A one-pot synthesis of water soluble highly fluorescent silica nanoparticles†
Received
27th October 2016
, Accepted 13th December 2016
First published on 14th December 2016
Abstract
We report a one-pot synthesis of water dispersible fluorescent silica nanoparticles (NPs) functionalized with terminal amine groups, starting from silicon tetrabromide (SiBr4) and aminopropyltriethoxy silane (APTES). The NPs range from 1 to 2 nm in diameter, and exhibit an intense blue emission with a quantum yield (QY) of around 34% in water. They were characterized using XRD, XPS, TEM and FTIR spectroscopy for structural analysis. A tentative mechanism explaining the origin of the NPs emission in the blue region is presented based on the distinctive features of their low temperature photoluminescence (PL), photoluminescence excitation (PLE) spectrum and time correlated single photon counting lifetime decay profiles. The outstanding PL QY and photostability of the NPs, together with their water dispersibility and biocompatibility, constitute a unique set of properties among existing silica NPs and enable the application of the NPs in various fields.
1. Introduction
Photoluminescent functionalized silica nanoparticles (NPs) are promising materials for biomedical applications, especially in optical imaging and drug delivery in view of their high photoluminescence quantum yields (PL QYs), outstanding photostability, colloidal stability and biocompatibility.1–4 Surface functionalization introduces an additional feature, providing opportunities to connect antibodies, polymers, dyes, and receptors to the silica surface.5 There are several methods available for the synthesis of silica NPs, in which the Stober method is the most commonly used. In this method, monodisperse silica spheres are grown via the controlled hydrolysis and condensation of tetraethyl orthosilicate (TEOS) in the presence of ammonia as a catalyst.6 The particles obtained in alkaline media are non-luminescent in nature. Organic or inorganic fluorophores can be entrapped within the silica matrix or framework via noncovalent interactions.7–9 The dye-loaded silica NPs are less toxic and more resistant against photobleaching compared to the corresponding free dyes.10 In addition, the outer surface of the dye-loaded NPs can be used for further functionalization.11 Dyed silica NPs emitting throughout the UV-visible to the NIR region are currently available, but their high tendency to release some of the entrapped fluorophores and photobleaching prevent their use in vivo for long term applications.12 Hybrid inorganic materials, such as quantum dot-7 or lanthanide-13 loaded silica NPs, have been used as photostable alternatives to dyed NPs. However, their practical applications, particularly in vivo, remain limited in view of their tedious, multistep synthesis and concerns related to their toxicity.14
Jakob et al. reported the synthesis of strongly emissive monodisperse silica spheres by calcination of aminopropyl silica obtained by co-mixing of TEOS and aminopropyltriethoxy silane (APTES) in an ammonia/water/ethanol solution.15 The optical properties of the resulting silica spheres vary, depending on the calcination temperature and the APTES to TEOS molar ratio. The highest PL QY (12%) with an emission maximum at 430 nm was achieved when a fraction of APTES was kept low (3%) and the calcination temperature was set at 400 °C. Interestingly, silica spheres prepared without APTES were almost non-luminescent. Later, Brites et al. reported a low temperature (45 °C) preparation of organosilica NPs using a modified Stober method using TEOS and APTES as the precursor.16 The resulting NPs emitted light between 430 and 500 nm with PL QY up to 15%. White light emitting silica nanostructures were obtained via acid catalyzed sol gel reaction of TEOS.17 In addition, Mahtab et al. reported the synthesis of magnetic silica NPs with green PL emission and a PL QY of around 7.1%.18 Here, we report a new synthesis of amine-functionalized silica NPs 1 to 2 nm in diameter via a solvothermal reaction between SiBr4 and APTES at 200 °C. The resulting NPs disperse readily in water, yielding colloidally stable dispersions that resist aggregation and photobleaching upon storage under bright ambient conditions for several months. The NPs emit an intense blue light with a PL QY around 34%, the highest QY value reported so far for silica NPs. These highly efficient, non-photobleaching, biocompatible blue-luminescent NPs outperform commercially available products used in light harvesting applications and pharmacokinetic measurements and, in view of their simple synthesis, could eventually replace them.
2. Materials and method
2.1. Materials
Water was purified and deionized using a Sartorius (Arium 611 UV) water purification system. Aminopropyltriethoxysilane (APTES, 99%) and silicon tetrabromide (SiBr4, 99.995%) were obtained from Sigma Aldrich and used as received. All other chemicals were purchased from Wako pure chemical industries Ltd (Japan) and used as received. Trypan blue and Dulbecco's modified Eagle’s medium (DMEM) were purchased from Gibco. The CCK-8 kit was received from Dojindo Molecular Technologies (Japan). NIH3T3 and Hek 293 cell lines were collected from the Riken Cell Bank (Japan). Fetal bovine serum (FBS) was purchased from Sigma Aldrich. Trypsin–EDTA (0.05%) and penicillin–streptomycin were purchased from Life Technologies.
2.2. Synthesis of silica NPs
A stirred mixture of ethylene glycol (10 mL) and water (5 mL), kept under a flow of argon at 25 °C was treated sequentially with SiBr4 (100 μL, 0.805 mmol) and APTES (3 mL, 0.128 mol). The solution was stirred for 30 minutes. The resulting milky suspension was transferred in a 30 mL-Teflon container. The sealed container was inserted in a stainless steel autoclave. The autoclave was brought to 200 °C with a heating rate of 4 °C min−1, kept at this temperature for 15 h and brought back to room temperature. The resultant yellowish-green solution was dispersed in water. The product was purified by dialysis (Spectra/Por Dialysis Membrane, MWCO: 3500) in deionized water for 15 h to remove ethylene glycol and other impurities. The NPs precipitated during dialysis. They were collected by centrifugation (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 20 min), washed with water (100 mL) several times, and dried under vacuum. The powder was insoluble in neutral water but was readily soluble in aqueous NaOH (1 N), yielding a solution having the same UV-visible absorption spectrum as the crude product, but a significantly reduced PL. Control syntheses were performed under the same protocol using either SiBr4 (100 μL, 0.805 mmol) alone or APTES (3 mL and 100 μL) alone dissolved in 10 mL of ethylene glycol in 5 mL of water and heated at 200 °C for 15 h, all other conditions being kept unaltered.
000 rpm, 20 min), washed with water (100 mL) several times, and dried under vacuum. The powder was insoluble in neutral water but was readily soluble in aqueous NaOH (1 N), yielding a solution having the same UV-visible absorption spectrum as the crude product, but a significantly reduced PL. Control syntheses were performed under the same protocol using either SiBr4 (100 μL, 0.805 mmol) alone or APTES (3 mL and 100 μL) alone dissolved in 10 mL of ethylene glycol in 5 mL of water and heated at 200 °C for 15 h, all other conditions being kept unaltered.
2.3. Characterization
Fourier Transform Infrared (FTIR) spectroscopy measurements were carried out on a JASCO FT/IR 4100 spectrometer, using powder samples placed in the sample holder. UV-visible absorption spectra were recorded on a JASCO V-650 spectrophotometer. X-ray photoelectron spectroscopy (XPS) was performed using a Shimadzu, ESCA3400 using MgKα (E = 1253.6 eV) radiation. The binding energy (BE) scale was calibrated to provide Pt4f7/2 = 71.1 eV. The X-ray source was operated at 10 mA and 12 kV. The core-level signals were obtained with a photoelectron take-off angle of 90° with respect to the sample surface. Data acquisition and processing were performed by a SUN Microsystems ULTRA 5 computer, using the VISION 2.0 processing package. The accuracy of the BE determined with respect to the standard value was within ±0.3 eV. X-ray diffraction (XRD) was carried out with a Rigaku Smart lab X-ray diffractometer, operated in ambient air. Dynamic light scattering (DLS) and zeta potential measurements were performed on aqueous NP dispersions kept at 25 °C using an ELSZ-2000 zeta-potential and particle size analyzer (Otsuka Electronics Co., Ltd). High resolution transmission electron microscopy (HR-TEM) and selected area electron diffraction (SAED) pattern measurements were performed with a JEOL JEM 2010, operated at an acceleration voltage of 200 kV. Samples for HR-TEM analysis were drop-cast on a copper grid from a very dilute aqueous particle suspension. PL and PLE spectra of aqueous silica NP dispersions were recorded using a NanoLog Horiba Jovin Yvon spectrofluorometer. Time-resolved fluorescence decay profiles were obtained with a time-correlated single photon counting (TCSPC) lifetime spectroscopy system (NanoLog, Horiba Jovin Ybon, Japan), equipped with pulse laser diodes (λem = 370 nm). The quality of the fit was assessed based on the χ2 value (∼1.0) and visual inspection of the residuals. Absolute PL QYs were estimated by the standardized integrating-sphere method using a Hamamatsu C9920-02 Photonics system, equipped with a 150 W xenon lamp. Dilute aqueous solutions with an absorbance in between 0.1 and 0.2 at 304 nm wavelength were taken in 10 mm2 quartz cuvettes and were placed into the instrument sample compartment.
2.4. Cytotoxicity assays
For cellular assays, silica NP suspensions were prepared by dilution of the particles in water (pH = 7.0).
Trypan blue test.
Fibroblasts non-cancerous murine cells (NIH3T3) were seeded in a 24-wells plate containing the DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 units per mL penicillin, and 100 mg mL−1 streptomycin at 37 °C in humidified air contains 5% CO2. After 1 day, the cells were treated with increasing concentrations of silica NP (25, 50, 100, 200, 300 and 500 μg mL−1) and further incubated for 24 h. The cells were washed thrice with PBS and trypsinized. Trypan blue (0.4% wt/vol, 100 μL) was added to an aliquot of the cell suspension (100 μL) and incubated for 5 min at room temperature. The viable cells were counted by bright field microscopy. The viability values of the silica NP treated cells are expressed as a percentage of the viability of control cells. All experiments were repeated 3 times.
CCK-8 assay.
The viability of the NIH3T3 cells was checked using a CCK-8 kit (Dojindo Molecular Technologies, Japan) according to the manufacturer’s instructions. Briefly, a NIH3T3 cell suspension (100 μL, 5000 cells per well) was dispersed in a 96-well plate containing DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 units per mL penicillin, and 100 mg mL−1 streptomycin. The plate was pre-incubated at 37 °C in humidified air containing 5% CO2 for 1 day. Then, silica NP suspensions (10 μL) were added into the wells to final concentrations of 25, 50, 100, 200, 300, and 500 μg mL−1. The plate was incubated for 24 h under the same conditions mentioned above. A CCK-8 solution (10 μL) was added to each well and the plate was incubated for 4 h in the incubator. Finally, the absorbance at 450 nm of each well was measured using an MTP-800 microplate reader (Corona Electric).
2.5. Cellular uptake and confocal microscopy
NIH3T3 cells were plated on glass cover slips at a concentration of 105 cells per mL. They were treated with the particle suspension in water (100 μg mL−1) and incubated for 24 h. They were washed thrice with PBS buffer (5 minute each time) and fixed with paraformaldehyde. Cells were imaged on a confocal microscope (TCS-SP5 confocal microscope, Leica Microsystems, λex = 405 nm).
3. Results and discussion
3.1. Synthesis and characterization of Silica NPs
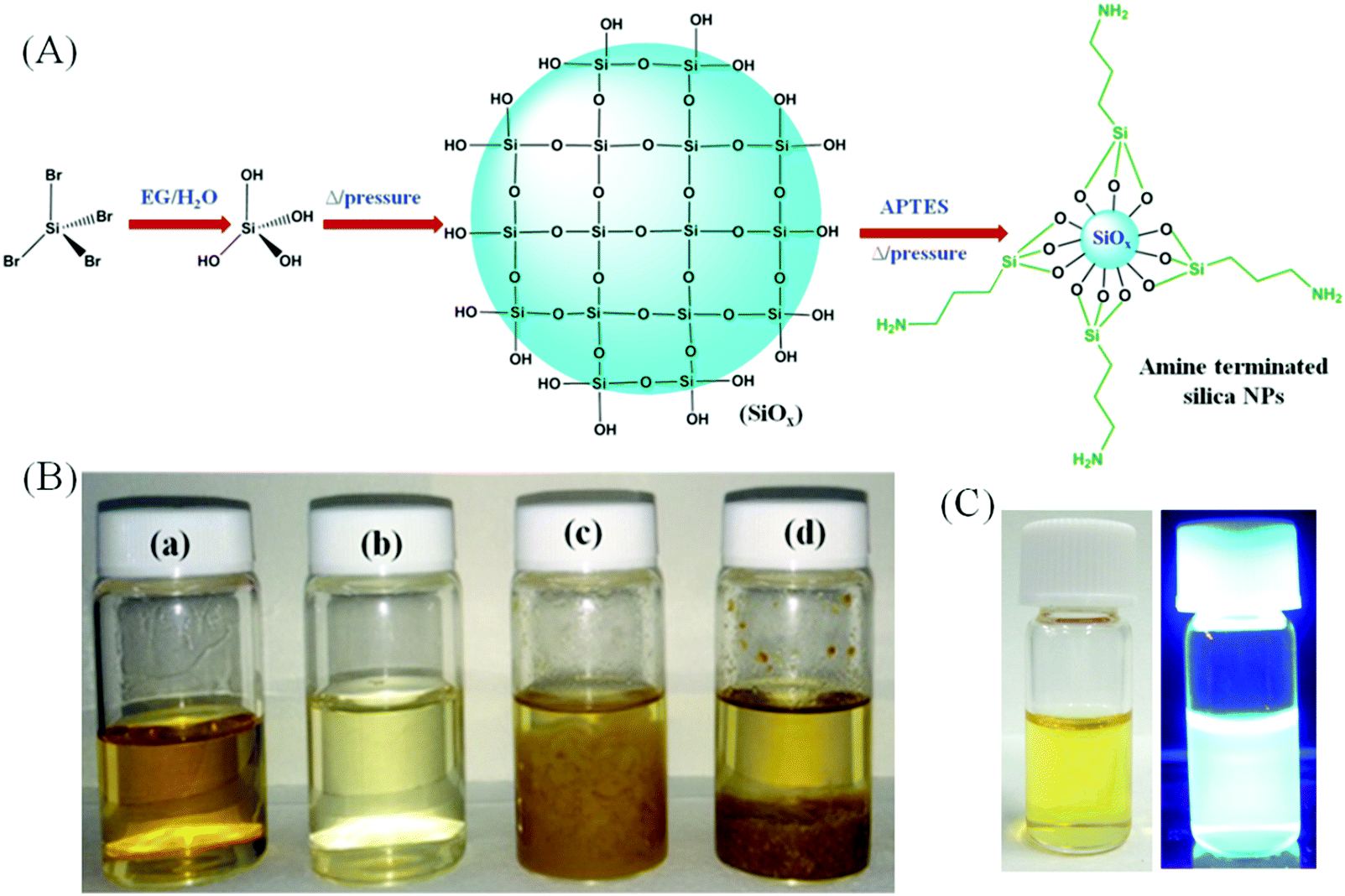
We report a solvothermal approach to obtain amorphous silica NPs through the reaction between SiBr4 and APTES in water as depicted schematically in Fig. 1A. It is well recognized that SiBr4 is readily hydrolyzed in contact with water to form Si(OH)4, eventually converted into silicon oxide. APTES has a strong tendency to form a covalently-bound monolayer on chemically grown silicon oxide.19,20 Thus, in this synthesis, SiBr4 immediately hydrolyzes to Si(OH)4 in the presence of an ethylene glycol (EG)/water mixture and, when kept under pressure at high temperature, it polymerizes to form –(Si–O)x stabilized in water by hydrolyzed APTES bound to the surface. The APTES grafting stops the growth of –(Si–O)x and introduces amine groups tethered to the –(Si–O)x surface. The resulting colloidal NPs emit an intense blue luminescence under UV irradiation. Their aqueous dispersions resist precipitation even after centrifugation at 100000 rpm.
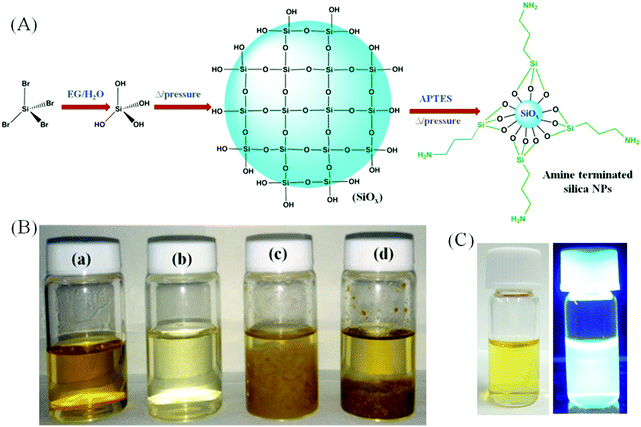 |
| | Fig. 1 (A) Schematic representation for the formation of silica NPs. (B) Photographs of the products, prepared by using (a) SiBr4 and APTES in the EG/water system, (b) SiBr4 alone in the EG/water system, (c) APTES alone (100 μL) in the EG/water system and (d) APTES alone (3 mL) in the EG/water system. (C) Photographs of silica NPs in water under (a) white light and (b) UV light. | |
The validity of the mechanism proposed is strengthened by our observations that the same procedure performed with either only SiBr4 or only APTES fails to produce emissive silica NPs, yielding exclusively non-emissive macroscopic aggregates. Fig. 1B presents photographs of aqueous dispersions of the products in water obtained using APTES and SiBr4 together or separately under the same conditions. Silica NPs form only when the reaction is conducted with both APTES and SiBr4 in a water/EG system. The resulting sample (vial a) is a clear liquid with a yellowish-green hue. However, when the reaction is executed with SiBr4 alone, a colorless liquid is formed together with some yellow precipitate (vial b). Using APTES alone, the process leads to the formation of a yellow liquid and a brownish floc, irrespective of the starting amount of APTES (vials c and d). Fig. 1C demonstrates that the silica NPs emit bright blue luminescence under UV irradiation.
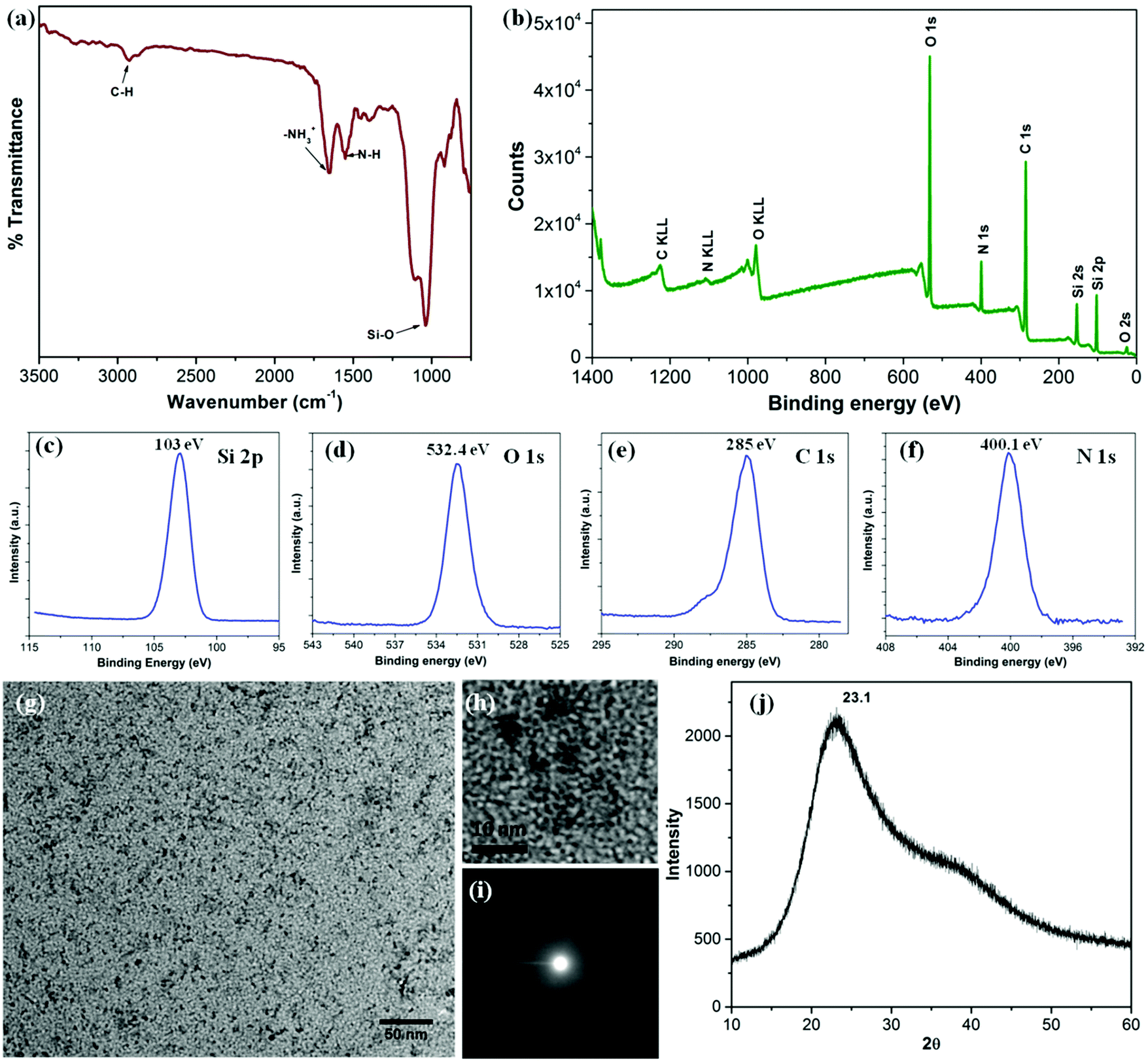
The silica nanoparticles were analyzed using FTIR spectroscopy and XPS in order to ascertain their composition and surface chemistry (Fig. 2). The FTIR spectrum shown in Fig. 2a presents a strong band at 1040 cm−1 characteristic of the Si–O linkage as well as a band at 1552 cm−1, typical of the amine vibrations. The presence of both bands is consistent with the mechanism proposed above, which involves first the formation of –(Si–O)x from SiBr4 followed by the reaction of –(Si–O)x with hydrolyzed APTES.21 The sharp band at 1655 cm−1 is assigned to the asymmetric –NH3+ deformation mode. The water molecules retained by siliceous materials could also account for this band. A small band at 2930 cm−1, assigned to C–H bond vibrations provides further evidence for the presence of APTES moieties.19,21 The presence of functional groups on the outer surface of the silica NPs and the mode of bonding in between the elements was confirmed by analysis of the XPS spectra shown in Fig. 2b. The wide XPS scan displays signals due to carbon (53.2%), oxygen (28.1%), silicon (10.6%) and nitrogen (8.1%). The prominent peaks for Si2s, Si2p, O1s, C1s and N1s are consistent with the formation of silica NPs bearing hydrolyzed APTES molecules on their surface. The Si2p spectrum (Fig. 2c) consists of a distinct peak centered at 103 eV, characteristic of Si–O bonds.22 The C1s peak (Fig. 2e) centered at 285 eV supports the existence of the C–C bonds of the hydrolyzed APTES moieties. Finally, the N1s spectrum (Fig. 2f) at 400.1 eV confirms the presence of primary amine N–H bonds.23Fig. 2g and h present low and high resolution TEM micrographs of the silica NPs, which are spherical in shape with diameters of 1–2 nm. The NPs are very small in size. They are uniformly distributed without signs of agglomeration. The corresponding SAED pattern (Fig. 2i) reveals that they are amorphous in nature, as confirmed by the X-ray diffraction pattern shown in Fig. 2j which presents a broad featureless signal centered at 2θ = 23.1. The average NP size calculated from the diffraction peak is around 1.2 nm, very close to the result obtains from TEM image. The hydrodynamic diameter of the NPs in water, determined by DLS, was 1.2 ± 0.2 nm, in excellent agreement with the TEM and XRD data (Fig. S1, ESI†). The zeta potential values of the particles dispersed in aqueous media of pH < 7 were positive in agreement with the presence of surface –NH3+ functionalities (Fig. S2, ESI†). Low positive zeta potential with very high stability in an aqueous medium could be explained by the presence over the surface of the silica NPs, of tethered EG groups responsible for charge shielding and preventing their agglomeration in solution.24,25
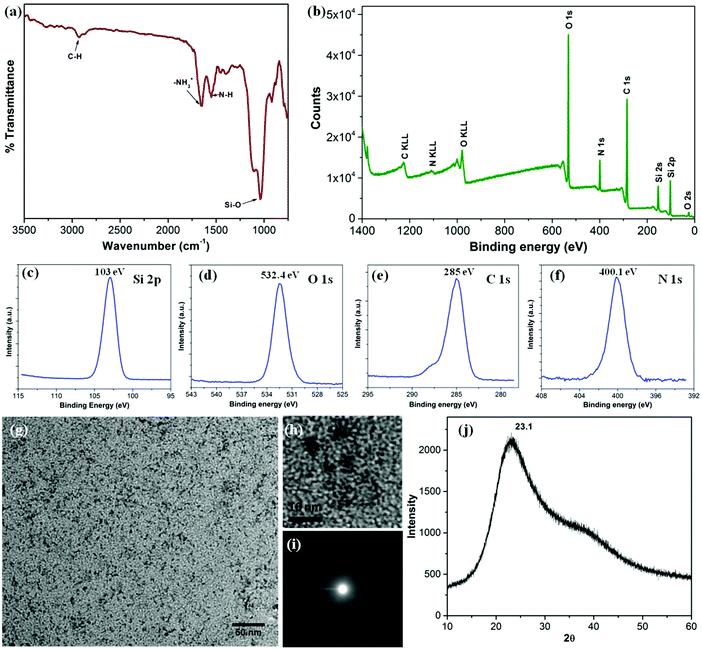 |
| | Fig. 2 (a) FTIR spectrum and (b) wide scan XPS spectrum of the silica NPs; the enlarged regions of the XPS spectrum of Si2p (c), O1s (d), C1s (e) and N1s (f). (g) TEM image, (h) HR-TEM image, (i) SAED pattern and (j) X-ray diffraction pattern of the silica NPs. | |
3.2. Optical properties
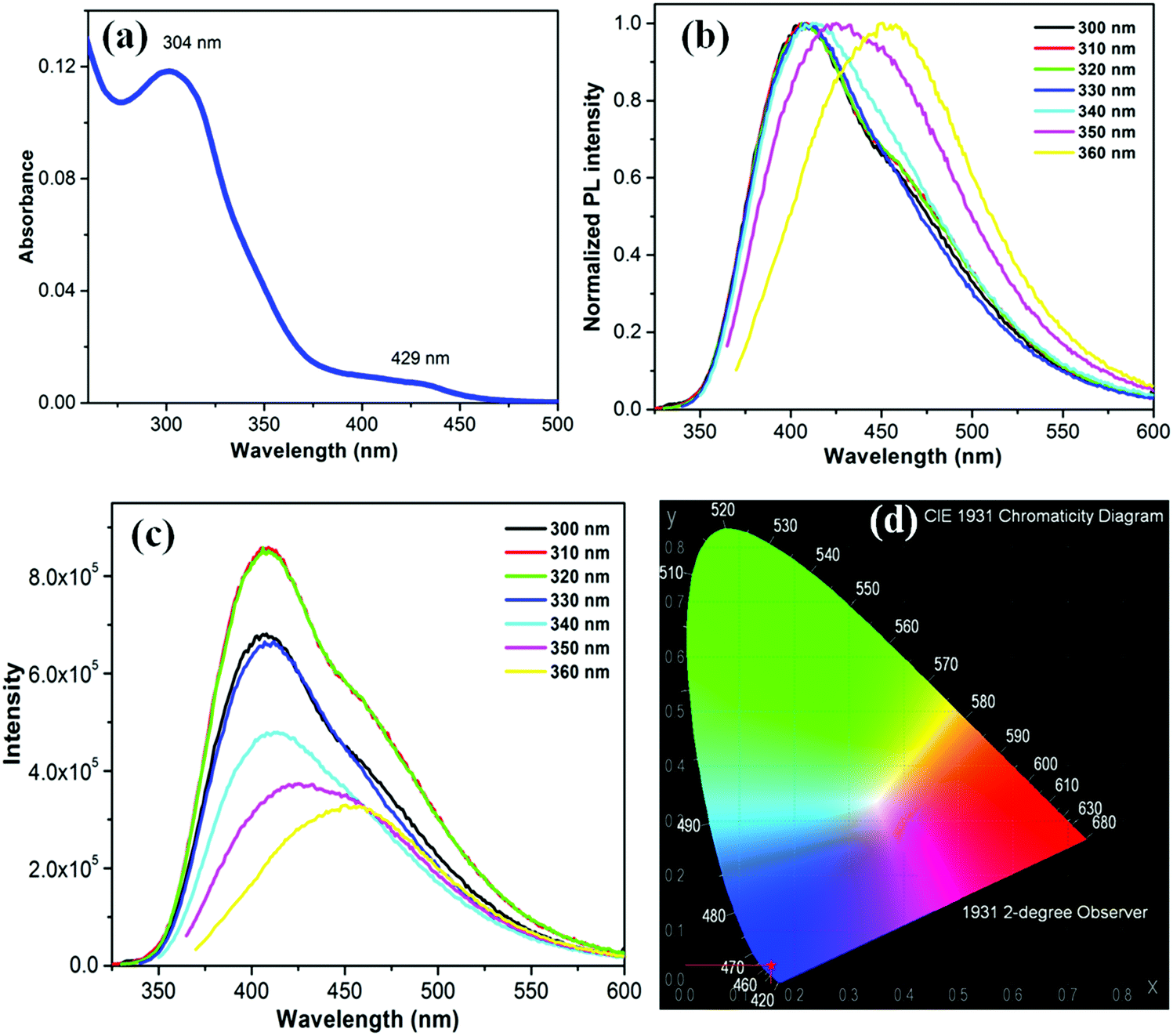
The UV-Vis absorption spectrum of the as-synthesized bright greenish yellow aqueous silica NPs dispersion in water consists of two distinct bands: a strong band centered at 304 nm (major) and a weak band around 429 nm (Fig. 3a). This strong and broad absorption permits excitation over a long wavelength range26 as shown in Fig. 3b, which presents the emission spectra of the silica NPs in water using excitation wavelength from 300 to 360 nm. The emission undergoes red shift with increasing excitation wavelength. This shift may be due to the presence of emissive traps associated with the oxygen or amine functionalities on the silica surface.27 The highest emission intensity, centered at 410 nm, was recorded upon excitation at 310 nm (Fig. 3c). The estimated PL full width half maximum (FWHM) is about 105 nm. The PL QY of this silica NP excited at 310 nm is approximately 34.3%, which is the highest value reported so far for emissive silica NPs. The PL QY value is about 31.2%, 34%, 27.1%, 25.3%, 24.5% and 23.2% upon excitation at 300, 320, 330, 340, 350 and 360 nm, respectively (Fig. S3, ESI†). The PL intensity is not influenced by changes in the pH of the medium from 1 to 14 (Fig. S4, ESI†). The PL QY of silica NP remained constant over the entire range. The emission underwent a small red shift for samples at pH 1. The NPs are highly photostable. No photobleaching was detected upon UV irradiation (4 Watt, 365 nm) over an 8 h period (Fig. S5, ESI†). The Commission Internationale de I'Eclairage (CIE) diagram shown in Fig. 3d, reveals the CIE coordinates (1931) x = 0.15262, y = 0.03345, corresponding to the blue region of the visible light. The origin of this blue luminescence is still a matter of debate as discussed in the next section.
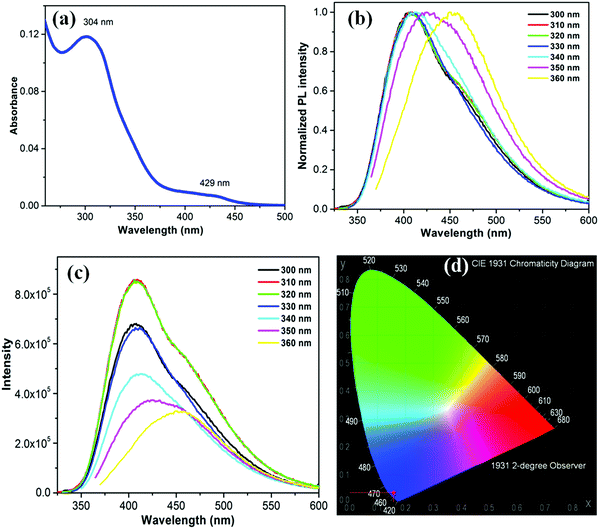 |
| | Fig. 3 (a) UV-visible spectrum, (b) normalized PL spectra at different excitation wavelength and (c) corresponding PL spectra of silica NP. (d) CIE diagram of silica NP, showing chromaticity coordinates of the sample. | |
3.3. Emission mechanism
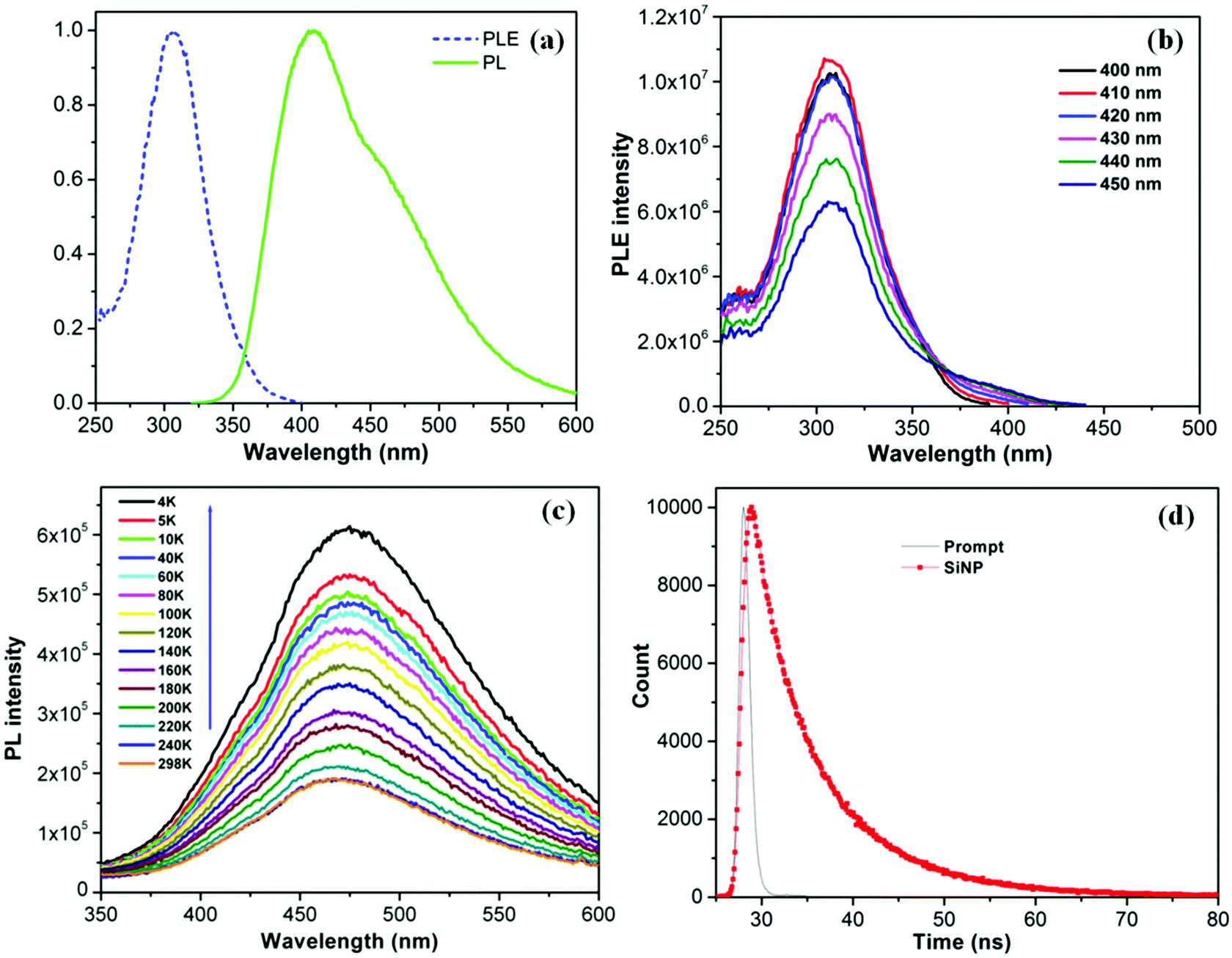
In order to determine the origin of this blue emission, we have carried out an extensive photophysical analysis that included PLE spectroscopy, low temperature PL emission spectroscopy, and TCSPC lifetime decay analysis. Fig. 4a displays the normalized PL and PLE spectra of the silica NP obtained, respectively, with an excitation wavelength of 310 nm and an emission wavelength of 410 nm. The PL spectrum is centered at 410 nm and the PLE spectrum has a maximum at 308 nm, confirming that the broad emission band corresponds to the PLE excitation energy.28,29 The highest PLE intensity has been achieved with a 410 nm emission. The PLE intensity gradually diminishes as the emission wavelength increases from 410 to 450 nm (Fig. 4b), indicating that the PL efficiency is a linear function of the PLE excitation. Interestingly, the PLE spectrum remains centered at 308 nm (Fig. S6, ESI†), irrespective of the emission wavelength monitored. Moreover the PLE spectra coincide well with the UV-visible absorption spectra of the NPs. The PL intensity of solid NPs deposited on quartz decreases with increasing temperature from 4 K to 298 K (Fig. 4c). Unlike the large red shifts in energy gap observed for crystalline particles,30 silica NPs exhibit a slight blue shift of 34 meV as the temperature passes from 4 K to 298 K. This shift is attributed to the enhanced interaction of electron–phonon scattering with increasing temperature.31 The result suggests that electron–phonon interactions and electron–surface defect scattering play an important role in the PL properties of the very small sized silica NPs, as discussed by Kwack et al.32 At high temperatures, several nonradiative channels are thermally activated due to the formation of surface trapping states caused by surface defects caused by impurities, such as carbon and oxygen atoms, and by the nitrogen centers on the surface.30,33,34 It has been reported that the structural disorder associated with the amorphous state enhances PL efficiency in amorphous particles.32,35 The PL lifetime decay profile of silica NP (Fig. 4d) was fitted to a bi-exponential function, with lifetimes τ1 = 3.97 ns and τ2 = 10.19 ns; the average life time is 4.27 ns. The local atomic distortions on the electronic structure due to the insertion of oxygen in amorphous silicon oxide are believed to be responsible for the high radiative recombination rate.36,37
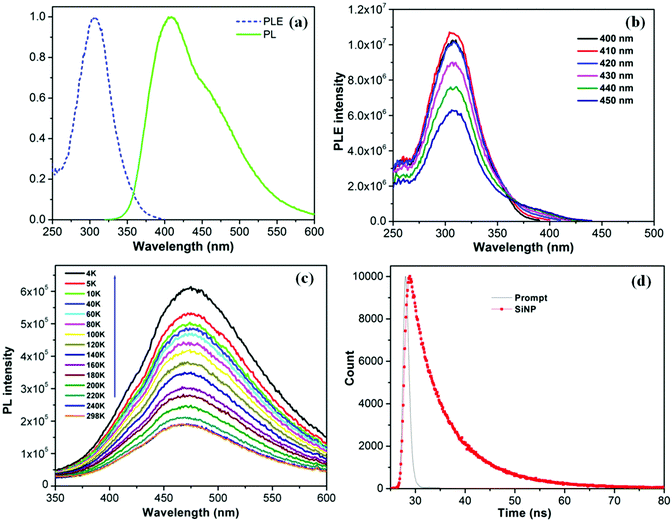 |
| | Fig. 4 (a) Normalized PLE and PL spectrum of silica NPs at 410 nm emission and 310 nm excitation respectively, (b) PLE spectrum of silica NPs at different emission wavelength. (c) Temperature dependent PL emission of silica NPs, deposited on a quartz surface. (d) TCSPC life time decay profile of the silica NPs. | |
3.4.
In vitro cytotoxicity and bio-imaging
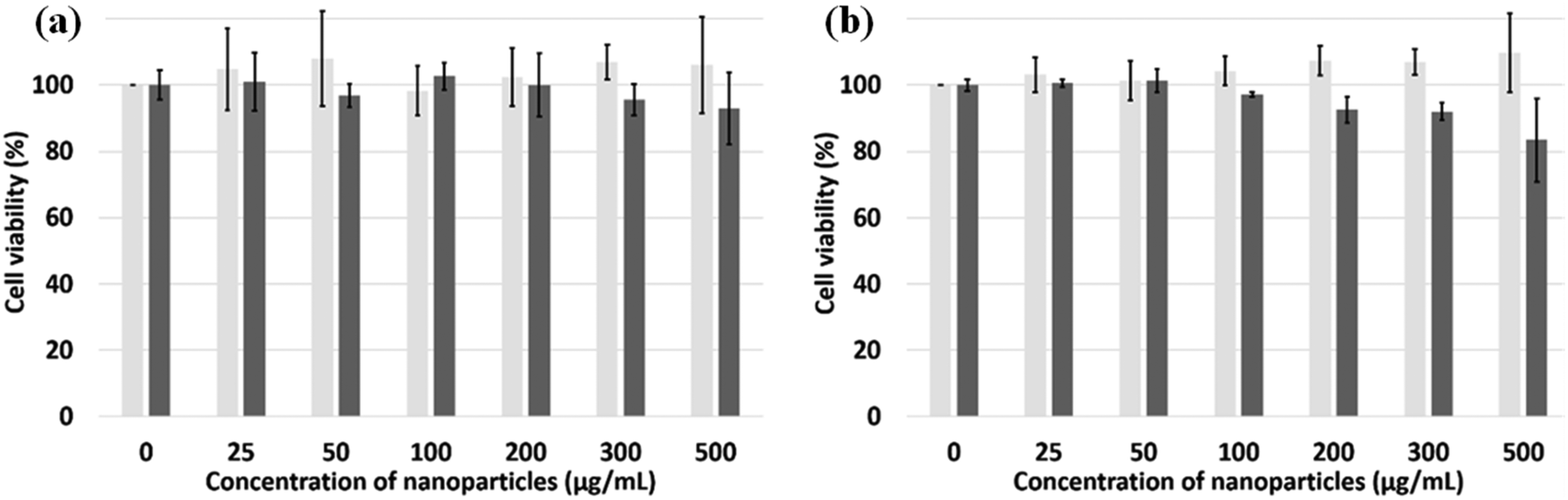
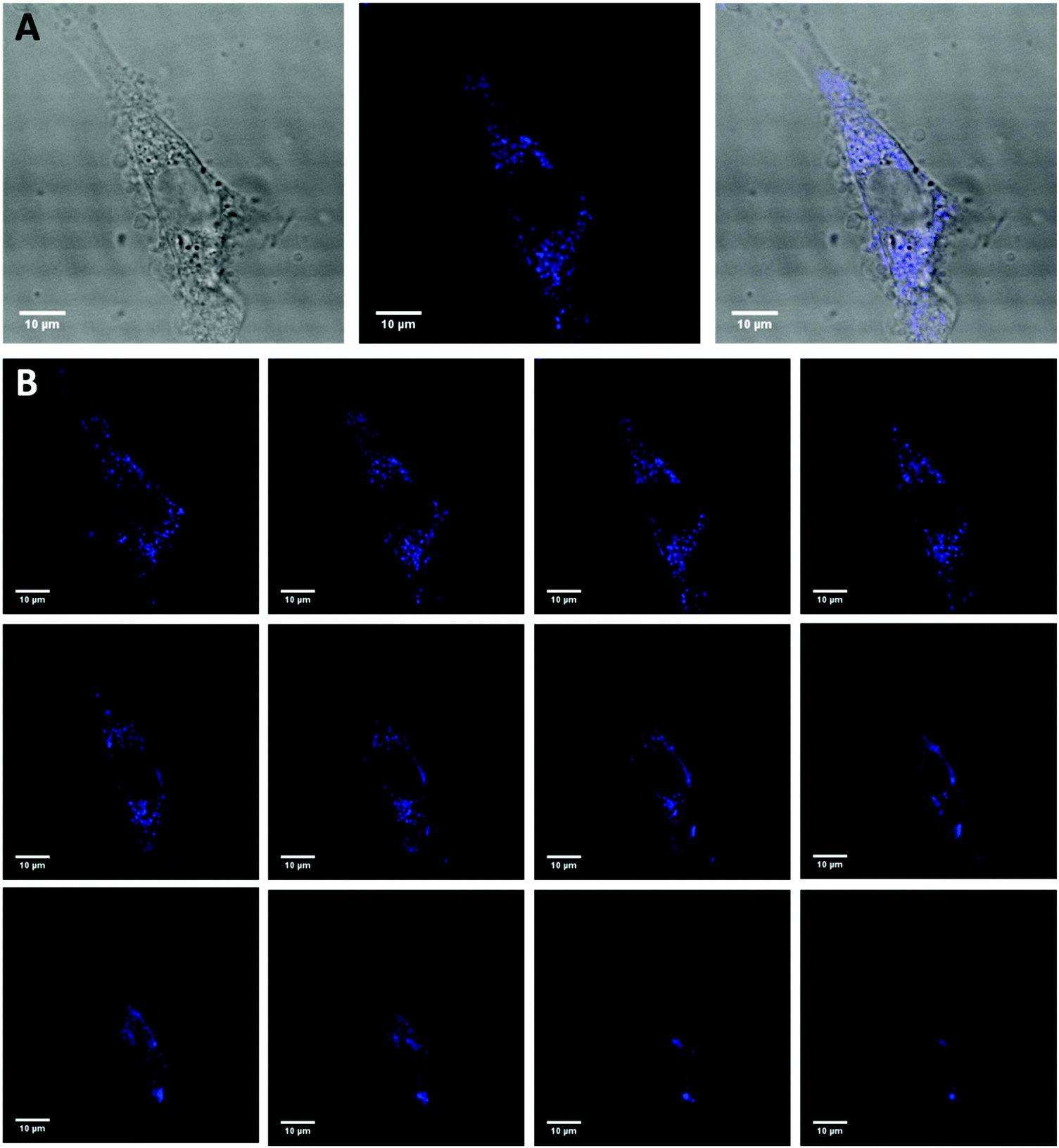
CCK-8 and trypan blue tests were performed on two cell lines, Hek-293 and NIH3T3, to assess the silica NPs cytotoxicity.38 The experimental results of both assays (Fig. 5a and b) indicate that the NPs are non toxic up to the concentration of 500 μg mL−1 for the Hek293 and NIH3T3 cell lines. To ascertain the intracellular uptake of these NPs, fluorescence imaging of NP-treated cells were carried out on NIH3T3 mouse embryonic fibroblast cells (Fig. 6A). After a 24 h-incubation with 100 μL (100 μg mL−1) of NPs, they are located in the cell cytoplasm and emit intense blue fluorescence light. Z-stack images in NIH3T3 cells shown in Fig. 6B confirm that the NPs are equally distributed over the interior of the cells. After insertion, the particles tend to accumulate at the edge of the nucleus but not inside.
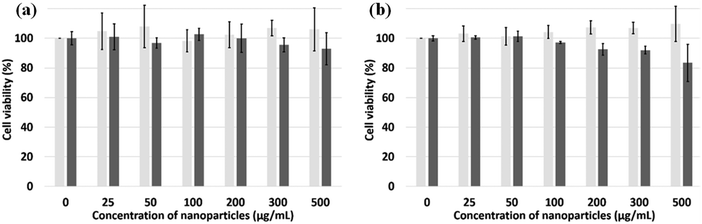 |
| | Fig. 5 Cell viability using CCK-8 assay (light grey, n = 6) and the trypan blue test (dark grey, n = 3). (a) Hek293 and (b) NIH3T3 cells incubated with silica NPs for 24 hours. | |
 |
| | Fig. 6 Confocal microscopy of NIH3T3 cells incubated with silica NP for 24 h in serum containing media (DMEM + 10% FBS) and then fixed with paraformaldehyde. (A) Differential interference contrast (DIC) image (left), confocal fluorescence image (middle) and overlapped (right) image of a cell representative of the experiment. (B) Individual Z-stacks (1.0 μm intervals) of the same cell are shown. | |
4. Conclusions
We prepared amine functionalized water-dispersible silica NPs 1–2 nm in diameter. The NPs emit intense blue PL without photobleaching. An elevated rate of radiative recombination due to localized surface states present in amorphous silica is believed to be responsible for the NPs high PL intensity with QY > 34%. The NPs are non-toxic. They are taken up by cells where they are readily detected by their bright blue luminescence upon excitation at 405 nm. The NPs should find applications in various fields in view of the ease of their preparation, their outstanding colloidal and photochemical stability, and their high PL QY.
Acknowledgements
This study was supported by the WPI-Program and NIMS Molecule & Material Synthesis Platform in “Nanotechnology Platform Project” both operated by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. NS would like to thank Kakenhi (No. 26390024) of MEXT, and the Sumitomo Foundation.
References
- X. He, H. Nie, K. Wang, W. Tan, X. Wu and P. Zhang, Anal. Chem., 2008, 80, 9597–9603 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, R. C. Huxford-Phillips and W. Lin, Chem. Soc. Rev., 2012, 41, 2673–2685 RSC.
- H. Ow, D. R. Larson, M. Srivastava, B. A. Baird, W. W. Webb and U. Wiesner, Nano Lett., 2005, 5, 113–117 CrossRef CAS PubMed.
- H. M. Liu, S. H. Wu, C. W. Lu, M. Yao, J. K. Hsiao, Y. Hung, Y. S. Lin, C. Y. Mou, C. S. Yang, D. M. Huang and Y. C. Chen, Small, 2008, 4, 619–626 CrossRef CAS PubMed.
- K. M. L. Taylor-Pashow, J. Della Rocca, R. C. Huxford and W. Lin, Chem. Commun., 2010, 46, 5832–5849 RSC.
- W. Stober, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- Y. Chan, J. P. Zimmer, M. Stroh, J. S. Steckel, R. K. Jain and M. G. Bawendi, Adv. Mater., 2004, 16, 2092–2097 CrossRef CAS.
- R. P. Bagwe, C. Yang, L. R. Hilliard and W. Tan, Langmuir, 2004, 20, 8336–8342 CrossRef CAS PubMed.
- A. Van Blaaderen and A. Vrij, Langmuir, 1992, 8, 2921–2931 CrossRef CAS.
- X.-H. Wang, A. R. Morales, T. Urakami, L.-F. Zhang, M. V. Bondar, M. Komatsu and K. D. Belfield, Bioconjugate Chem., 2011, 22, 1438–1450 CrossRef CAS PubMed.
- L. Xue, B. Li, Q. Fei, G. Feng, Y. Huan and Z. Shi, Nanotechnology, 2010, 21, 215502 CrossRef PubMed.
- A. Burns, H. Owand and U. Weisner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- L. D. Carlos, R. A. S. Ferreira, V. de Zea Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509–534 CrossRef CAS PubMed.
- B. Prasad, N. Nikolskaya, D. Connolly, T. Smith, S. J. Byrne, V. Gerard, Y. Gun'ko and Y. Rochev, J. Nanobiotechnol., 2010, 8, 1 CrossRef PubMed.
- A. M. Jakob and T. A. Schmedake, Chem. Mater., 2006, 18, 3173–3175 CrossRef CAS.
- C. D. S. Brites, V. T. Freitas, R. A. S. Ferreira, A. Millan, F. Palacio and L. D. Carlos, Langmuir, 2012, 28, 8190–8196 CrossRef CAS PubMed.
- G. L. Davies, J. E. McCarthy, A. Rakovich and Y. K. Gunko, J. Mater. Chem., 2012, 22, 7358–7365 RSC.
- F. Mahtab, Y. Yu, J. W. Y. Lam, J. Liu, B. Zhang, P. Lu, X. Zhang and B. Z. Tang, Adv. Funct. Mater., 2011, 21, 1733–1740 CrossRef CAS.
- N. H. N. Kamarudin, A. A. Jalil, S. Triwahyono, N. F. M. Salleh, A. H. Karim, R. R. Mukti, B. H. Hameed and A. Ahmad, Microporous Mesoporous Mater., 2013, 180, 235–241 CrossRef CAS.
- V. R. Rai and S. Agarwal, Chem. Mater., 2011, 23, 2312–2316 CrossRef CAS.
- R. M. Pasternack, S. R. Amy and Y. J. Chabal, Langmuir, 2008, 24, 12963–12971 CrossRef CAS PubMed.
-
(a) B. Ghosh, Y. Masuda, Y. Wakayama, Y. Imanaka, J. Inoue, K. Hashi, K. Deguchi, H. Yamada, Y. Sakka, S. Ohki, T. Shimizu and N. Shirahata, Adv. Funct. Mater., 2014, 24, 7151–7160 CAS;
(b)
J. F. Moulder, W. F. Stickle, P. E. Sobel and K. D. Bomben, Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, Perkin-Elmer Corporation, Eden Prairie, MN, 2nd edn, 1992 Search PubMed.
- B. M. Gallant, X. W. Gu, D. Z. Chen, J. R. Greer and N. S. Lewis, ACS Nano, 2015, 9, 5143–5153 CrossRef CAS PubMed.
- H. Petersen, P. M. Fechner, A. L. Martin, K. Kunath, S. Stolnik, C. J. Roberts, D. Fischer, M. C. Davies and T. Kissel, Bioconjugate Chem., 2002, 13, 845–854 CrossRef CAS PubMed.
- S. Yamamoto, E. Yuba, A. Harada and K. Kono, Langmuir, 2015, 31, 8583–8588 CrossRef CAS PubMed.
- A. Paul and A. Samanta, J. Chem. Sci., 2006, 118, 335–340 CrossRef CAS.
- Y. Yang, J. Cui, M. Zheng, C. Hu, S. Tan, Y. Xiao, Q. Yang and Y. Liu, Chem. Commun., 2012, 48, 380 RSC.
- D. S. English, L. E. Pell, Z. Yu, P. F. Barbara and B. A. Korgel, Nano Lett., 2002, 2, 681–685 CrossRef CAS.
- R. Xie and X. Peng, J. Am. Chem. Soc., 2009, 131, 10645–10651 CrossRef CAS PubMed.
- B. Ghosh, M. Takeguchi, J. Nakamura, Y. Nemoto, T. Hamaoka, S. Chandra and N. Shirahata, Sci. Rep., 2016, 6, 36951 CrossRef CAS PubMed.
- P. Yu, X. Wen, Y. R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 6567–6571 CAS.
- H. S. Kwack, Y. Sun and Y. H. Cho, Appl. Phys. Lett., 2003, 83, 2901–2903 CrossRef CAS.
- K. S. Min, K. V. Shcheglov, C. M. Yang, H. A. Atwater, M. L. Brongersman and A. Polman, Appl. Phys. Lett., 1996, 69, 2033 CrossRef CAS.
- T. Brankova, V. Bekiari and P. Lianos, Chem. Mater., 2003, 15, 1855 CrossRef CAS.
- N. M. Park, T. S. Kim and S. J. Park, Appl. Phys. Lett., 2001, 78, 2575 CrossRef CAS.
- M. J. Estes and G. Moddel, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 14633–14642 CrossRef CAS.
- A. Puzder, A. J. Williamson, J. C. Grossman and G. Galli, J. Chem. Phys., 2002, 117, 6721–6729 CrossRef CAS.
- S. Chandra, B. Ghosh, G. Beaune, U. Nagarajan, T. Yasui, J. Nakamura, T. Tsuruoka, Y. Baba, N. Shirahata and F. M. Winnik, Nanoscale, 2016, 8, 9009–9019 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb02813f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Françoise M.
Winnik
*abc
*a and
Françoise M.
Winnik
*abc