters reduced overnight biofilm formation by ∼92%, which is slightly less efficacious than the Dover™ silver-coated catheter.
Unlike the abovementioned coatings which are based on bactericidal and anti-adhesive properties, another strategy targets the quorum-sensing (QS) process involved in biofilm development. The QS process allows bacteria to communicate through specific signal molecules and coordinate their group behavior to form biofilms.203 Thiazolidinedione-8 (TZD-8), an anti-quorum-sensing molecule, was incorporated into polymeric varnishes based on ethylcellulose and/or Eudragit® and painted on latex or silicone catheter segments.204 The coating can be retained on the catheter for 8 days in PBS at 37 °C, and TZD-containing ethylcellulose films can inhibit biofilm formation by C. albicans over 24 h by ∼60%. In another study, coatings comprising acylase, a negatively charged QS disrupting enzyme, were built on silicone urinary catheters using a layer-by-layer technique.205 Silicone catheter segments were first treated with aminopropyl triethoxysilane solution to introduce amino groups on the surface followed by the deposition of the first enzyme layer and subsequently a positively charged polyethylenimine layer. A total of 10 bilayers were built on the catheter with the outermost layer being acylase. Using a dynamic model of a catheterized bladder,205 the coated catheter was shown to reduce P. aeruginosa biofilm formation over 7 days by 45 to 80% (depending on the part of the catheter) compared to the untreated catheter.
4.4 Coatings for intravascular catheters
Two recent articles compared the effectiveness of antimicrobial-impregnated CVCs versus uncoated commercial catheters on bloodstream infections.206,207 In the first study, a randomized controlled trial was carried out with 1485 children admitted to English pediatric ICUs to compare commercially available CVCs impregnated with minocycline and rifampicin, or heparin, or a standard CVC.206 The results showed that the primary outcome, time for the first bloodstream infection, did not differ between impregnated (antibiotic or heparin) and standard CVCs. However, reduction in the risk of bloodstream infections (a secondary outcome) was found to be lower with the antibiotic-impregnated catheters than with either heparin-impregnated or standard catheters. There was no significant difference between standard and heparin-impregnated catheters. The second study reviewed evidence from 57 studies carried out with 16784 catheters and 11 types of antimicrobial impregnation in adult patients cared for in hospital settings. The antimicrobial catheters were mainly chlorhexidine–silver sulfadiazine, minocycline–rifampin/rifampicin, heparin or silver–platinum–carbon impregnated catheters.207 The participants with impregnated catheters had 2% lower rates of CRBSI compared to those with standard non-impregnated catheters, and there was a 9% lower probability of bacterial colonization on the impregnated catheters. However, the reduction in bacterial colonization was significant only in studies conducted in ICUs, and not in hematological and oncological units or in patients who required CVCs for long-term total parenteral nutrition. There were no significant differences between the impregnated and non-impregnated groups in the rates of thrombosis/thrombophlebitis or adverse effects at the insertion site. Current guidelines do not recommend the routine use of commercially available coated catheters for patients who require central venous access. A meta-analysis of 12 studies with a total of 2854 patients concluded that silver-impregnated CVCs are not associated with lower rates of bacterial colonization and do not reduce the incidence of CRBSI as compared to standard CVCs.208 Recently, an ex vivo study showed that an antimicrobial coating based on a thin layer of noble metal (silver, gold, palladium) alloy developed by Bactiguard® has a lower hemolysis rate than the chlorhexidine–silver sulfadiazine coating.209 The noble metal alloy coating also demonstrated lower potential for blood coagulation activation and 90% reduction in microbial adhesion compared to the unmodified surface.
The use of NO-releasing polymers for intravascular catheters is deemed promising since NO is not only an antimicrobial agent, but also a potent vasodilator and inhibitor of platelet activation210,211 that is released from vascular endothelium.212 NO-releasing intravascular catheters have been fabricated by Meyerhoff’s group via a dip coating process using stainless steel mandrels with SNAP in Elast-eon™ E2As polymer as the active layer sandwiched between the top and base coats of E2As. The E2As base and top coats were employed to reduce any initial burst of NO from leaching of SNAP from the surface of the catheters.213 These catheters implanted in sheep veins for 7 days showed ∼70% reduction in the thrombus area and 90% reduction in bacterial adhesion compared to control catheters prepared without SNAP. Ethylene oxide treatment of the catheter under humid conditions resulted in a loss of 19% of the loaded SNAP. The same group also fabricated NO-releasing catheters using the Elast-eon™ E2As polymer with diazeniumdiolated dibutylhexanediamine (DBHD/NONO) and poly(lactic-co-glycolic acid) (PLGA) additives using a similar dip coating process.214 The trilayer catheter comprised DBHD/NONO with various ratios of PLGA and E2As between the top and base coats of E2As. Hydrolysis of the PLGA additives with ester end groups produced lactic/glycolic acid species which balanced the production of a lipophilic DBHD amine byproduct from the NO release reaction and modulated the NO release at >0.5 × 10−10 mol min−1 cm−2 over 14 days. After 9 days of catheter implantation in the jugular veins of rabbits, the NO-releasing catheters reduced the thrombus area by a factor of 7 in addition to 95% reduction in bacterial adhesion.
Strategies to modify the surfaces of intravascular catheters with non-leaching moieties to inhibit bacterial colonization and thrombosis focused primarily on the use of anti-adhesive polymers. A zwitterionic sulfobetaine monomer, N,N-dimethyl-N-methacryloxyethyl-N-(3-sulfopropyl) ammonium, graft polymerized on an ozone-pretreated polyurethane vascular catheter improved the anti-thrombogenicity of the surface as shown by the inhibition of platelet adhesion after 2 h of exposure to platelet rich plasma.215 In another study, the intraluminal and extraluminal surfaces of peripherally inserted central catheters (PICCs) were modified with zwitterionic polymeric sulfobetaine (polySB) via pretreatment with tert-butylperoxy 2-ethylhexyl carbonate followed by immersion in a polymerization solution of 10% N-(3-sulfopropyl)-N-methacryloxyethyl-N,N-dimethylammonium betaine containing 5 mM Fe(II) gluconate at 40 °C for 16 hours (Fig. 8a).216 Using in vitro assays with human and bovine blood, and a canine model, the polySB-modified catheters were shown to reduce platelet attachment and activation, and formation of thrombi compared to unmodified PICCs or commercial PICCs (Fig. 8b–d). Due to the anti-adhesive nature of the polySB coating, the number of adhering microorganisms on the surface of serum-preconditioned polySB-modified PICCs after 24 h showed a 97 to 99.9% reduction as compared to similarly treated unmodified PICCs (Fig. 8e).
 | ||
| Fig. 8 (a) PolySB modification of a PICC surface. The zwitterionic polySB modification coordinates both free and bound water molecules to create a hydrophilic surface. (b) Adherent platelet and (c) accumulated thrombus on commercial and polySB-modified PICCs exposed to bovine blood in a flow loop system for 60 to 120 min at 37 °C. PICCs were exposed to serum for 60 days prior to blood exposure. Data from serum-exposed polySB-modified PICCs were normalized to serum-exposed commercial PICCs. Error bars indicated the standard error of the mean (SEM) for each set of samples (n = 15). *** P < 0.001, Student’s t test. (d) Images of explanted catheters and associated vessels after 4 h in a canine thrombus model. Representative image is shown for n = 3 animals. (e) Adhesion of microorganisms onto external surfaces of serum-preconditioned unmodified and polySB-modified PICC samples after 24 h. Data are average log10 CFU cm−2 ± SEM. *** P < 0.001, Student’s t test. Adapted from ref. 216 with permission from The American Society for the Advancement of Science. | ||
Methylcellulose (MeCe), a methylated polysaccharide, was deemed to have good anti-adhesive properties since it is hydrophilic, it contains primarily hydrogen bond acceptors, and its overall electrical charge is neutral.217 A MeCe derivative with alkene groups was used to modify the septum and the catheter (made of silicone) of commercial totally implantable venous access ports (TIVAPs) via 1-step hydrosilylation in water.218 An in vitro bacterial assay showed that after 24 hours of static contact, there was 2log reduction in P. aeruginosa and S. aureus adhesion to MeCe-modified surfaces compared with the unmodified surface. Using a rat model with inoculated P. aeruginosa and S. aureus, 66 to 89% of uncoated TIVAPs lost patency due to clogging within 24 hours of biofilm formation whereas the corresponding figures for the MeCe-coated TIVAPs were 0 to 22% (depending on the bacterium).
Unlike the hydrophilic anti-adhesive polymer coatings described above, an omniphobic coating was developed as an alternate strategy to repel bacteria and whole blood.219 Polymeric tubing and flat substrates were first pre-treated with oxygen plasma and then immediately immersed in 5% v/v tridecafluoro-1,1,2,2-tetrahydrooctyl trichlorosilane solution. The covalently tethered perfluorocarbon (TP) on the material surface was then coated with a thin liquid layer of perfluorodecalin (LP), a US Food and Drug Administration-approved blood substitute, to form a two-component tethered-liquid perfluorocarbon (TLP) coating (Fig. 9a). Fresh human blood pumped through TLP-coated polyvinyl chloride (PVC) loops resulted in a reduction in thrombus weight by about 4-fold after 2 h compared to uncoated control loops. The anti-thrombogenic properties of the TLP coating were also tested in a pig model, which showed that in non-heparinized animals, 4 out of 5 control (uncoated) circuits occluded, whereas TLP circuits remained patent for 8 h (Fig. 9b). Biofilm formation by P. aeruginosa (105 CFU mL−1 and incubated at 25 °C) in TLP-coated PVC medical tubing for up to 6.5 weeks was 8-fold lower compared to control tubing (Fig. 9c).
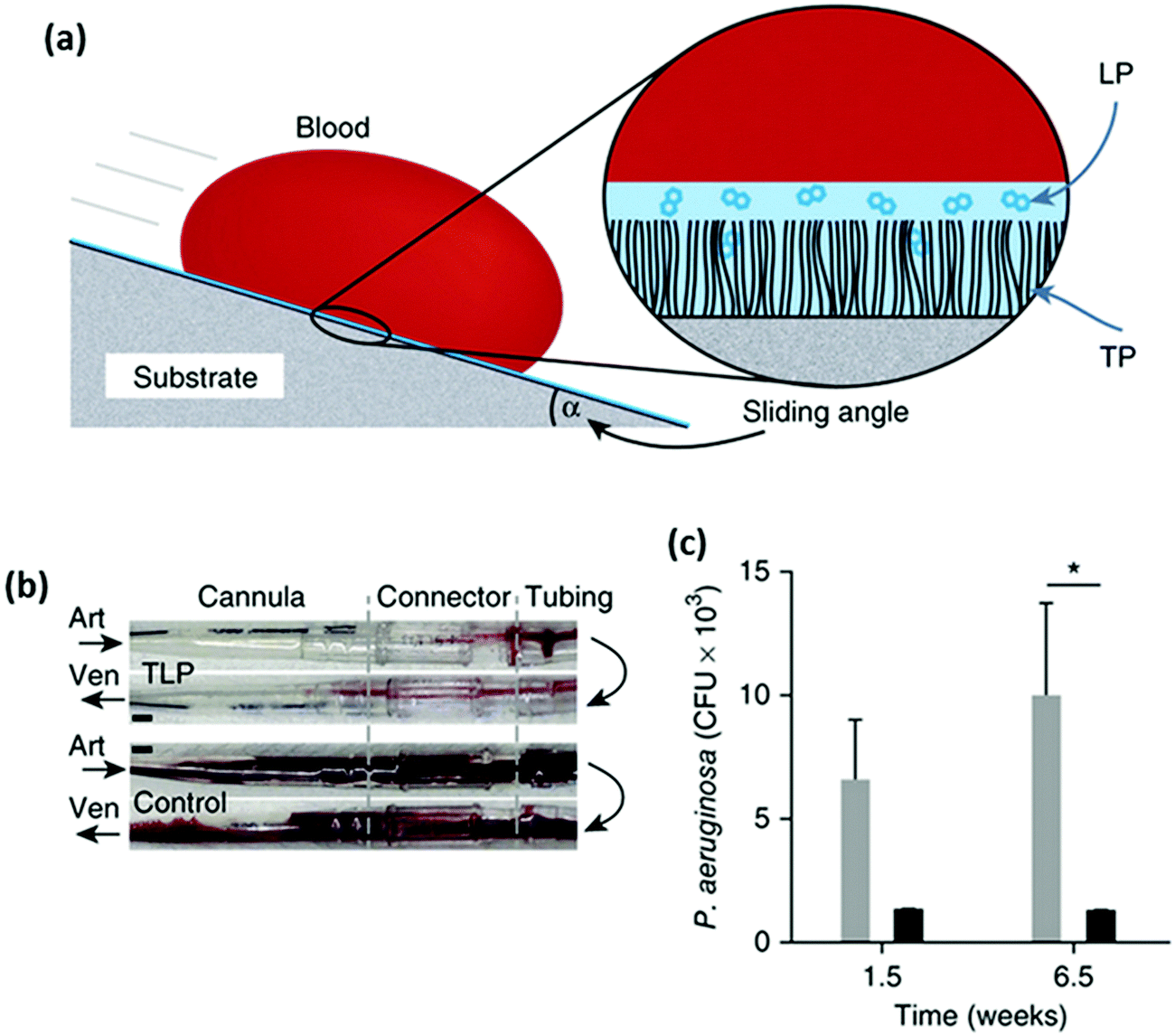 | ||
| Fig. 9 (a) Schematic of blood repellency on TLP surfaces showing the TP bound to a substrate through plasma activation and silane treatment, which then allows a stable film of LP to adhere to the surface and (b) photographs of polyurethane cannulae, polycarbonate connectors and PVC tubing of TLP (top) and control (bottom) circuits after 8 h of blood flow. Arrows indicate the direction of blood flow through an arterial (Art) or a venous (Ven) cannula. Increased thrombus is visible in the control circuit, and (c) biofilm formation on control PVC (gray bars) and TLP PVC (black bars) for 1.5 and 6.5 weeks. (*P < 0.05, two-way ANOVA with Bonferroni’s multiple comparisons test, SEM). Adapted from ref. 219 with permission from Nature Publishing Group. | ||
4.5 Coatings for PD catheters
Unlike the large scale clinical trials conducted with antimicrobial urinary and intravascular catheters reported above, such trials with PD catheters are lacking. An early prospective randomized clinical trial of 86 patients showed that there was no reduction in catheter-related infectious complications when catheters pretreated with 5% tridodecylmethylammonium chloride were used in place of untreated catheters.220 In another clinical trial with 195 patients, the placement of a silver ring on the catheter at the insertion site resulted in no reduction in the incidence of catheter-related infection relative to controls over a 12 month period.221 Similarly, in another trial with 139 patients, silver-ion implanted peritoneal dialysis catheters resulted in no clinical effect in reducing dialysis-related infections compared to the unmodified catheters.222Combinations of antimicrobial agents rather than single agents have been tested in in vitro and animal models. Silicone tubing can be readily impregnated with rifampicin, trimethoprim and triclosan by swelling the tubing in a chloroform solution of these antimicrobial compounds.99 The combination of three antimicrobial agents was deemed to offer higher protection against the emergence of resistant strains than the individual agents. In vitro challenge to establish the period of antimicrobial activity under flow conditions was carried out with weekly inoculation of methicillin-resistant S. aureus (MRSA) and E. coli, and the results showed that plain tubing was colonized within 48 h while the antimicrobials-impregnated tubing resisted colonization for 90 days. In another study, catheters comprising silicone tubing impregnated with chlorhexidine/triclosan with two acrylic felt cuffs impregnated with chlorhexidine/silver sulfadiazine/triclosan were tested in vitro and in rats.223In vitro tests showed that the catheter segments demonstrated antimicrobial activity for 10 days against S. aureus and S. epidermidis, 5 days for E. coli and C. albicans and only 1 day for P. aeruginosa. The impregnated cuffs showed >10 days of inhibition against the Gram-positive organisms and at least 5 days against Gram-negative organisms and C. albicans. Implantation of the catheters in rats was carried out followed by inoculation of the exit site with S aureus. Seven days post-implantation, none of the impregnated catheters were colonized intraperitoneally whereas 100% of the control catheters were colonized. At the exit site, 12.5% of the impregnated catheters were colonized compared to 100% colonization of the controls. Over 24 days, the impregnated catheters resulted in no adverse effect on healing compared to the control catheters in non-inoculated rats.
Surface modification of PD catheters to inhibit omental wrapping has also been carried out in vitro and in animal models. The performance of commercial regular peritoneal silicone catheters and commercial polyurethane catheters coated with heparin implanted in rats with daily infusion of PD fluid was monitored over 5 weeks.224 By the end of this period, 57% of the animals with the regular catheter suffered from omental wrapping around the catheter tip whereas 20% of the animals with the heparin-coated catheter had the same problem. While the mechanism by which the heparin-coated catheter inhibited omental wrapping was not established, these catheters were reported to be less adhesive as compared with conventional silicone catheters. Covalent grafting of anti-adhesive coatings onto surfaces of biomaterials such as silicone and polyurethane has been investigated as a potential means to prevent bacterial colonization as well as omental wrapping of PD catheters. A tetraether lipid-based coating based on caldarchaeol isolated from the archaebacterium Thermoplasma acidophilum with additional functionalities to improve the anti-adhesive property has been developed.225 The head-groups (hydroxyl groups) of caldarchaeol were first activated with cyanuric chloride and then reacted with silicone pretreated with aminopropyl trimethoxysilane. The cyanuric chloride group of the tetraether lipid monolayer on the silicone surface was further modified with PEG or negatively charged taurine or a combination of PEG and sultone. The lipid coating combined with PEG and sultone resulted in the highest reduction of colonization (80% reduction) of a mixed culture of S. aureus and S. epidermidis in a peritoneal dialysis medium under flow conditions for 20 h. The tetraether lipid-based coatings showed no significant cytotoxic effect, and did not significantly influence leukocyte, erythrocyte and platelet numbers or increased activation of the coagulation system compared to the commercially available polytetrafluoroethylene control.
Another potential anti-adhesive coating for peritoneal dialysis catheters is based on the natural polysaccharide, agarose.134 Acrylated agarose (from the reaction of agarose with acryloyl chloride) was covalently grafted and crosslinked on oxygen plasma or ozone pretreated silicone sheets or PD catheter surfaces to form a coating of micron thickness (Fig. 10A). In in vitro experiments, this coating achieved a ∼2log reduction in S. aureus biofilm formation and ∼3log reduction in E. coli and P. aeruginosa biofilm formation over 48 h compared to pristine silicone (Fig. 10B). Cell and platelet adhesion and protein adsorption were also reduced by ≥90%. To further improve the hemocompatibility of the coating and inhibit the activation of platelets, 2.6 μg cm−1 heparin was co-immobilized with agarose (Fig. 10C). The amount of heparin was controlled since excessive amounts of heparin resulted in increased bacterial adhesion compared to that of the agarose-grafted surface. The coatings pose no significant cytotoxicity to mammalian cells, and are highly stable to frictional and bending forces, immersion in lysozyme aqueous solution for extended periods and autoclaving at 121 °C for 20 min.
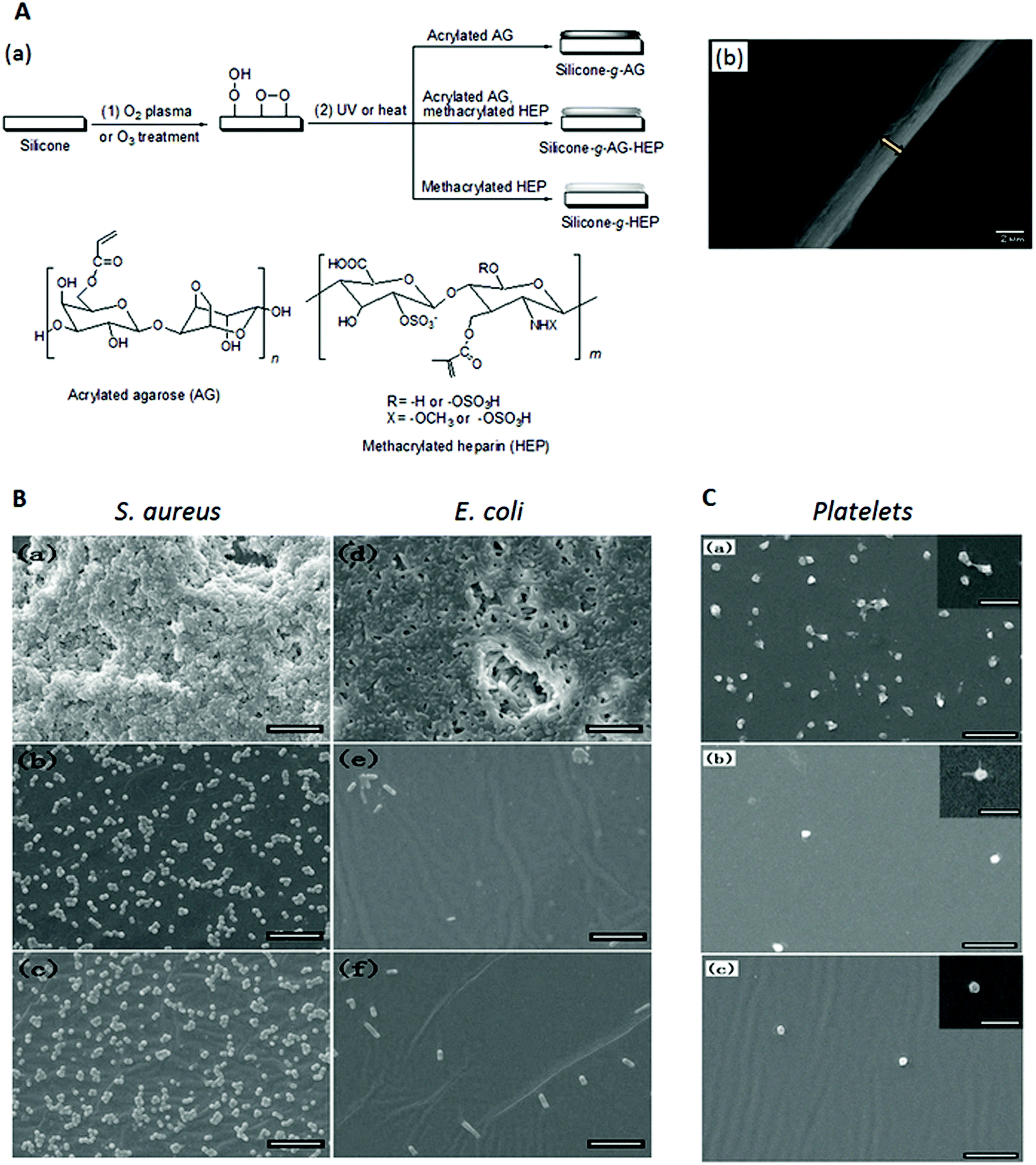 | ||
| Fig. 10 (A) (a) Schematic illustration of the modification of a silicone surface via oxygen plasma or ozone treatment (Step (1)), and UV or heat-induced immobilization and crosslinking of acrylated agarose and methacrylated heparin (Step (2)) and (b) scanning electron microscopy image of the cross-section of an agarose-grafted PD catheter segment. (B) Scanning electron microscopy images of (a–c) S. aureus and (d–f) E. coli biofilms on (a and d) pristine silicone films, and silicone films grafted with (b and e) agarose and (c and f) agarose and heparin after incubation in growth medium containing 107 bacterial cells per mL for 48 h. (C) Scanning electron microscopy images of the (a) pristine silicone film and the silicone film grafted with (b) agarose and (c) agarose and heparin after incubation with platelet-rich plasma for 1 h. Scale bar in (B) and (C) is 10 μm and scale bar in (C) insets is 5 μm. Adapted from ref. 134 with permission from John Wiley and Sons. | ||
5. Conclusion and perspectives
Microbial colonization of indwelling biomedical devices is a recognized major complication contributing to healthcare-associated infections. The three classes of indwelling catheters, intravascular catheters, urinary catheters and peritoneal dialysis catheters, are similarly plagued by this problem in addition to other problems related to their interaction with biological entities specific to the in vivo environment where they are used. Since these complications are related to the surfaces of the catheters, technology that can control the surface bioactivity without compromising the catheter design, material bulk properties and function would be a major advancement. So far, clinical trials with commercial antimicrobial catheters have shown a lack of success. For example, although silver is a well-established bactericidal agent, silver coatings were not shown to reduce catheter-related infections for all three classes of catheters. Unfortunately, the promising performance of other types of antimicrobial coatings under laboratory conditions was also not translated into clinical success.A number of key factors must be considered in the design of indwelling catheter coatings which have to be biocompatible, antimicrobial and non-thrombogenic. For coatings with eluting agents, e.g. to either kill the microorganisms or disrupt their quorum sensing process, the coating has to maintain a high and yet safe concentration of the agents in the vicinity of the catheter throughout the duration of use of the catheter. This strategy is feasible only for short-term applications rather than for long-term indwelling catheters like the peritoneal dialysis catheter due to the limited capacity of the coating or catheter material acting as a reservoir for the agents. The immobilization of an antimicrobial agent on the catheter surface may prolong its activity as long as it is not masked by surface deposits. Since anti-adhesive coatings cannot be 100% effective, any microorganism that managed to adhere will proliferate. On the other hand, biomolecules and dead microorganisms will accumulate on bactericidal coatings especially those containing cationic moieties. Thus, a combination of anti-adhesive and bactericidal moieties is likely to be more effective than either component alone but the proportion of the different moieties has to be carefully optimized for different biological environments e.g. blood versus urine.
While this review has focused on strategies for chemical modification of catheter surfaces to alleviate catheter-related complications, other non-chemical approaches such as those based on the modification of surface topography or use of acoustic energy or electrical current should also be further explored. Embossing the Sharklet micropattern on polyurethane has been shown to reduce bacterial colonization and platelet interactions after simulated vascular exposure.226 Application of acoustic waves can reduce viable microbial counts on surfaces, diminish biofilm formation in vitro and prolong catheter sterility in a rabbit model.227 However, certain levels of acoustic energy dose and/or frequency can lead to the opposite effect of promoting biofilm formation, illustrating the complexity of the phenomena. Passage of low electrical current through electrodes placed in the lumen of catheters on which a biofilm had been grown resulted in a reduction of the biofilm.228 The safety aspects of this method as well as how it can be applied for preventing biofilm formation on both intraluminal and extraluminal surfaces of catheters have to be considered.
Laboratory tests to assess the efficacy of catheter coatings are usually carried out with short catheter segments over a few hours to a few days. The conditions of these tests do not reflect the complicated milieu encountered by catheters inserted into the body. Thus, it is not surprising that despite the myriad promising antimicrobial and non-thrombogenic coatings reported in the literature, coatings that can minimize complications from indwelling catheters clinically are still lacking. There is an urgent need for methodology for designing and executing in vitro and preclinical trials that can better mimic the actual conditions of application of these catheters. Furthermore, the feasibility of whether techniques developed for modifying short catheter segments or flat sheets in the laboratory can be economically scaled up to full length catheters covering both intraluminal and extraluminal surfaces is an important consideration. These challenges offer opportunities for innovation but to achieve advancement in this field, close collaboration is needed between materials scientists, biologists and clinicians to establish a better understanding of the multifactorial variables governing the behavior of different types of catheters in the in vivo environment, and to establish long-term in vivo models for studying how to mediate such behavior.
References
- Oak Brook, IL: Joint Commission Resources, http://https://www.jointcommission.org/assets/1/18/CLABSI_Monograph.pdf, accessed September, 2016.
- E. P. Dellinger, Surg. Infect.: Sel. Antibiot. Ther., 2016, 17, 422–426 CrossRef PubMed
.
- Centers for Disease Control and Prevention, http://www.cdc.gov/hai/surveillance/, accessed Spetember, 2016.
- European Center for Disease Control and Prevention, http://ecdc.europa.eu/en/healthtopics/healthcare-associated_infections/pages/index.aspx, accessed September, 2016.
- V. D. Rosenthal, D. G. Maki, Y. Mehta, H. Leblebicioglu, Z. A. Memish, H. H. Al-Mousa, H. Balkhy, B. J. Hu, C. Alvarez-Moreno, E. A. Medeiros, A. Apisarnthanarak, L. Raka, L. E. Cuellar, A. Ahmed, J. A. Navoa-Ng, A. A. El-Kholy, S. S. Kanj, I. Bat-Erdene, W. Duszynska, N. Van Truong, L. N. Pazmino, L. C. See-Lum, R. Fernández-Hidalgo, G. Di-Silvestre, F. Zand, S. Hlinkova, V. Belskiy, H. Al-Rahma, M. T. Luque-Torres, N. Bayraktar, Z. Mitrev, V. Gurskis, D. Fisher, I. B. Abu-Khader, K. Berechid, A. Rodríguez-Sánchez, F. G. Horhat, O. Requejo-Pino, N. Hadjieva, N. Ben-Jaballah, E. García-Mayorca, L. Kushner-Dávalos, S. Pasic, L. E. Pedrozo-Ortiz, E. Apostolopoulou, N. Mejía, M. O. Gamar-Elanbya, K. Jayatilleke, M. de Lourdes-Dueñas and G. Aguirre-Avalos, Am. J. Infect. Control, 2014, 42, 942–956 CrossRef PubMed
- E. Zimlichman, D. Henderson, O. Tamir, C. Franz, P. Song, C. K. Yamin, C. Keohane, C. R. Denham and D. W. Bates, JAMA Intern. Med., 2013, 173, 2039–2046 CrossRef PubMed
- Centers for Disease Control and Prevention, http://www.cdc.gov/hai/pdfs/progress-report/hai-progress-report.pdf, accessed September, 2016.
- M. A. Dudeck, J. R. Edwards, K. Allen-Bridson, C. Gross, P. J. Malpiedi, K. D. Peterson, D. A. Pollock, L. M. Weiner and D. M. Sievert, Am. J. Infect. Control, 2015, 43, 206–221 CrossRef PubMed
- R. E. Helm, J. D. Klausner, J. D. Klemperer, L. M. Flint and E. Huang, J. Infus. Nurs., 2015, 38, 189–203 CrossRef PubMed
- L. Hadaway, J. Infus. Nurs., 2012, 35, 230–240 CrossRef PubMed
- D. Frasca, C. Dahyot-Fizelier and O. Mimoz, Crit. Care, 2010, 14, 212 CrossRef PubMed
- R. O. Darouiche, Clin. Infect. Dis., 2001, 33, 1567–1572 CrossRef CAS PubMed
- S. Saint, R. N. Olmsted, M. G. Fakih, C. P. Kowalski, S. R. Watson, A. E. Sales and S. L. Krein, The Joint Commission Journal on Quality and Patient Safety, 2009, 35, 449–455 CrossRef PubMed
- S. Öncü, H. Özsüt, A. Yildirim, P. Ay, N. Çakar, H. Eraksoy and S. Çalangu, Ann. Clin. Microbiol. Antimicrob., 2003, 2, 1–6 CrossRef
- H. S. Chen, F. D. Wang, M. Lin, Y. C. Lin, L. J. Huang and C. Y. Liu, J. Microbiol., Immunol. Infect., 2006, 39, 231–236 Search PubMed
- R. Vilela, A. D. N. Jácomo and A. T. Tresoldi, Clinics, 2007, 62, 537–544 CrossRef PubMed
- J. Crouzet, X. Bertrand, A. G. Venier, M. Badoz, C. Flusson and D. Talon, J. Hosp. Infect., 2007, 67, 253–257 CrossRef CAS PubMed
- T. M. Hooton, S. F. Bradley, D. D. Cardenas, R. Colgan, S. E. Geerlings, J. C. Rice, S. Saint, A. J. Schaeffer, P. A. Tambayh, P. Tenke and L. E. Nicolle, Clin. Infect. Dis., 2010, 50, 625–663 CrossRef PubMed
- M. Leone, J. Albanese, F. Garnier, C. Sapin, K. Barrau, M. C. Bimar and C. Martin, Intensive Care Med., 2003, 29, 929–932 CrossRef PubMed
- P. Gastmeier, S. Stamm-Balderjahn, S. Hansen, F. Nitzschke-Tiemann, I. Zuschneid, K. Groneberg and H. Ruden, Infect. Control. Hosp. Epidemiol., 2005, 26, 357–361 CrossRef PubMed
- D. Pavithra and M. Doble, Biomed. Mater., 2008, 3, 034003 CrossRef CAS PubMed
- E. E. MacKintosh, J. D. Patel, R. E. Marchant and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2006, 78A, 836–842 CrossRef CAS PubMed
-
T. Boland, R. A. Latour and F. J. Stutzenberger, in Handbook of bacterial adhesion: principles, methods, and applications, ed. Y. H. An and R. J. Friedman, Humana Press Inc., Totowa, NJ 2000, pp. 29–41 Search PubMed
- R. M. Donlan, Clin. Infect. Dis., 2001, 33, 1387–1392 CrossRef CAS PubMed
- S. L. Percival, L. Suleman, C. Vuotto and G. Donelli, J. Med. Microbiol., 2015, 64, 323–334 CrossRef PubMed
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CAS
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS PubMed
- B. Schachter, Nat. Biotechnol., 2003, 21, 361–365 CrossRef CAS PubMed
- H. Ceri, M. E. Olson, C. Stremick, R. R. Read, D. Morck and A. Buret, J. Clin. Microbiol., 1999, 37, 1771–1776 CAS
- I. Williams, W. A. Venables, D. Lloyd, F. Paul and I. Critchley, Microbiology, 1997, 143, 2407–2413 CrossRef CAS PubMed
- S. D. Aaron, W. Ferris, K. Ramotar, K. Vandemheen, F. Chan and R. Saginur, J. Clin. Microbiol., 2002, 40, 4172–4179 CrossRef CAS PubMed
- K. Smith and I. S. Hunter, J. Med. Microbiol., 2008, 57, 966–973 CrossRef CAS PubMed
- J. A. Otter, K. Vickery, J. T. Walker, E. D. Pulcini, P. Stoodley, S. D. Goldenberg, J. A. G. Salkeld, J. Chewins, S. Yezli and J. D. Edgeworth, J. Hosp. Infect., 2015, 89, 16–27 CrossRef CAS PubMed
- J. N. Anderl, J. Zahller, F. Roe and P. S. Stewart, Antimicrob. Agents Chemother., 2003, 47, 1251–1256 CrossRef CAS PubMed
- I. G. Duguid, E. Evans, M. R. W. Brown and P. Gilbert, J. Antimicrob. Chemother., 1992, 30, 803–810 CrossRef CAS PubMed
- T. K. Wood, S. J. Knabel and B. W. Kwan, Appl. Environ. Microbiol., 2013, 79, 7116–7121 CrossRef CAS PubMed
- T. J. Marrie and J. W. Costerton, J. Clin. Microbiol., 1984, 19, 687–693 CAS
- T. J. Marrie, J. Nelligan and J. W. Costerton, Circulation, 1982, 66, 1339–1341 CrossRef CAS PubMed
- P. K. T. Li and K. M. Chow, Nat. Rev. Nephrol., 2012, 8, 77–88 CrossRef CAS PubMed
- B. Adam, G. S. Baillie and L. J. Douglas, J. Med. Microbiol., 2002, 51, 344–349 CrossRef PubMed
- E. Carlson, Infect. Immun., 1982, 38, 921–924 CAS
- R. M. Donlan, Clin. Infect. Dis., 2011, 52, 1038–1045 CrossRef PubMed
- D. Lebeaux, J. M. Ghigo and C. Beloin, Microbiol. Mol. Biol. Rev., 2014, 78, 510–543 CrossRef CAS PubMed
- M. T. McCann, B. F. Gilmore and S. P. Gorman, J. Pharm. Pharmacol., 2008, 60, 1551–1571 CrossRef CAS PubMed
- J. Janzen, B. M. Buurman, L. Spanjaard, T. M. de Reijke, A. Goossens and S. E. Geerlings, BMJ Quality & Safety, 2013, 22, 984–988 Search PubMed
- J. Meddings, M. A. M. Rogers, M. Macy and S. Saint, Clin. Infect. Dis., 2010, 51, 550–560 CrossRef PubMed
- P. J. van den Broek, J. C. Wille, B. H. B. van Benthem, R. J. M. Perenboom, M. E. van den Akker-van Marle and B. S. Niel-Weise, BMC Urol., 2011, 11, 10 CrossRef PubMed
- Y. Y. Chen, M. M. Chi, Y. C. Chen, Y. J. Chan, S. S. Chou and F. D. Wang, Am. J. Respir. Crit. Care Med., 2013, 22, 105–114 Search PubMed
- L. Mody, J. Meddings, B. S. Edson, S. E. McNamara, B. W. Trautner, N. D. Stone, S. L. Krein and S. Saint, Clin. Infect. Dis., 2015, 61, 86–94 CrossRef PubMed
- M. M. Tiwari, M. E. Charlton, J. R. Anderson, E. D. Hermsen and M. E. Rupp, Am. J. Infect. Control, 2012, 40, 51–54 CrossRef PubMed
- A. L. Flores-Mireles, J. N. Walker, M. Caparon and S. J. Hultgren, Nat. Rev. Microbiol., 2015, 13, 269–284 CrossRef CAS PubMed
- American Urological Association, http://www.auanet.org/advnews/press_releases/article.cfm?articleNo=304, accessed November, 2016.
- B. W. Trautner, Curr. Opin. Infect. Dis., 2010, 23, 76–82 CrossRef PubMed
- S. M. Jacobsen, D. J. Stickler, H. L. T. Mobley and M. E. Shirtliff, Clin. Microbiol. Rev., 2008, 21, 26–59 CrossRef CAS PubMed
- D. G. Maki and P. A. Tambyah, Emerging Infect. Dis., 2001, 7, 342–347 CrossRef CAS PubMed
- D. M. Siddiq and R. O. Darouiche, Nat. Rev. Urol., 2012, 9, 305–314 CrossRef CAS PubMed
- L. E. Nicolle, Antimicrob. Resist Infect. Control, 2014, 3, 23 CrossRef PubMed
- D. J. Stickler and R. C. L. Feneley, Spinal Cord, 2010, 48, 784–790 CrossRef CAS PubMed
- S. D. Morgan, D. Rigby and D. J. Stickler, Urol. Res., 2009, 37, 89–93 CrossRef CAS PubMed
- D. Stickler, L. Ganderton, J. King, J. Nettleton and C. Winters, Urol. Res., 1993, 21, 407–411 CrossRef CAS PubMed
- D. C. McGee and M. K. Gould, N. Engl. J. Med., 2003, 348, 1123–1133 CrossRef PubMed
- J. R. Mehall, D. A. Saltzman, R. J. Jackson and S. D. Smith, Crit. Care Med., 2002, 30, 908–912 CrossRef PubMed
- A. L. Casey, L. A. Mermel, P. Nightingale and T. S. J. Elliott, Lancet Infect. Dis., 2008, 8, 763–776 CrossRef CAS PubMed
- A. Besarab and R. Pandey, Clin. J. Am. Soc. Nephrol., 2011, 6, 227–234 CrossRef PubMed
- D. J. Mayo, J. Infus. Nurs., 2001, 24, S13–S22 Search PubMed
- M. R. Crain, M. G. Horton and M. W. Mewissen, Am. J. Roentgenol., 1998, 171, 341–346 CrossRef CAS PubMed
- J. L. Baskin, C. H. Pui, U. Reiss, J. A. Wilimas, M. L. Metzger, R. C. Ribeiro and S. C. Howard, Lancet, 2009, 374, 159–169 CrossRef
- R. K. Peel and J. H. Turney, Nephrol., Dial., Transplant., 2003, 18, 1026 CrossRef
- I. Radd, W. Costerton, U. Sabharwal, M. Sacilowski, E. Anaissie and G. P. Bodey, J. Infect. Dis., 1993, 168, 400–407 CrossRef
- L. A. Mermel, Clin. Infect. Dis., 2011, 52, 211–212 CrossRef PubMed
- L. A. Mermel, Ann. Intern. Med., 2000, 132, 391–402 CrossRef CAS PubMed
- J. M. Walz, S. G. Memtsoudis and S. O. Heard, J. Intensive Care Med., 2010, 25, 131–138 CrossRef PubMed
- Centers for Disease Control and Prevention, http://www.cdc.gov/hicpac/BSI/BSI-guidelines-2011.html, 2011.html, Accessed September, 2016.
- H. Shah, W. Bosch, K. M. Thompson and W. C. Hellinger, Neurohospitalist, 2013, 3, 144–151 CrossRef PubMed
- L. Lorente, A. Jimenez, M. Santana, J. L. Iribarren, J. J. Jimenez, M. M. Martin and M. L. Mora, Crit. Care Med., 2007, 35, 2424–2427 CrossRef PubMed
- R. S. Boersma, K. S. G. Jie, A. Verbon, E. C. M. van Pampus and H. C. Schouten, Ann. Oncol., 2008, 19, 433–442 CrossRef CAS PubMed
- M. Y. Chan, J. I. Weitz, Y. Merhi, R. A. Harrington and R. C. Becker, J. Thromb. Thrombolysis, 2009, 28, 366–380 CrossRef PubMed
- H. K. Thomsen, K. Kjeldsen and J. F. Hansen, Cathet. Cardiovasc. Diagn., 1977, 3, 351–358 CrossRef CAS PubMed
- M. G. Bourassa, M. Cantin, E. B. Sandborn and E. Pederson, Circulation, 1976, 53, 992–996 CrossRef CAS PubMed
- H. Miyagaki, K. Nakajima, J. Hara, M. Yamasaki, Y. Kurokawa, H. Miyata, S. Takiguchi, Y. Fujiwara, M. Mori and Y. Doki, Clin. Nutr., 2012, 31, 48–52 CrossRef PubMed
- I. H. Jaffer, J. C. Fredenburgh, J. Hirsh and J. I. Weitz, J. Thromb. Haemostasis, 2015, 13, S72–S81 CrossRef PubMed
- C. Wall, J. Moore and J. Thachil, J. Intensive Care Soc., 2015, 17, 160–167 CrossRef
- S. F. Mohammad, ASAIO J., 2000, 46, S63–S68 CrossRef CAS PubMed
- T. J. Foster and D. McDevitt, FEMS Microbiol. Lett., 1994, 118, 199–205 CrossRef CAS PubMed
- L. G. Harris, S. J. Foster and R. G. Richards, Eur. Cells Mater., 2002, 4, 39–60 CrossRef CAS PubMed
- L. C. Xu and C. A. Siedlecki, J. Biomater. Nanobiotechnol., 2012, 3, 487–498 CrossRef CAS
- S. W. Kerrigan, N. Clarke, A. Loughman, G. Meade, T. J. Foster and D. Cox, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 335–340 CrossRef CAS PubMed
- D. Cox, S. W. Kerrigan and S. P. Watson, J. Thromb. Haemostasis, 2011, 9, 1097–1107 CrossRef CAS PubMed
- C. J. van Rooden, E. F. Schippers, R. M. Y. Barge, F. R. Rosendaal, H. F. L. Guiot, F. J. M. van der Meer, A. E. Meinders and M. V. Huisman, J. Clin. Oncol., 2005, 23, 2655–2660 CrossRef PubMed
- C. Fourtounas, Hippokratia, 2011, 15, 15–20 Search PubMed
- K. Chaudhary, H. Sangha and R. Khanna, Clin. J. Am. Soc. Nephrol., 2011, 6, 447–456 CrossRef PubMed
- T. Yip, S. L. Lui and W. K. Lo, Int. J. Nephrol., 2013, 2013, 940106 CAS
- A. Berger, J. Edelsberg, G. W. Inglese, S. K. Bhattacharyya and G. Oster, Am. J. Manag. Care, 2009, 15, 509–518 Search PubMed
- G. Abraham, S. Varughese, M. Mathew and M. Vijayan, Clin. Kidney J., 2015, 8, 310–317 CrossRef PubMed
- R. Sinnakirouchenan and J. L. Holley, Adv. Chronic. Kidney Dis., 2011, 18, 428–432 CrossRef PubMed
- L. R. Lukowsky, R. Mehrotra, L. Kheifets, O. A. Arah, A. R. Nissenson and K. Kalantar-Zadeh, Clin. J. Am. Soc. Nephrol., 2013, 8, 619–628 CrossRef PubMed
- S. J. Nessim, Semin. Nephrol., 2011, 31, 199–212 CrossRef PubMed
- J. H. Crabtree and R. J. Burchette, Am. J. Surg., 2005, 190, 4–8 CrossRef PubMed
- R. Bayston, L. E. Fisher and K. Weber, Biomaterials, 2009, 30, 3167–3173 CrossRef CAS PubMed
- M. Rudnicki, J. Kerschbaum, J. Hausdorfer, G. Mayer and P. Konig, Peritoneal Dial. Int., 2010, 30, 541–548 CrossRef PubMed
- P. K. T. Li, C. C. Szeto, B. Piraino, J. Bernardini, A. E. Figueiredo, A. Gupta, D. W. Johnson, E. J. Kuijper, W. C. Lye, W. Salzer, F. Schaefer and D. G. Struijk, Peritoneal Dial. Int., 2010, 30, 393–423 CrossRef PubMed
- M. Pihl, J. R. Davies, A. C. Johansson and G. Svensater, Peritoneal Dial. Int., 2013, 33, 51–59 CrossRef CAS PubMed
- J. A. Akoh, World J. Nephrol., 2012, 1, 106–122 CrossRef PubMed
- J. Matuszkiewicz-Rowinska, Peritoneal Dial. Int., 2009, 29, S161–S165 CAS
- J. R. Li, C. H. Cheng, K. Y. Chiu, C. L. Cheng, C. R. Yang, H. C. Ho, J. L. Ko and Y. C. Ou, Peritoneal Dial. Int., 2013, 33, 46–50 CrossRef PubMed
- J. Y. Xie, H. Ren, K. Kiryluk and N. Chen, Am. J. Kidney Dis., 2010, 56, 1006–1011 CrossRef PubMed
- M. Moreiras-Plaza and N. Caceres-Alvarado, Am. J. Kidney Dis., 2004, 44, e28–30 CrossRef PubMed
- J. S. Jheng, C. T. Chang and C. C. Huang, Kidney Int., 2012, 82, 827 CrossRef PubMed
- Y. H. Goh, Peritoneal Dial. Int., 2008, 28, 626–631 Search PubMed
- C. Platell, D. Cooper, J. M. Papadimitriou and J. C. Hall, World J. Gastroenterol., 2000, 6, 169–176 Search PubMed
- A. Peppelenbosch, W. H. M. van Kuijk, N. D. Bouvy, F. M. van der Sande and J. H. M. Tordoir, NDT Plus, 2008, 1, iv23–iv28 Search PubMed
- G. Chiarelli, M. Beaulieu, M. Cozzolino, S. Singh, M. Kiaii, P. Taylor, A. Levin, D. Brancaccio and M. Gallieni, Peritoneal Dial. Int., 2008, 28, 585–590 Search PubMed
- P. G. Lan, P. A. Clayton, J. Saunders, K. R. Polkinghorne and P. L. Snelling, Peritoneal Dial. Int., 2015, 35, 306–315 CrossRef CAS PubMed
- Y. H. An and R. J. Friedman, J. Biomed. Mater. Res., 1998, 43, 338–348 CrossRef CAS PubMed
- S. F. Chen, L. Y. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS
- N. Hadjesfandiari, K. Yu, Y. Mei and J. N. Kizhakkedathu, J. Mater. Chem. B, 2014, 2, 4968–4978 RSC
- J. Zheng, L. Y. Li, H. K. Tsao, Y. J. Sheng, S. F. Chen and S. Y. Jiang, Biophys. J., 2005, 89, 158–166 CrossRef CAS PubMed
- S. Herrwerth, W. Eck, S. Reinhardt and M. Grunze, J. Am. Chem. Soc., 2003, 125, 9359–9366 CrossRef CAS PubMed
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. De Gennes, J. Colloid Interface Sci., 1991, 142, 149–158 CrossRef CAS
- S. I. Jeon and J. D. Andrade, J. Colloid Interface Sci., 1991, 142, 159–166 CrossRef CAS
- B. Gottenbos, D. W. Grijpma, H. C. van der Mei, J. Feijen and H. J. Busscher, J. Antimicrob. Chemother., 2001, 48, 7–13 CrossRef CAS PubMed
- A. Terada, K. Okuyama, M. Nishikawa, S. Tsuneda and M. Hosomi, Biotechnol. Bioeng., 2012, 109, 1745–1754 CrossRef CAS PubMed
- O. Rzhepishevska, S. Hakobyan, R. Ruhal, J. Gautrot, D. Barbero and M. Ramstedt, Biomater. Sci., 2013, 1, 589–602 RSC
- M. Lorenzetti, I. Dogsa, T. Stosicki, D. Stopar, M. Kalin, S. Kobe and S. Novak, ACS Appl. Mater. Interfaces, 2015, 7, 1644–1651 CAS
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed
- H. B. Zhang and M. Chiao, J. Med. Biol. Eng., 2015, 35, 143–155 CrossRef PubMed
- J. Wu, C. Zhao, R. D. Hu, W. F. Lin, Q. M. Wang, J. Zhao, S. M. Bilinovich, T. C. Leeper, L. Y. Li, H. M. Cheung, S. F. Chen and J. Zheng, Acta Biomater., 2014, 10, 751–760 CrossRef CAS PubMed
- R. S. Kane, P. Deschatelets and G. M. Whitesides, Langmuir, 2003, 19, 2388–2391 CrossRef CAS
- G. Cheng, Z. Zhang, S. F. Chen, J. D. Bryers and S. Y. Jiang, Biomaterials, 2007, 28, 4192–4199 CrossRef CAS PubMed
- A. L. Hook, C.-Y. Chang, J. Yang, J. Luckett, A. Cockayne, S. Atkinson, Y. Mei, R. Bayston, D. J. Irvine, R. Langer, D. G. Anderson, P. Williams, M. C. Davies and M. R. Alexander, Nat. Biotechnol., 2012, 30, 868–875 CrossRef CAS PubMed
- O. Sanni, C. Y. Chang, D. G. Anderson, R. Langer, M. C. Davies, P. M. Williams, P. Williams, M. R. Alexander and A. L. Hook, Adv. Healthcare Mater., 2015, 4, 695–701 CrossRef CAS PubMed
- V. Gadenne, L. Lebrun, T. Jouenne and P. Thebault, Colloids Surf., B, 2013, 112, 229–236 CrossRef CAS PubMed
- G. A. Junter, P. Thebault and L. Lebrun, Acta Biomater., 2016, 30, 13–25 CrossRef CAS PubMed
- M. Li, K. G. Neoh, E. T. Kang, T. Lau and E. Chiong, Adv. Funct. Mater., 2014, 24, 1631–1643 CrossRef CAS
- M. Morra and C. Cassineli, J. Biomater. Sci., Polym. Ed., 1999, 10, 1107–1124 CrossRef CAS PubMed
- R. J. Heath, J. R. Rubin, D. R. Holland, E. L. Zhang, M. E. Snow and C. O. Rock, J. Biol. Chem., 1999, 274, 11110–11114 CrossRef CAS PubMed
- I. Chopra and M. Roberts, Microbiol. Mol. Biol. Rev., 2001, 65, 232–260 CrossRef CAS PubMed
- W. Wehrli, J. Infect. Dis., 1983, 5, S407–S411 CAS
- H. Y. Cheung, M. M. K. Wong, S. H. Cheung, L. Y. Liang, Y. W. Lam and S. K. Chiu, PLoS One, 2012, 7, e36659 CAS
- J. R. Morones-Ramirez, J. A. Winkler, C. S. Spina and J. J. Collins, Sci. Transl. Med., 2013, 5, 190ra81 CrossRef PubMed
- B. Li, K. V. Brown, J. C. Wenke and S. A. Guelcher, J. Controlled Release, 2010, 145, 221–230 CrossRef CAS PubMed
- J. M. Schierholz, H. Steinhauser, A. F. E. Rump, R. Berkels and G. Pulverer, Biomaterials, 1997, 18, 839–844 CrossRef CAS PubMed
- L. E. Fisher, A. L. Hook, W. Ashraf, A. Yousef, D. A. Barrett, D. J. Scurr, X. Y. Chen, E. F. Smith, M. Fay, C. D. J. Parmenter, R. Parkinson and R. Bayston, J. Controlled Release, 2015, 202, 57–64 CrossRef CAS PubMed
- D. Campoccia, L. Montanaro and C. R. Arciola, Biomaterials, 2013, 34, 8533–8554 CrossRef CAS PubMed
- M. Cloutier, D. Mantovani and F. Rosei, Trends Biotechnol., 2015, 33, 637–652 CrossRef CAS PubMed
- D. I. Andersson and D. Hughes, Nat. Rev. Microbiol., 2014, 12, 465–478 CrossRef CAS PubMed
- P. H. Mc Cay, A. A. Ocampo-Sosa and G. T. A. Fleming, Microbiol. SGM, 2010, 156, 30–38 CrossRef CAS PubMed
- M. Lazaro-Diez, S. Remuzgo-Martinez, C. Rodriguez-Mirones, F. Acosta, J. M. Icardo, L. Martinez-Martinez and J. Ramos-Vivas, PLoS One, 2016, 11, e0147569 Search PubMed
- A. Cavallaro, A. Mierczynska, M. Barton, P. Majewski and K. Vasilev, Biofouling, 2016, 32, 13–24 CrossRef CAS PubMed
- K. Vasilev, J. Cook and H. J. Griesser, Expert Rev. Med. Devices, 2009, 6, 553–567 CrossRef PubMed
- Y. Xue, H. N. Xiao and Y. Zhang, Int. J. Mol. Sci., 2015, 16, 3626–3655 CrossRef CAS PubMed
- J. C. Tiller, C. J. Liao, K. Lewis and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5981–5985 CrossRef CAS PubMed
- H. Murata, R. R. Koepsel, K. Matyjaszewski and A. J. Russell, Biomaterials, 2007, 28, 4870–4879 CrossRef CAS PubMed
- Z. L. Shi, K. G. Neoh and E. T. Kang, Ind. Eng. Chem. Res., 2007, 46, 439–445 CrossRef CAS
- L. Cen, K. G. Neoh and E. T. Kang, Langmuir, 2003, 19, 10295–10303 CrossRef CAS
- S. B. Lee, R. R. Koepsel, S. W. Morley, K. Matyjaszewski, Y. J. Sun and A. J. Russell, Biomacromolecules, 2004, 5, 877–882 CrossRef CAS PubMed
- K. Kalinov, M. Ignatova, V. Maximova, I. Rashkov and N. Manolova, Eur. Polym. J., 2014, 50, 18–29 CrossRef CAS
- M. W. Huh, I. K. Kang, D. H. Lee, W. S. Kim, D. H. Lee, L. S. Park, K. E. Min and K. H. Seo, J. Appl. Polym. Sci., 2001, 81, 2769–2778 CrossRef CAS
- D. Mitra, M. Li, R. Wang, Z. H. Tang, E. T. Kang and K. G. Neoh, Ind. Eng. Chem. Res., 2016, 55, 9603–9613 CrossRef CAS
- R. Wang, K. G. Neoh and E. T. Kang, J. Colloid Interface Sci., 2015, 438, 138–148 CrossRef CAS PubMed
- X. Ding, C. Yang, T. P. Lim, L. Y. Hsu, A. C. Engler, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2012, 33, 6593–6603 CrossRef CAS PubMed
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed
- B. Bechinger and K. Lohner, Biochim. Biophys. Acta, 2006, 1758, 1529–1539 CrossRef CAS PubMed
- K. Glinel, A. M. Jonas, T. Jouenne, J. Leprince, L. Galas and W. T. S. Huck, Bioconjugate Chem., 2009, 20, 71–77 CrossRef CAS PubMed
- R. E. W. Hancock and H. G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed
- C. L. Friedrich, D. Moyles, T. J. Beveridge and R. E. W. Hancock, Antimicrob. Agents Chemother., 2000, 44, 2086–2092 CrossRef CAS PubMed
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 CrossRef CAS PubMed
- V. I. Band and D. S. Weiss, Antibiotics, 2015, 4, 18–41 CrossRef CAS PubMed
- M. Gabriel, K. Nazmi, E. C. Veerman, A. V. N. Amerongen and A. Zentner, Bioconjugate Chem., 2006, 17, 548–550 CrossRef CAS PubMed
- M. Bagheri, M. Beyermann and M. Dathe, Antimicrob. Agents Chemother., 2009, 53, 1132–1141 CrossRef CAS PubMed
- A. K. Muszanska, E. T. J. Rochford, A. Gruszka, A. A. Bastian, H. J. Busscher, W. Norde, H. C. van der Mei and A. Herrmann, Biomacromolecules, 2014, 15, 2019–2026 CrossRef CAS PubMed
- W. Aoki and M. Ueda, Pharmaceuticals, 2013, 6, 1055–1081 CrossRef PubMed
- R. Pickard, T. Lam, G. MacLennan, K. Starr, M. Kilonzo, G. McPherson, K. Gillies, A. McDonald, K. Walton, B. Buckley, C. Glazener, C. Boachie, J. Burr, J. Norrie, L. Vale, A. Grant and J. N'Dow, Lancet, 2012, 380, 1927–1935 CrossRef CAS
- T. B. L. Lam, M. I. Omar, E. Fisher, K. Gillies and S. MacLennan, Cochrane Database Syst. Rev., 2014, 23, CD004013 Search PubMed
- K. D. Mandakhalikar, R. R. Chua and P. A. Tambyah, Curr. Treat. Options Infect. Dis., 2016, 8, 24–41 CrossRef
- L. Islas, C. Alvarez-Lorenzo, B. Magarinos, A. Concheiro, L. F. del Castillo and G. Burillo, Int. J. Pharm., 2015, 488, 20–28 CrossRef CAS PubMed
- J. L. Pugach, V. DiTizio, M. W. Mittelman, A. W. Bruce, F. DiCosmo and A. E. Khoury, J. Urol., 1999, 162, 883–887 CrossRef CAS PubMed
- G. Segev, T. Bankirer, D. Steinberg, M. Duvdevani, N. K. Shapur, M. Friedman and E. Lavy, J. Vet. Intern. Med., 2013, 27, 39–46 CrossRef CAS PubMed
- L. E. Fisher, A. L. Hook, W. Ashraf, A. Yousef, D. A. Barrett, D. J. Scurr, X. Chen, E. F. Smith, M. Fay, C. D. J. Parmenter, R. Parkinson and R. Bayston, J. Controlled Release, 2015, 202, 57–64 CrossRef CAS PubMed
- D. E. Carey and P. J. McNamara, Front. Microbiol., 2015, 5, 780 Search PubMed
- K. M. Crofton, K. B. Paul, M. J. De Vito and J. M. Hedge, Environ. Toxicol. Pharmacol., 2007, 24, 194–197 CrossRef CAS PubMed
- M. F. Yueh, K. Taniguchi, S. J. Chen, R. M. Evans, B. D. Hammock, M. Karin and R. H. Tukey, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17200–17205 CrossRef CAS PubMed
- R. Thomas, K. R. Soumya, J. Mathew and E. K. Radhakrishnan, J. Photochem. Photobiol., B, 2015, 149, 68–77 CrossRef CAS PubMed
- Y. Yao, Y. Ohko, Y. Sekiguchi, A. Fujishima and Y. Kubota, J. Biomed. Mater. Res., Part B, 2008, 85B, 453–460 CrossRef CAS PubMed
- R. Wang, K. G. Neoh, E. T. Kang, P. A. Tambyah and E. Chiong, J. Biomed. Mater. Res., Part B, 2015, 103, 519–528 CrossRef PubMed
- M. L. Jones, J. G. Ganopolsky, A. Labbé, C. Wahl and S. Prakash, Appl. Microbiol. Biotechnol., 2010, 88, 401–407 CrossRef CAS PubMed
- D. O. Schairer, J. S. Chouake, J. D. Nosanchuk and A. J. Friedman, Virulence, 2012, 3, 271–279 CrossRef PubMed
- G. Regev-Shoshani, M. Ko, C. Miller and Y. Av-Gay, Antimicrob. Agents Chemother., 2010, 54, 273–279 CrossRef CAS PubMed
- A. R. Ketchum, M. P. Kappler, J. F. Wu, C. W. Xi and M. E. Meyerhoff, J. Mater. Chem. B, 2016, 4, 422–430 RSC
- A. Colletta, J. F. Wu, Y. Q. Wo, M. Kappler, H. Chen, C. W. Xi and M. E. Meyerhoff, ACS Biomater. Sci. Eng., 2015, 1, 416–424 CrossRef CAS PubMed
- W. L. Fu, T. Forster, O. Mayer, J. J. Curtin, S. M. Lehman and R. M. Donlan, Antimicrob. Agents Chemother., 2010, 54, 397–404 CrossRef CAS PubMed
- S. M. Lehman and R. M. Donlan, Antimicrob. Agents Chemother., 2015, 59, 1127–1137 CrossRef PubMed
- L. D. R. Melo, P. Veiga, N. Cerca, A. M. Kropinski, C. Almeida, J. Azeredo and S. Sillankorva, Front. Microbiol., 2016, 7, 1024 Search PubMed
- K. A. Hughes, I. W. Sutherland and M. V. Jones, Microbiology, 1998, 144, 3039–3047 CrossRef CAS PubMed
- L. Carson, S. P. Gorman and B. F. Gilmore, FEMS Immunol. Med. Microbiol., 2010, 59, 447–455 CrossRef CAS PubMed
- E. Jończyk, M. Kłak, R. Midzybrodzki and A. Górski, Folia Microbiol., 2011, 56, 191–200 CrossRef PubMed
- V. C. Thompson, P. J. Adamson, J. Dilag, D. Liyanage, K. Srikantharajah, A. Blok, A. V. Ellis, D. L. Gordon and I. Koper, RSC Adv., 2016, 6, 53303–53309 RSC
- A. Vaterrodt, B. Thallinger, K. Daumann, D. Koch, G. M. Guebitz and M. Ulbricht, Langmuir, 2016, 32, 1347–1359 CrossRef CAS PubMed
- B. Thallinger, M. Brandauer, P. Burger, C. Sygmund, R. Ludwig, K. Ivanova, J. Kun, D. Scaini, M. Burnet, T. Tzanov, G. S. Nyanhongo and G. M. Guebitz, J. Biomed. Mater. Res., Part B, 2016, 104, 1448–1456 CrossRef CAS PubMed
- B. Mishra, A. Basu, R. R. Y. Chua, R. Saravanan, P. A. Tambyah, B. Ho, M. W. Chang and S. S. J. Leong, J. Mater. Chem. B, 2014, 2, 1706–1716 RSC
- X. Li, P. Li, R. Saravanan, A. Basu, B. Mishra, S. H. Lim, X. D. Su, P. A. Tambyah and S. S. J. Leong, Acta Biomater., 2014, 10, 258–266 CrossRef CAS PubMed
- K. Lim, R. R. Y. Chua, B. Ho, P. A. Tambyah, K. Hadinoto and S. S. J. Leong, Acta Biomater., 2015, 15, 127–138 CrossRef CAS PubMed
- Y. H. Li and X. L. Tian, Sensors, 2012, 12, 2519–2538 CrossRef CAS PubMed
- J. Shenderovich, M. Feldman, D. Kirmayer, A. Al-Quntar, D. Steinberg, E. Lavy and M. Friedman, Int. J. Pharm., 2015, 485, 164–170 CrossRef CAS PubMed
- K. Ivanova, M. M. Fernandes, E. Mendoza and T. Tzanov, Appl. Microbiol. Biotechnol., 2015, 99, 4373–4385 CrossRef CAS PubMed
- R. E. Gilbert, Q. Mok, K. Dwan, K. Harron, T. Moitt, M. Millar, P. Ramnarayan, S. M. Tibby, D. Hughes, C. Gamble and C. T. Investigators, Lancet, 2016, 387, 1732–1742 CrossRef
- N. M. Lai, N. Chaiyakunapruk, N. A. Lai, E. O'Riordan, W. S. C. Pau and S. Saint, Cochrane Database Syst. Rev., 2016, 3, CD007878 Search PubMed
- Y. M. Chen, A. P. Dai, Y. Shi, Z. J. Liu, M. F. Gong and X. B. Yin, Int. J. Infect. Dis., 2014, 29, 279–286 CrossRef CAS PubMed
- M. V. Homann, D. Johansson, H. Wallen and J. Sanchez, J. Biomed. Mater. Res., Part B, 2016, 104, 1359–1365 CrossRef PubMed
- M. W. Radomski, R. M. J. Palmer and S. Moncada, Br. J. Pharmacol., 1987, 92, 639–646 CrossRef CAS PubMed
- G. R. Wang, Y. Zhu, P. V. Halushka, T. M. Lincoln and M. E. Mendelsohn, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4888–4893 CrossRef CAS
- M. W. Vaughn, L. Kuo and J. C. Liao, Am. J. Physiol. Heart Circ. Physiol., 1998, 274, H2163–H2176 CAS
- E. J. Brisbois, R. P. Davis, A. M. Jones, T. C. Major, R. H. Bartlett, M. E. Meyerhoff and H. Handa, J. Mater. Chem. B, 2015, 3, 1639–1645 RSC
- E. J. Brisbois, T. C. Major, M. J. Goudie, M. E. Meyerhoff, R. H. Bartlett and H. Handa, Acta Biomater., 2016, 44, 304–312 CrossRef CAS PubMed
- Y. L. Yuan, F. Ai, X. P. Zang, W. Zhuang, J. Shen and S. C. Lin, Colloids Surf., B, 2004, 35, 1–5 CrossRef CAS PubMed
- R. S. Smith, Z. Zhang, M. Bouchard, J. Li, H. S. Lapp, G. R. Brotske, D. L. Lucchino, D. Weaver, L. A. Roth, A. Coury, J. Biggerstaff, S. Sukavaneshvar, R. Langer and C. Loose, Sci. Transl. Med., 2012, 4, 153ra132 Search PubMed
- W. Mussard, N. Kebir, I. Kriegel, M. Esteve and V. Semetey, Angew. Chem., Int. Ed., 2011, 50, 10871–10874 CrossRef CAS PubMed
- A. Chauhan, A. Bernardin, W. Mussard, I. Kriegel, M. Esteve, J. M. Ghigo, C. Beloin and V. Semetey, J. Infect. Dis., 2014, 210, 1347–1356 CrossRef PubMed
- D. C. Leslie, A. Waterhouse, J. B. Berthet, T. M. Valentin, A. L. Watters, A. Jain, P. Kim, B. D. Hatton, A. Nedder, K. Donovan, E. H. Super, C. Howell, C. P. Johnson, T. L. Vu, D. E. Bolgen, S. Rifai, A. R. Hansen, M. Aizenberg, M. Super, J. Aizenberg and D. E. Ingber, Nat. Biotechnol., 2014, 32, 1134–1140 CrossRef CAS PubMed
- S. Z. Trooskin, R. A. Harvey, T. W. J. Lennard and R. S. Greco, Peritoneal Dial. Int., 1990, 10, 57–59 CAS
- W. Pommer, M. Brauner, H. J. Westphale, R. Brunkhorst, R. Kramer, D. Bundschu, B. Hoffken, H. B. Steinhauer, E. Schumann, F. M. Luttgen, E. Schillinger-Pokorny, F. Schaefer, R. Wende, G. Offner, S. Nather, B. Osten, M. Zimmering, J. H. H. Ehrich, M. Kehn, U. Mansmann and C. Grosse-Siestrup, Am. J. Kidney Dis., 1998, 32, 752–760 CrossRef CAS PubMed
- J. H. Crabtree, R. J. Burchette, R. A. Siddiqi, L. L. Huen, L. L. Hadnott and A. Fishman, Peritoneal Dial. Int., 2003, 23, 368–374 CAS
- C. Y. Kim, A. Kumar, L. Sampath, K. Sokol and S. Modak, Am. J. Kidney Dis., 2002, 39, 165–173 CrossRef PubMed
- M. Zareie, E. D. Keuning, P. M. t. Wee, R. H. J. Beelen and J. v. d. Born, Adv. Perit. Dial., 2004, 20, 150–154 Search PubMed
- M. Frant, P. Stenstad, H. Johnsen, K. Dolling, U. Rothe, R. Schmid and K. Liefeith, Materwiss. Werksttech., 2006, 37, 538–545 CrossRef CAS
- R. M. May, C. M. Magin, E. E. Mann, M. C. Drinker, J. C. Fraser, C. A. Siedlecki, A. B. Brennan and S. T. Reddy, Clin. Transl. Med., 2015, 4, 9 CrossRef PubMed
- N. Dror, M. Mandel, Z. Hazan and G. Lavie, Sensors, 2009, 9, 2538–2554 CrossRef CAS PubMed
- P. Voegele, J. Badiola, S. M. Schmidt-Malan, M. J. Karau, K. E. Greenwood-Quaintance, J. N. Mandrekar and R. Patel, Antimicrob. Agents Chemother., 2016, 60, 1476–1480 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2017 |